Imbalance of ER and Mitochondria Interactions: Prelude to Cardiac Ageing and Disease?



{kind=link}
{kind=link}
Abstract
:1. Introduction
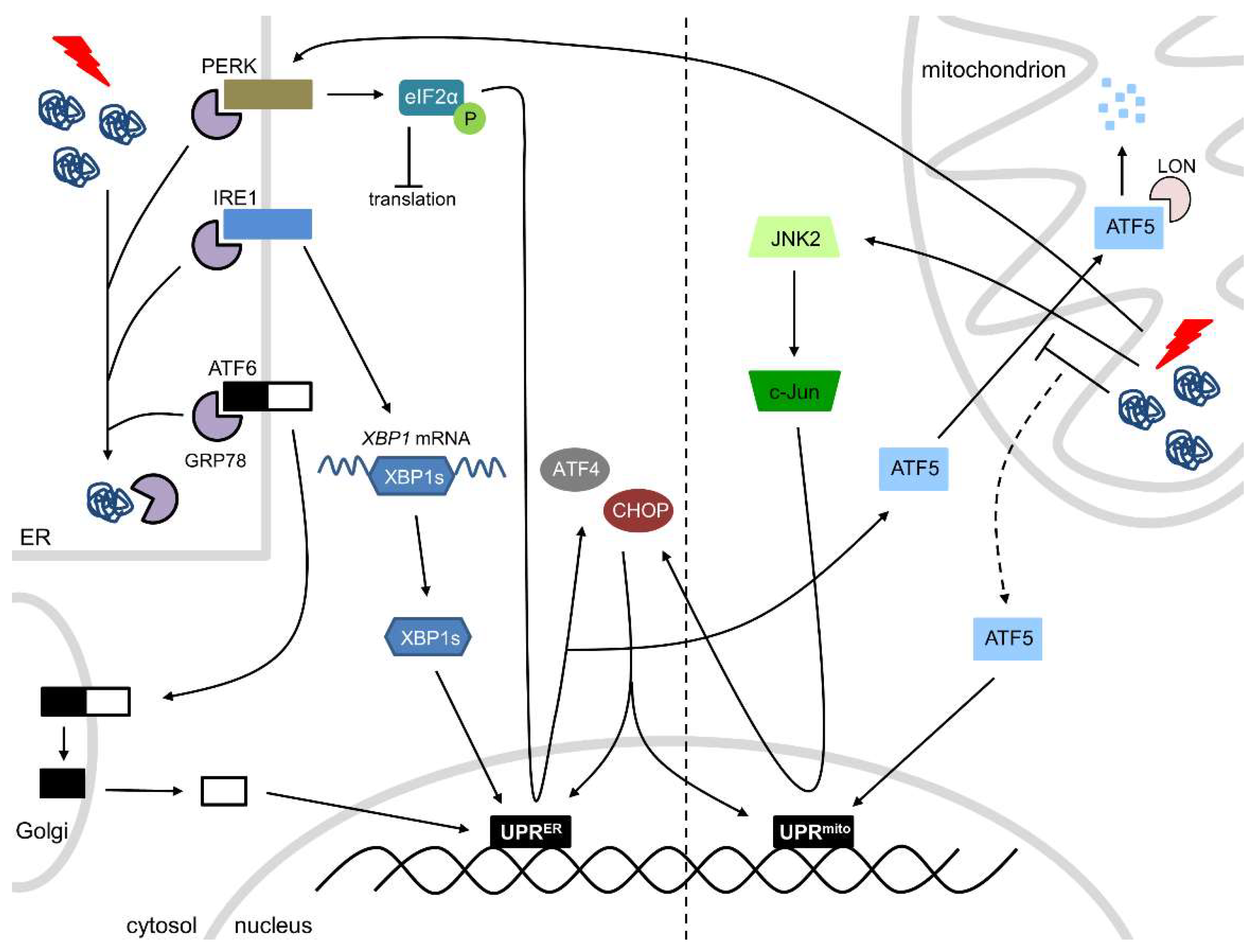
2. Role of the ER in Cardiac Health
3. Role of Mitochondria in Cardiac Health
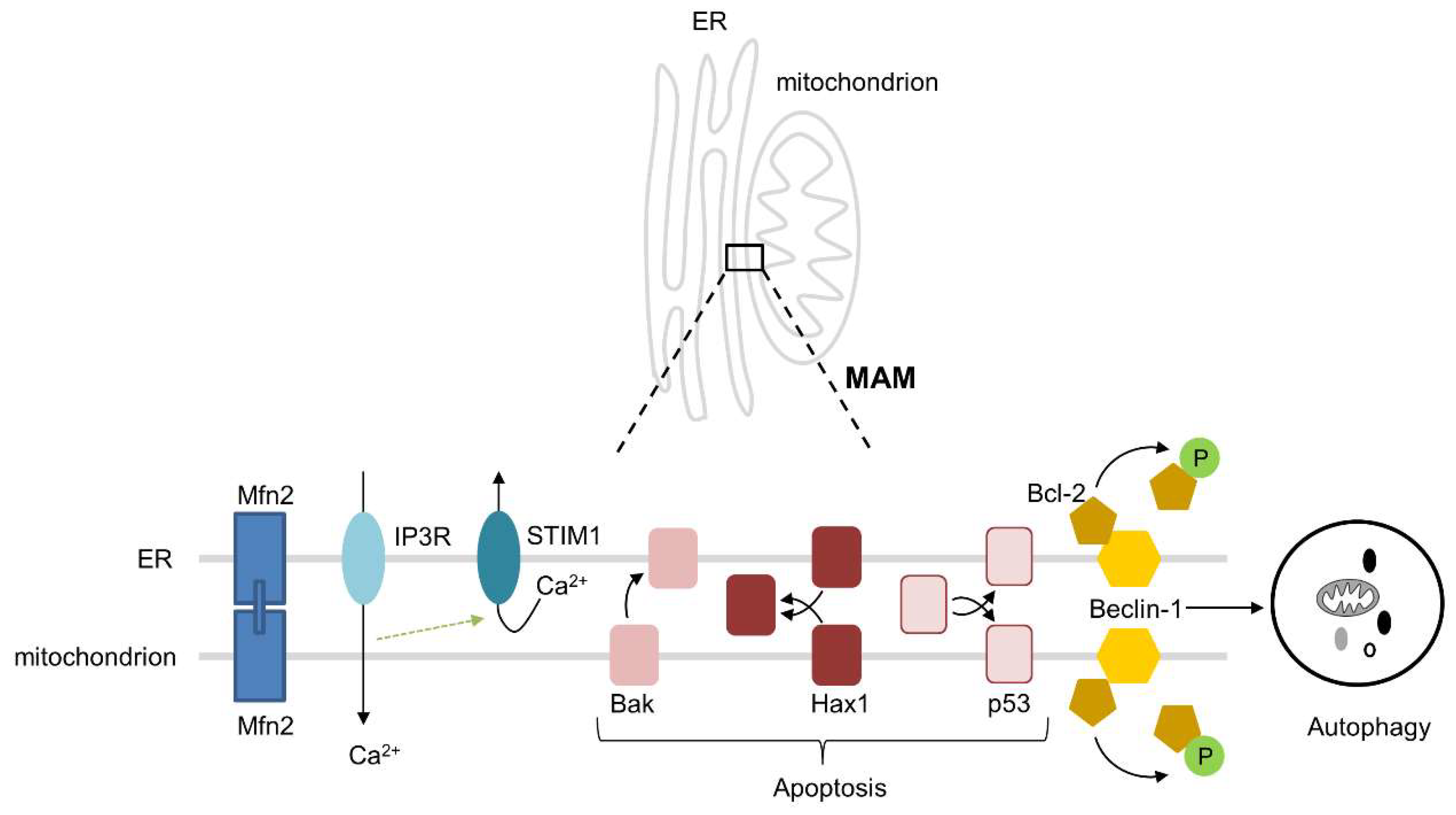
4. Interactions between the ER and Mitochondria
5. Interactions between the ER and Mitochondria in Cardiac Ageing and Cardiac Disease
5.1. Alterations of Interactions between the ER and Mitochondria in Cardiac Ageing
5.2. Alterations of Interactions between the ER and Mitochondria in Myocardial Ischemia/Reperfusion
5.3. Alterations of Interactions between the ER and Mitochondria in AF
5.4. Alteractions of Interactions between the ER and Mitochondria in (Inherited) Cardiomyopathy
5.5. Alterations of Interactions between the ER and Mitochondria in Diabetic Cardiomyopathy
6. ER and Mitochondria Interactions as Therapeutic Targets
6.1. Lifestyle Interventions—Caloric Restriction and Exercise
6.2. Pharmacological Interventions—Improved Interactions between the ER and Mitochondria
7. Conclusions
Funding
Conflicts of Interest
References
- Piepoli, M.F.; Hoes, A.W.; Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; Cooney, M.T.; Corra, U.; Cosyns, B.; Deaton, C.; et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts)Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur. Heart J. 2016, 37, 2315–2381. [Google Scholar] [CrossRef] [PubMed]
- Camm, A.J.; Lip, G.Y.; De Caterina, R.; Savelieva, I.; Atar, D.; Hohnloser, S.H.; Hindricks, G.; Kirchhof, P.; Guidelines, E.S.C.C.f.P. 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: An update of the 2010 ESC Guidelines for the management of atrial fibrillation. Developed with the special contribution of the European Heart Rhythm Association. Eur. Heart J. 2012, 33, 2719–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirchhof, P.; Benussi, S.; Kotecha, D.; Ahlsson, A.; Atar, D.; Casadei, B.; Castella, M.; Diener, H.C.; Heidbuchel, H.; Hendriks, J.; et al. 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Europace 2016, 18, 1609–1678. [Google Scholar] [CrossRef] [PubMed]
- Lesyuk, W.; Kriza, C.; Kolominsky-Rabas, P. Cost-of-illness studies in heart failure: A systematic review 2004–2016. Bmc Cardiovasc. Disord. 2018, 18, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henning, R.H.; Brundel, B. Proteostasis in cardiac health and disease. Nat. Rev. Cardiol. 2017, 14, 637–653. [Google Scholar] [CrossRef] [PubMed]
- Wiersma, M.; Henning, R.H.; Brundel, B.J. Derailed Proteostasis as a Determinant of Cardiac Aging. Can. J. Cardiol. 2016, 32, 1166.e11-1166.e20. [Google Scholar] [CrossRef]
- Meijering, R.A.; Zhang, D.; Hoogstra-Berends, F.; Henning, R.H.; Brundel, B.J. Loss of proteostatic control as a substrate for atrial fibrillation: A novel target for upstream therapy by heat shock proteins. Front Physiol. 2012, 3, 36. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Su, H.; Ranek, M.J. Protein quality control and degradation in cardiomyocytes. J. Mol. Cell Cardiol. 2008, 45, 11–27. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, D.; Wiersma, M.; Brundel, B. Role of Autophagy in Proteostasis: Friend and Foe in Cardiac Diseases. Cells 2018, 7, 279. [Google Scholar] [CrossRef] [Green Version]
- Akerfelt, M.; Morimoto, R.I.; Sistonen, L. Heat shock factors: Integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell Biol. 2010, 11, 545–555. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glembotski, C.C.; Arrieta, A.; Blackwood, E.A. Unfolding the Roles of Mitochondria as Therapeutic Targets for Heart Disease. J. Am. Coll. Cardiol. 2019, 73, 1807–1810. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.M.; Oster, M.E.; Hebert, D.N. Protein Quality Control in the Endoplasmic Reticulum. Protein J. 2019, 38, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, T.; Benedetti, C.; Urano, F.; Clark, S.G.; Harding, H.P.; Ron, D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J. Cell Sci. 2004, 117, 4055–4066. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.T.; Hoogenraad, N.J. A mitochondrial specific stress response in mammalian cells. Embo. J. 2002, 21, 4411–4419. [Google Scholar] [CrossRef]
- Groenendyk, J.; Sreenivasaiah, P.K.; Kim, D.H.; Agellon, L.B.; Michalak, M. Biology of endoplasmic reticulum stress in the heart. Circ. Res. 2010, 107, 1185–1197. [Google Scholar] [CrossRef] [Green Version]
- Ghaemmaghami, S.; Huh, W.K.; Bower, K.; Howson, R.W.; Belle, A.; Dephoure, N.; O’Shea, E.K.; Weissman, J.S. Global analysis of protein expression in yeast. Nature 2003, 425, 737–741. [Google Scholar] [CrossRef]
- Ouyang, Y.B.; Xu, L.J.; Emery, J.F.; Lee, A.S.; Giffard, R.G. Overexpressing GRP78 influences Ca2+ handling and function of mitochondria in astrocytes after ischemia-like stress. Mitochondrion 2011, 11, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Gething, M.J. Role and regulation of the ER chaperone BiP. Semin. Cell Dev. Biol. 1999, 10, 465–472. [Google Scholar] [CrossRef]
- Simmen, T.; Lynes, E.M.; Gesson, K.; Thomas, G. Oxidative protein folding in the endoplasmic reticulum: Tight links to the mitochondria-associated membrane (MAM). Biochim. Biophys. Acta. 2010, 1798, 1465–1473. [Google Scholar] [CrossRef] [Green Version]
- Gross, E.; Sevier, C.S.; Heldman, N.; Vitu, E.; Bentzur, M.; Kaiser, C.A.; Thorpe, C.; Fass, D. Generating disulfides enzymatically: Reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc. Natl. Acad. Sci. USA 2006, 103, 299–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Kitakaze, M. ER stress in cardiovascular disease. J. Mol. Cell Cardiol. 2010, 48, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, V. Sarcoplasmic reticulum: Structural determinants and protein dynamics. Int. J. Biochem. Cell Biol. 2011, 43, 1075–1078. [Google Scholar] [CrossRef] [PubMed]
- Nixon, G.F.; Mignery, G.A.; Somlyo, A.V. Immunogold localization of inositol 1,4,5-trisphosphate receptors and characterization of ultrastructural features of the sarcoplasmic reticulum in phasic and tonic smooth muscle. J. Muscle Res. Cell Motil. 1994, 15, 682–700. [Google Scholar] [CrossRef]
- Otsu, K.; Willard, H.F.; Khanna, V.K.; Zorzato, F.; Green, N.M.; MacLennan, D.H. Molecular cloning of cDNA encoding the Ca2+ release channel (ryanodine receptor) of rabbit cardiac muscle sarcoplasmic reticulum. J. Biol. Chem. 1990, 265, 13472–13483. [Google Scholar]
- Stutzmann, G.E.; Mattson, M.P. Endoplasmic reticulum Ca(2+) handling in excitable cells in health and disease. Pharm. Rev. 2011, 63, 700–727. [Google Scholar] [CrossRef] [Green Version]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. Embo. Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, R.J. Stress signaling from the lumen of the endoplasmic reticulum: Coordination of gene transcriptional and translational controls. Genes Dev. 1999, 13, 1211–1233. [Google Scholar] [CrossRef] [Green Version]
- Jovaisaite, V.; Mouchiroud, L.; Auwerx, J. The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J. Exp. Biol. 2014, 217, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Valera-Alberni, M.; Canto, C. Mitochondrial stress management: A dynamic journey. Cell Stress 2018, 2, 253–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radke, S.; Chander, H.; Schafer, P.; Meiss, G.; Kruger, R.; Schulz, J.B.; Germain, D. Mitochondrial protein quality control by the proteasome involves ubiquitination and the protease Omi. J. Biol. Chem. 2008, 283, 12681–12685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mottis, A.; Jovaisaite, V.; Auwerx, J. The mitochondrial unfolded protein response in mammalian physiology. Mamm. Genome. 2014, 25, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qureshi, M.A.; Haynes, C.M.; Pellegrino, M.W. The mitochondrial unfolded protein response: Signaling from the powerhouse. J. Biol. Chem. 2017, 292, 13500–13506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shpilka, T.; Haynes, C.M. The mitochondrial UPR: Mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 2018, 19, 109–120. [Google Scholar] [CrossRef]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, 170–185. [Google Scholar] [CrossRef]
- Murley, A.; Nunnari, J. The Emerging Network of Mitochondria-Organelle Contacts. Mol. Cell 2016, 61, 648–653. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; Brini, M.; Murgia, M.; Pozzan, T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 1993, 262, 744–747. [Google Scholar] [CrossRef]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased ER–mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 2011, 124, 2143–2152. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Csordas, G.; Jowdy, C.; Schneider, T.G.; Csordas, N.; Wang, W.; Liu, Y.; Kohlhaas, M.; Meiser, M.; Bergem, S.; et al. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca(2+) crosstalk. Circ. Res. 2012, 111, 863–875. [Google Scholar] [CrossRef] [Green Version]
- Gordaliza-Alaguero, I.; Canto, C.; Zorzano, A. Metabolic implications of organelle-mitochondria communication. Embo. Rep. 2019, 20, 47928. [Google Scholar] [CrossRef] [PubMed]
- Collins, H.E.; He, L.; Zou, L.; Qu, J.; Zhou, L.; Litovsky, S.H.; Yang, Q.; Young, M.E.; Marchase, R.B.; Chatham, J.C. Stromal interaction molecule 1 is essential for normal cardiac homeostasis through modulation of ER and mitochondrial function. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, 1231–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, H.; Matsui, Y.; Sadoshima, J. The role of autophagy in mediating cell survival and death during ischemia and reperfusion in the heart. Antioxid. Redox Signal 2007, 9, 1373–1381. [Google Scholar] [CrossRef]
- Wei, Y.; Sinha, S.; Levine, B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 2008, 4, 949–951. [Google Scholar] [CrossRef] [Green Version]
- Pattingre, S.; Bauvy, C.; Carpentier, S.; Levade, T.; Levine, B.; Codogno, P. Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J. Biol. Chem. 2009, 284, 2719–2728. [Google Scholar] [CrossRef] [Green Version]
- Gelmetti, V.; De Rosa, P.; Torosantucci, L.; Marini, E.S.; Romagnoli, A.; Di Rienzo, M.; Arena, G.; Vignone, D.; Fimia, G.M.; Valente, E.M. PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 2017, 13, 654–669. [Google Scholar] [CrossRef] [Green Version]
- Celardo, I.; Costa, A.C.; Lehmann, S.; Jones, C.; Wood, N.; Mencacci, N.E.; Mallucci, G.R.; Loh, S.H.; Martins, L.M. Mitofusin-mediated ER stress triggers neurodegeneration in pink1/parkin models of Parkinson’s disease. Cell Death Dis. 2016, 7, 2271. [Google Scholar] [CrossRef] [Green Version]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, J.D.; Kaufman, R.J. ER stress and its functional link to mitochondria: Role in cell survival and death. Cold Spring Harb. Perspect. Biol. 2011, 3, 4424. [Google Scholar] [CrossRef]
- Zong, W.X.; Li, C.; Hatzivassiliou, G.; Lindsten, T.; Yu, Q.C.; Yuan, J.; Thompson, C.B. Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J. Cell Biol. 2003, 162, 59–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelwahid, E.; Li, H.; Wu, J.; Irioda, A.C.; de Carvalho, K.A.; Luo, X. Endoplasmic reticulum (ER) stress triggers Hax1-dependent mitochondrial apoptotic events in cardiac cells. Apoptosis 2016, 21, 1227–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regula, K.M.; Kirshenbaum, L.A. p53 activates the mitochondrial death pathway and apoptosis of ventricular myocytes independent of de novo gene transcription. J. Mol. Cell Cardiol. 2001, 33, 1435–1445. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Ishikawa, Y. Apoptosis in heart failure. -The role of the beta-adrenergic receptor-mediated signaling pathway and p53-mediated signaling pathway in the apoptosis of cardiomyocytes. Circ. J. 2011, 75, 1811–1818. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Thompson, J.; Hu, Y.; Das, A.; Lesnefsky, E.J. Cardiac Specific Knockout of p53 Decreases ER Stress-Induced Mitochondrial Damage. Front Cardiovasc. Med. 2019, 6, 10. [Google Scholar] [CrossRef]
- Dhingra, R.; Vasan, R.S. Age as a risk factor. Med. Clin. North Am. 2012, 96, 87–91. [Google Scholar] [CrossRef] [Green Version]
- Gude, N.A.; Broughton, K.M.; Firouzi, F.; Sussman, M.A. Cardiac ageing: Extrinsic and intrinsic factors in cellular renewal and senescence. Nat. Rev. Cardiol. 2018, 15, 523–542. [Google Scholar] [CrossRef]
- Steenman, M.; Lande, G. Cardiac aging and heart disease in humans. Biophys. Rev. 2017, 9, 131–137. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Chen, Q.; Hoppel, C.L. Mitochondrial Metabolism in Aging Heart. Circ. Res. 2016, 118, 1593–1611. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Sanz, C.; Ruiz-Meana, M.; Miro-Casas, E.; Nunez, E.; Castellano, J.; Loureiro, M.; Barba, I.; Poncelas, M.; Rodriguez-Sinovas, A.; Vazquez, J.; et al. Defective sarcoplasmic reticulum-mitochondria calcium exchange in aged mouse myocardium. Cell Death Dis. 2014, 5, 1573. [Google Scholar] [CrossRef] [Green Version]
- Paz Gavilan, M.; Vela, J.; Castano, A.; Ramos, B.; del Rio, J.C.; Vitorica, J.; Ruano, D. Cellular environment facilitates protein accumulation in aged rat hippocampus. Neuro. Biol. Aging 2006, 27, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.G.; Ramaiah, K.V. Reduced eIF2alpha phosphorylation and increased proapoptotic proteins in aging. Biochem. Biophys. Res. Commun. 2007, 355, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Nuss, J.E.; Choksi, K.B.; DeFord, J.H.; Papaconstantinou, J. Decreased enzyme activities of chaperones PDI and BiP in aged mouse livers. Biochem. Biophys. Res. Commun. 2008, 365, 355–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, Y.J.; Hsu, S.C.; Hsu, C.P.; Chen, Y.H.; Chang, Y.L.; Sadoshima, J.; Huang, S.M.; Tsai, C.S.; Lin, C.Y. Sirtuin 1 protects the aging heart from contractile dysfunction mediated through the inhibition of endoplasmic reticulum stress-mediated apoptosis in cardiac-specific Sirtuin 1 knockout mouse model. Int. J. Cardiol. 2017, 228, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wang, S.; Hu, S.; Chen, Y.; Ren, J. ER-Mitochondria Microdomains in Cardiac Ischemia-Reperfusion Injury: A Fresh Perspective. Front Physiol. 2018, 9, 755. [Google Scholar] [CrossRef] [PubMed]
- Paillard, M.; Tubbs, E.; Thiebaut, P.A.; Gomez, L.; Fauconnier, J.; Da Silva, C.C.; Teixeira, G.; Mewton, N.; Belaidi, E.; Durand, A.; et al. Depressing mitochondria-reticulum interactions protects cardiomyocytes from lethal hypoxia-reoxygenation injury. Circulation 2013, 128, 1555–1565. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Zhang, Y.; Cui, M.; Jin, L.; Wang, Y.; Lv, F.; Liu, Y.; Zheng, W.; Shang, H.; Zhang, J.; et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat. Med. 2016, 22, 175–182. [Google Scholar] [CrossRef]
- Zhu, H.; Jin, Q.; Li, Y.; Ma, Q.; Wang, J.; Li, D.; Zhou, H.; Chen, Y. Melatonin protected cardiac microvascular endothelial cells against oxidative stress injury via suppression of IP3R-[Ca(2+)]c/VDAC-[Ca(2+)]m axis by activation of MAPK/ERK signaling pathway. Cell Stress Chaperones 2018, 23, 101–113. [Google Scholar] [CrossRef]
- Raturi, A.; Gutierrez, T.; Ortiz-Sandoval, C.; Ruangkittisakul, A.; Herrera-Cruz, M.S.; Rockley, J.P.; Gesson, K.; Ourdev, D.; Lou, P.H.; Lucchinetti, E.; et al. TMX1 determines cancer cell metabolism as a thiol-based modulator of ER-mitochondria Ca2+ flux. J. Cell Biol. 2016, 214, 433–444. [Google Scholar] [CrossRef] [Green Version]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Huan, L.; Yin, J.; Qin, M.; Zhang, Z.; Zhang, Z.; Zhang, J.; Wang, S. Role of microRNA-130a in myocardial hypoxia/reoxygenation injury. Exp. Med. 2017, 13, 759–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, D.; Chen, R.; Zhang, L.; Rao, Z.; Ruan, Y.; Li, L.; Chu, M.; Zhang, Y. Sulodexide attenuates endoplasmic reticulum stress induced by myocardial ischaemia/reperfusion by activating the PI3K/Akt pathway. J. Cell Mol. Med. 2019, 23, 5063–5075. [Google Scholar] [CrossRef] [PubMed]
- Wiersma, M.; Meijering, R.A.M.; Qi, X.Y.; Zhang, D.; Liu, T.; Hoogstra-Berends, F.; Sibon, O.C.M.; Henning, R.H.; Nattel, S.; Brundel, B. Endoplasmic Reticulum Stress Is Associated With Autophagy and Cardiomyocyte Remodeling in Experimental and Human Atrial Fibrillation. J. Am. Heart Assoc. 2017, 6, 6458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Jiang, Q.; Ding, X.; Xu, W.; Wang, D.W.; Chen, M. The ER stress-mediated mitochondrial apoptotic pathway and MAPKs modulate tachypacing-induced apoptosis in HL-1 atrial myocytes. PLoS ONE 2015, 10, e0117567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.; Santulli, G.; Reiken, S.R.; Yuan, Q.; Osborne, B.W.; Chen, B.X.; Marks, A.R. Mitochondrial oxidative stress promotes atrial fibrillation. Sci. Rep. 2015, 5, 11427. [Google Scholar] [CrossRef] [Green Version]
- Wiersma, M.; van Marion, D.M.S.; Wust, R.C.I.; Houtkooper, R.H.; Zhang, D.; Groot, N.M.S.; Henning, R.H.; Brundel, B. Mitochondrial Dysfunction Underlies Cardiomyocyte Remodeling in Experimental and Clinical Atrial Fibrillation. Cells 2019, 8, 1202. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Sun, Y.; Zhuang, Q.; Song, Y.; Wu, B.; Jia, Z.; Pan, H.; Zhou, H.; Hu, S.; Zhang, B.; et al. Mitochondrial dysfunction caused by m.2336T>C mutation with hypertrophic cardiomyopathy in cybrid cell lines. Mitochondrion 2019, 46, 313–320. [Google Scholar] [CrossRef]
- Kopajtich, R.; Nicholls, T.J.; Rorbach, J.; Metodiev, M.D.; Freisinger, P.; Mandel, H.; Vanlander, A.; Ghezzi, D.; Carrozzo, R.; Taylor, R.W.; et al. Mutations in GTPBP3 Cause a Mitochondrial Translation Defect Associated with Hypertrophic Cardiomyopathy, Lactic Acidosis, and Encephalopathy. Am. J. Hum. Genet. 2014, 95, 708–720. [Google Scholar] [CrossRef] [Green Version]
- Hashem, S.I.; Murphy, A.N.; Divakaruni, A.S.; Klos, M.L.; Nelson, B.C.; Gault, E.C.; Rowland, T.J.; Perry, C.N.; Gu, Y.; Dalton, N.D.; et al. Impaired mitophagy facilitates mitochondrial damage in Danon disease. J. Mol. Cell Cardiol. 2017, 108, 86–94. [Google Scholar] [CrossRef]
- Towbin, J.A. Inherited cardiomyopathies. Circ. J. 2014, 78, 2347–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, G.; Gullmets, J.; Virtanen, L.; Li, S.P.; Keinanen, A.; Shimi, T.; Mauermann, M.; Helio, T.; Kaartinen, M.; Ollila, L.; et al. Deleterious assembly of the lamin A/C mutant p.S143P causes ER stress in familial dilated cardiomyopathy. J. Cell Sci. 2016, 129, 2732–2743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christiansen, L.B.; Dela, F.; Koch, J.; Hansen, C.N.; Leifsson, P.S.; Yokota, T. Impaired cardiac mitochondrial oxidative phosphorylation and enhanced mitochondrial oxidative stress in feline hypertrophic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, 1237–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.R.; Zech, A.T.L.; Geertz, B.; Reischmann-Dusener, S.; Osinska, H.; Prondzynski, M.; Kramer, E.; Meng, Q.; Redwood, C.; van der Velden, J.; et al. Activation of Autophagy Ameliorates Cardiomyopathy in Mybpc3-Targeted Knockin Mice. Circ. Heart Fail. 2017, 10, 4140. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, E.; Levitas, A.; Singh, S.R.; Braiman, A.; Ofir, R.; Etzion, S.; Sheffield, V.C.; Etzion, Y.; Carrier, L.; Parvari, R. PLEKHM2 mutation leads to abnormal localization of lysosomes, impaired autophagy flux and associates with recessive dilated cardiomyopathy and left ventricular noncompaction. Hum. Mol. Genet. 2015, 24, 7227–7240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef]
- Yaras, N.; Ugur, M.; Ozdemir, S.; Gurdal, H.; Purali, N.; Lacampagne, A.; Vassort, G.; Turan, B. Effects of diabetes on ryanodine receptor Ca release channel (RyR2) and Ca2+ homeostasis in rat heart. Diabetes 2005, 54, 3082–3088. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Yu, X.; Li, T.; Wu, J.; Zhao, Y.; Liu, J.; Sun, A.; Dong, S.; Wu, J.; Zhong, X.; et al. Exogenous H2S regulates endoplasmic reticulum-mitochondria cross-talk to inhibit apoptotic pathways in STZ-induced type I diabetes. Am. J. Physiol. Endocrinol. Metab. 2017, 312, 190–203. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Lu, Q.; Ding, Y.; Wu, Y.; Qiu, Y.; Wang, P.; Mao, X.; Huang, K.; Xie, Z.; Zou, M.H. Hyperglycemia-Driven Inhibition of AMP-Activated Protein Kinase alpha2 Induces Diabetic Cardiomyopathy by Promoting Mitochondria-Associated Endoplasmic Reticulum Membranes In Vivo. Circulation 2019, 139, 1913–1936. [Google Scholar] [CrossRef]
- Xu, J.; Zhou, Q.; Xu, W.; Cai, L. Endoplasmic reticulum stress and diabetic cardiomyopathy. Exp. Diabetes Res. 2012, 2012, 827971. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhang, T.; Dai, H.; Liu, G.; Wang, H.; Sun, Y.; Zhang, Y.; Ge, Z. Involvement of endoplasmic reticulum stress in myocardial apoptosis of streptozocin-induced diabetic rats. J. Clin. Biochem. Nutr. 2007, 41, 58–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ljubkovic, M.; Gressette, M.; Bulat, C.; Cavar, M.; Bakovic, D.; Fabijanic, D.; Grkovic, I.; Lemaire, C.; Marinovic, J. Disturbed Fatty Acid Oxidation, Endoplasmic Reticulum Stress and Apoptosis in Left Ventricle of Patients with Type 2 Diabetes Mellitus. Diabetes 2019, 68, 1924–1933. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending healthy life span--from yeast to humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Ahn, N.; Jung, S.; Park, S. Effects of intermittent ladder-climbing exercise training on mitochondrial biogenesis and endoplasmic reticulum stress of the cardiac muscle in obese middle-aged rats. Korean J. Physiol. Pharm. 2017, 21, 633–641. [Google Scholar] [CrossRef] [Green Version]
- Bozi, L.H.; Jannig, P.R.; Rolim, N.; Voltarelli, V.A.; Dourado, P.M.; Wisloff, U.; Brum, P.C. Aerobic exercise training rescues cardiac protein quality control and blunts endoplasmic reticulum stress in heart failure rats. J. Cell Mol. Med. 2016, 20, 2208–2212. [Google Scholar] [CrossRef] [Green Version]
- Bourdier, G.; Flore, P.; Sanchez, H.; Pepin, J.L.; Belaidi, E.; Arnaud, C. High-intensity training reduces intermittent hypoxia-induced ER stress and myocardial infarct size. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, 279–289. [Google Scholar] [CrossRef] [Green Version]
- Cheang, W.S.; Wong, W.T.; Zhao, L.; Xu, J.; Wang, L.; Lau, C.W.; Chen, Z.Y.; Ma, R.C.; Xu, A.; Wang, N.; et al. PPARdelta Is Required for Exercise to Attenuate Endoplasmic Reticulum Stress and Endothelial Dysfunction in Diabetic Mice. Diabetes 2017, 66, 519–528. [Google Scholar] [CrossRef] [Green Version]
- Staerk, L.; Wang, B.; Preis, S.R.; Larson, M.G.; Lubitz, S.A.; Ellinor, P.T.; McManus, D.D.; Ko, D.; Weng, L.C.; Lunetta, K.L.; et al. Lifetime risk of atrial fibrillation according to optimal, borderline, or elevated levels of risk factors: Cohort study based on longitudinal data from the Framingham Heart Study. BMJ 2018, 361, 1453. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhang, Y.; Liu, X.; Zuo, J.; Wang, K.; Liu, W.; Ge, J. Exogenous taurine attenuates mitochondrial oxidative stress and endoplasmic reticulum stress in rat cardiomyocytes. Acta. Biochim. Biophys. Sin. 2013, 45, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Thompson, J.; Hu, Y.; Das, A.; Lesnefsky, E.J. Metformin attenuates ER stress-induced mitochondrial dysfunction. Transl. Res. 2017, 190, 40–50. [Google Scholar] [CrossRef]
- Arumugam, S.; Thandavarayan, R.A.; Veeraveedu, P.T.; Nakamura, T.; Arozal, W.; Sari, F.R.; Giridharan, V.V.; Soetikno, V.; Palaniyandi, S.S.; Harima, M.; et al. Beneficial effects of edaravone, a novel antioxidant, in rats with dilated cardiomyopathy. J. Cell Mol. Med. 2012, 16, 2176–2185. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Zhang, D.; Brundel, B.J.J.M.; Wiersma, M. Imbalance of ER and Mitochondria Interactions: Prelude to Cardiac Ageing and Disease? Cells 2019, 8, 1617. https://doi.org/10.3390/cells8121617
Li J, Zhang D, Brundel BJJM, Wiersma M. Imbalance of ER and Mitochondria Interactions: Prelude to Cardiac Ageing and Disease? Cells. 2019; 8(12):1617. https://doi.org/10.3390/cells8121617
Chicago/Turabian StyleLi, Jin, Deli Zhang, Bianca J. J. M. Brundel, and Marit Wiersma. 2019. "Imbalance of ER and Mitochondria Interactions: Prelude to Cardiac Ageing and Disease?" Cells 8, no. 12: 1617. https://doi.org/10.3390/cells8121617
APA StyleLi, J., Zhang, D., Brundel, B. J. J. M., & Wiersma, M. (2019). Imbalance of ER and Mitochondria Interactions: Prelude to Cardiac Ageing and Disease? Cells, 8(12), 1617. https://doi.org/10.3390/cells8121617