Diastereomeric Recognition of 5’,8-cyclo-2’-Deoxyadenosine Lesions by Human Poly(ADP-ribose) Polymerase 1 in a Biomimetic Model

 ,
, 
 , ,
, , 

 and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. General Methods for Synthesis and Purification of Oligonucleotides (ODNs)
2.2. Preparation of Double Stranded Oligonucleotide Substrates
2.3. Circular Dichroism
2.4. Fluorescence Spectroscopy
2.5. Immunoblotting Analyses of PARP Protein-DNA Complex
2.6. Gel Mobility Shift Assay
3. Results and Discussion
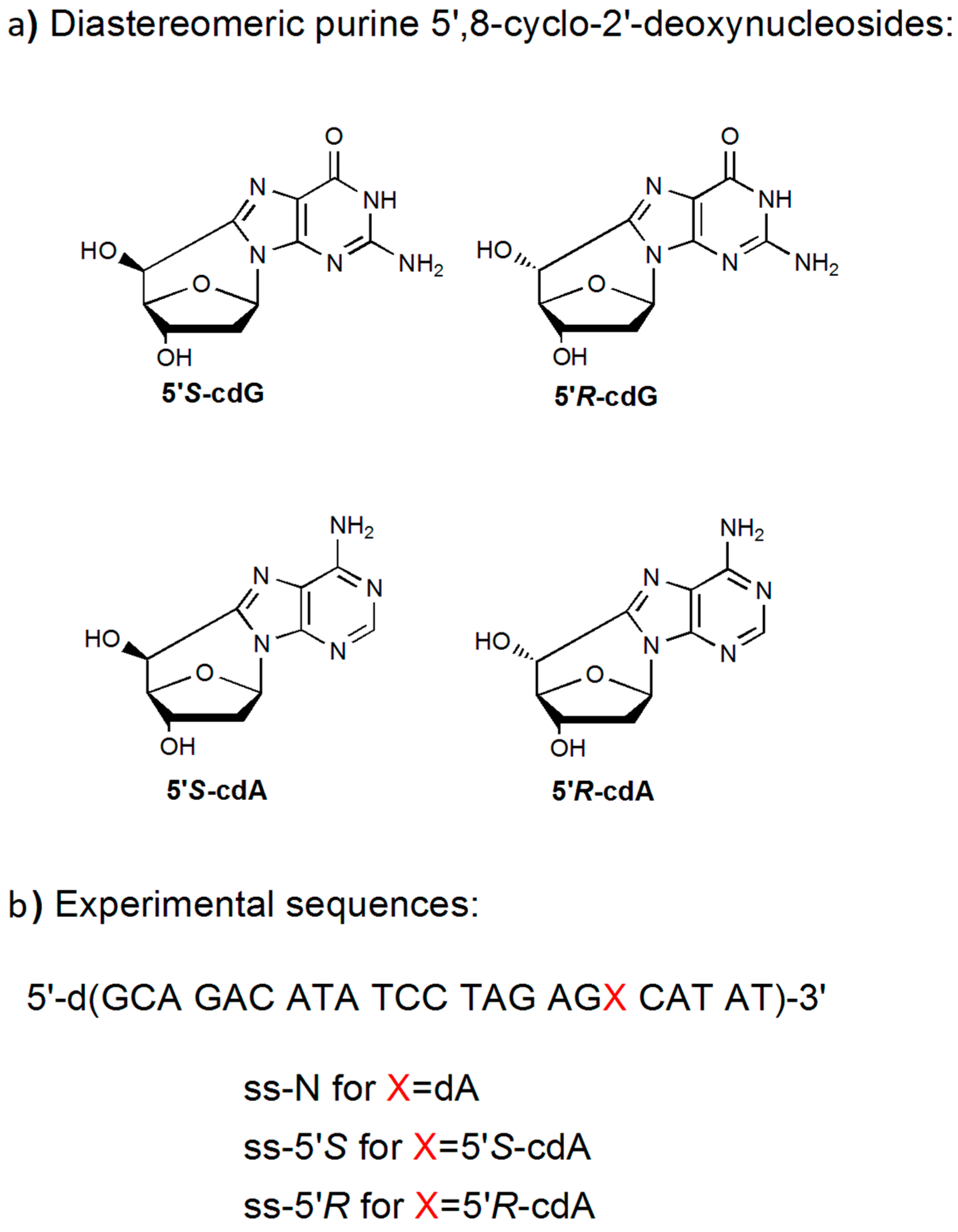
3.1. Synthesis and Characterization of 23-Mer Oligonucleotides Containing dA and cdA Lesions
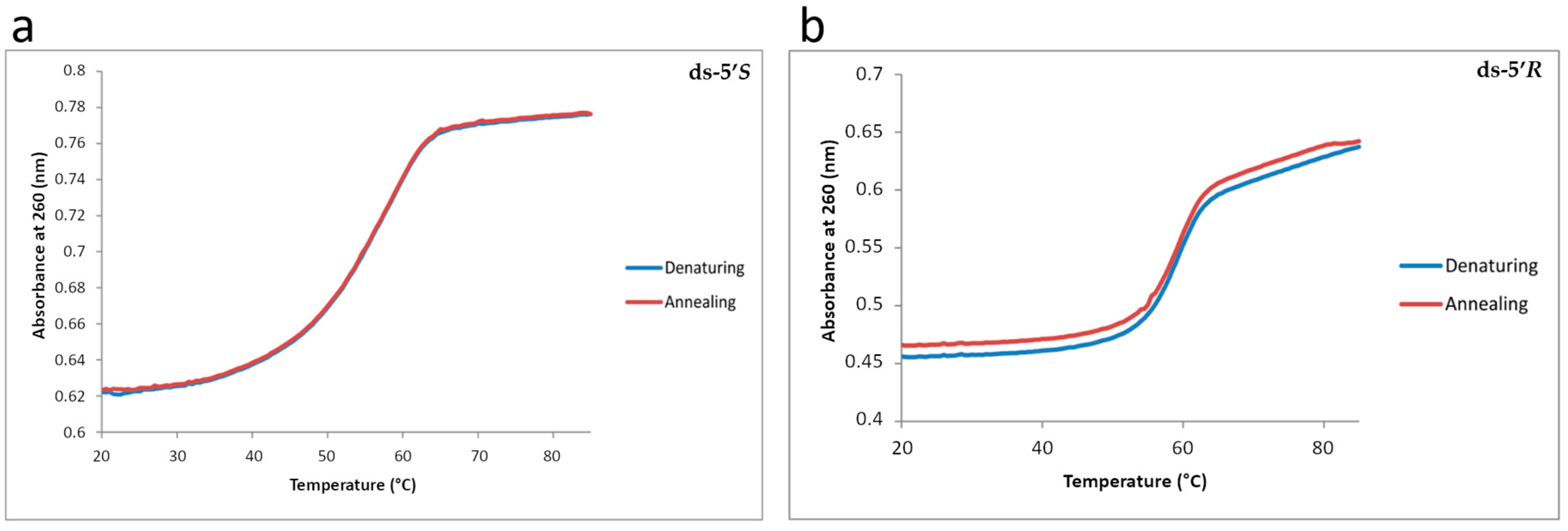
3.2. Thermal Stability of Modified Oligonucleotide Duplexes by Diastereomeric cdA Lesions
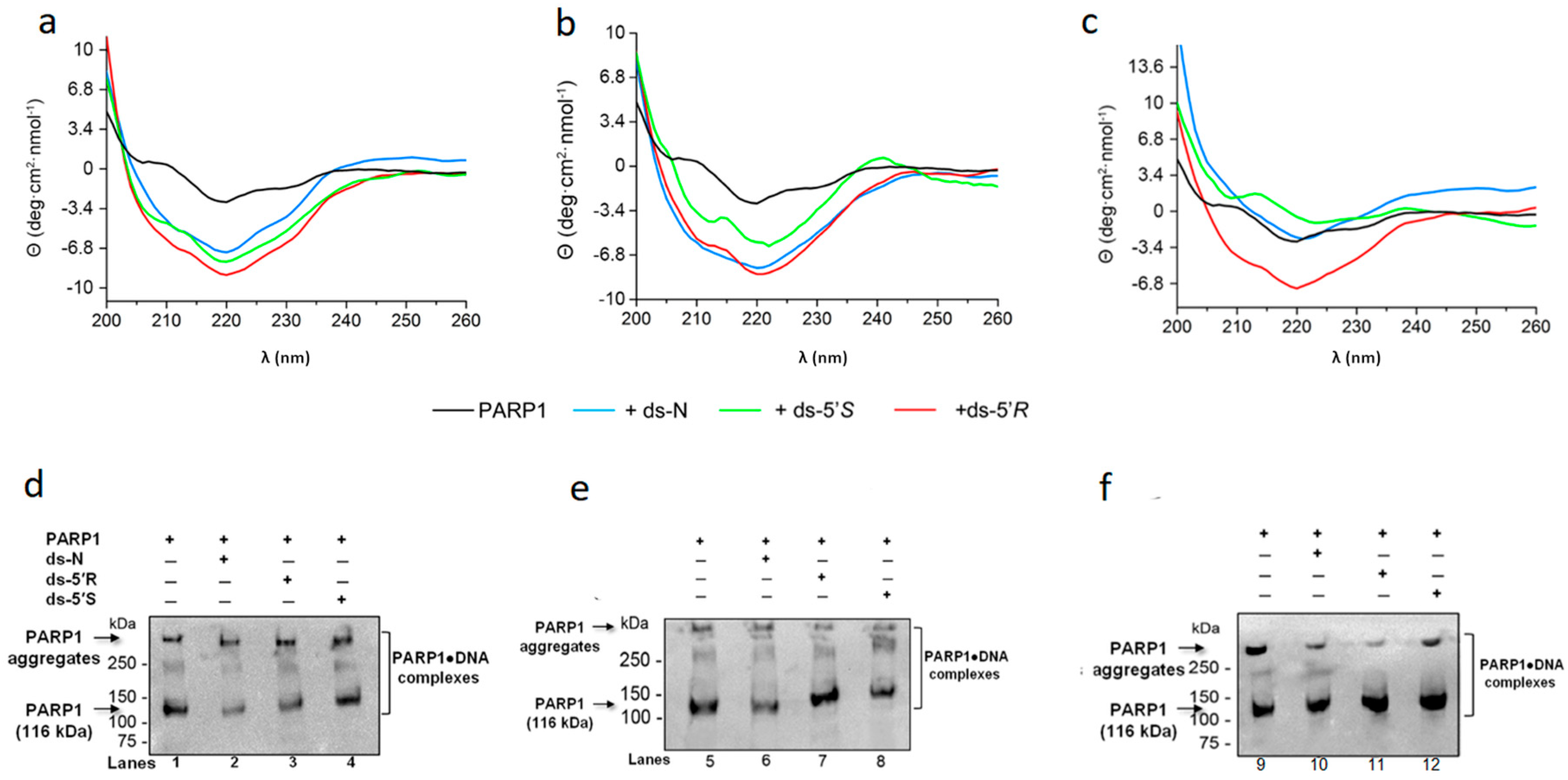
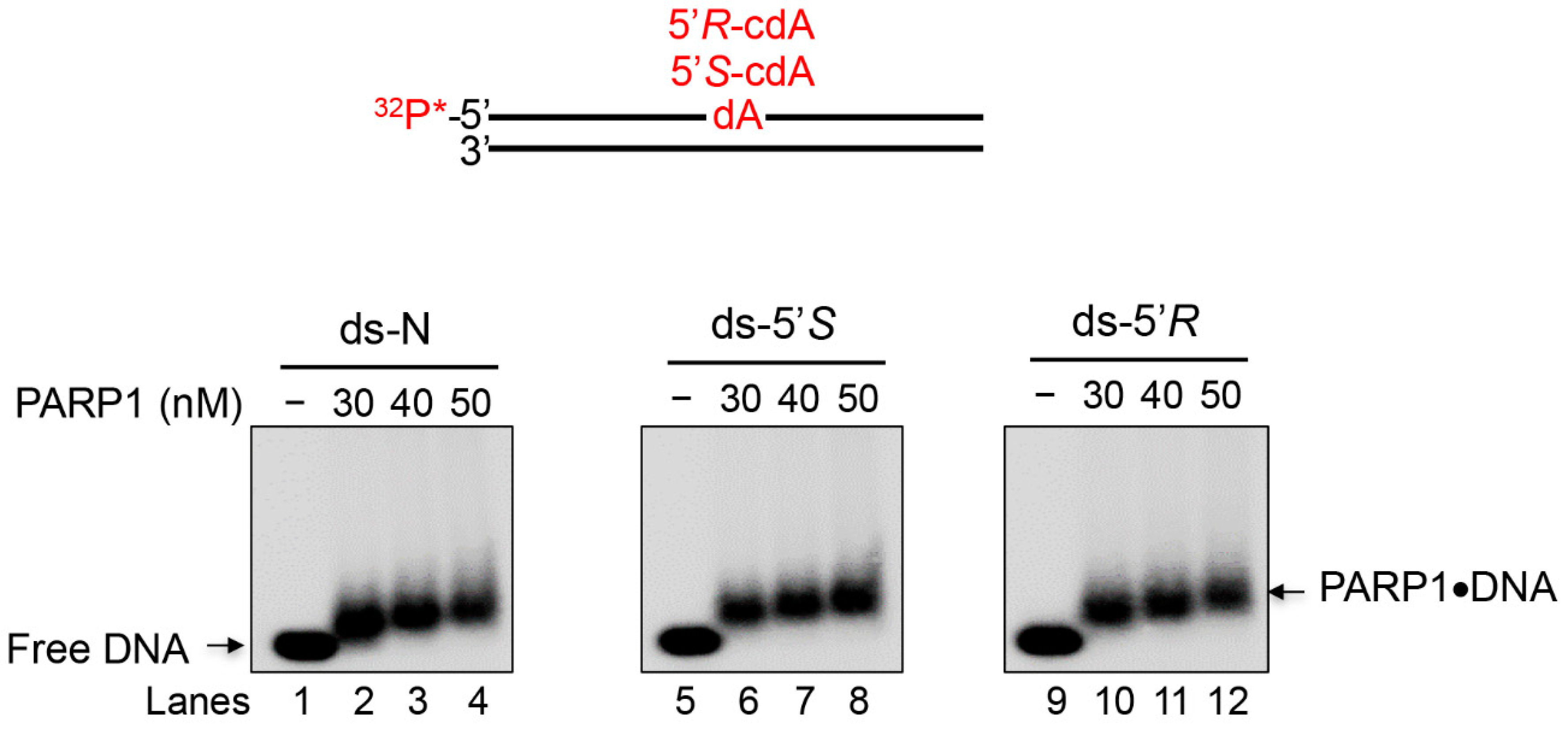
3.3. PARP1 Recognizes an Undamaged dA and cdA Diastereomeric Lesions in ds-Oligonucleotides
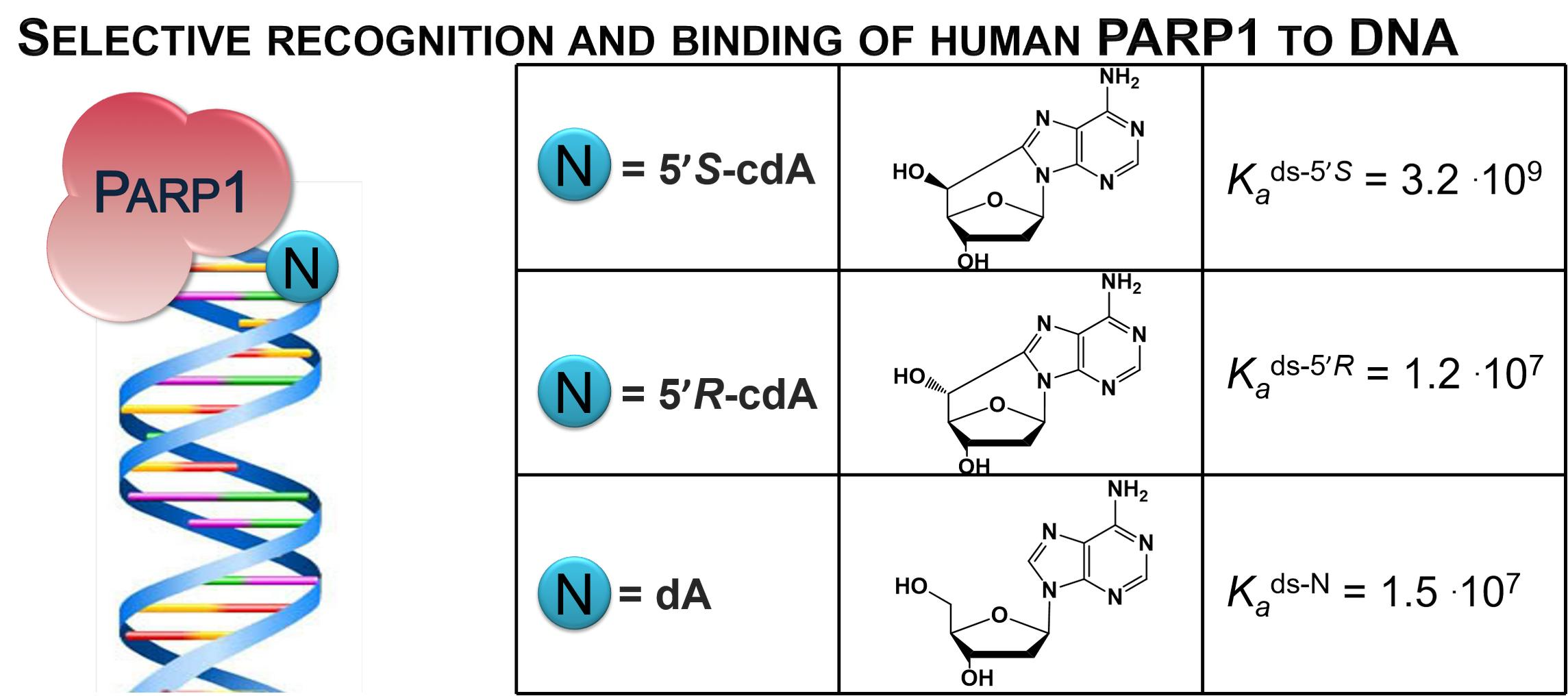
3.4. PARP1 Binding to Undamaged dA and cdA Lesions with Different Affinities
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chatgilialoglu, C.; Ferreri, C.; Terzidis, M.A. Purine 5’,8-cyclonucleoside lesions: Chemistry and biology. Chem. Soc. Rev. 2011, 40, 1368–1382. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C.; Krokidis, M.G.; Papadopoulos, K.; Terzidis, M.A. Purine 5’,8-cyclo-2’-deoxynucleoside lesions in irradiated DNA. Radiat. Phys. Chem. 2016, 128, 75–81. [Google Scholar] [CrossRef]
- Yuan, B.; Wang, J.; Cao, H.; Sun, R.; Wang, Y. High-throughput analysis of the mutagenic and cytotoxic properties of DNA lesions by next-generation sequencing. Nucleic Acids Res. 2011, 39, 5945–5954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, C.; Dai, X.; Yuan, B.; Wang, J.; Wang, J.; Brooks, P.J. A quantitative assay for assessing the effects of DNA lesions on transcription. Nat. Chem. Biol. 2012, 8, 817–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaruga, P.; Dizdaroglu, M. 8,5’-Cyclopurine-2’deoxynucleosides in DNA: Mechanisms of formation, measurement, repair and biological effects. DNA Repair 2008, 7, 1413–1417. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.J. The 8,5’-cyclopurine-2’-deoxynucleosides: Candidate neurodegenerative DNA lesions in xeroderma pigmentosum, and unique probes of transcription and nucleotide excision repair. DNA Repair 2008, 7, 1168–1179. [Google Scholar] [CrossRef] [PubMed]
- Kirkali, G.; de Souza-Pinto, N.C.; Jaruga, P.; Bohr, V.A.; Dizdaroglu, M. Accumulation of (5’S)-8,5’-cyclo-2’-deoxyadenosine in organs of Cockayne syndrome complementation group B gene knockout mice. DNA Repair 2009, 8, 274–278. [Google Scholar] [CrossRef]
- Wang, J.; Clauson, C.L.; Robbins, P.D.; Niedernhofer, L.J.; Wang, Y. The oxidative DNA lesions 8,5’-cyclopurines accumulate with aging in a tissue-specific manner. Aging Cell 2012, 11, 714–716. [Google Scholar] [CrossRef]
- Krokidis, M.G.; Terzidis, M.A.; Efthimiadou, E.; Zervou, S.; Kordas, G.; Papadopoulos, K.; Hiskia, A.; Kletsas, D.; Chatgilialoglu, C. Purine 5’,8-cyclo-2’-deoxynucleoside lesions: Formation by radical stress and repair in human breast epithelial cancer cells. Free Radic. Res. 2017, 51, 470–482. [Google Scholar] [CrossRef]
- Dizdaroglu, M. Free-radical-induced formation of an 8,5’-cyclo-2’-deoxyguanosine moiety in deoxyribonucleic acid. Biochem. J. 1986, 1, 247–254. [Google Scholar] [CrossRef]
- Belmadoui, N.; Boussicault, F.; Guerra, M.; Ravanat, J.L.; Chatgilialoglu, C.; Cadet, J. Radiation-induced formation of purine 5’,8-cyclonucleosides in isolated and cellular DNA: High stereospecificity and modulating effect of oxygen. Org. Biomol. Chem. 2010, 8, 3211–3219. [Google Scholar] [CrossRef] [PubMed]
- Terzidis, M.A.; Ferreri, C.; Chatgilialoglu, C. Radiation-induced formation of purine lesions in single and double stranded DNA: Revised quantification. Front. Chem. 2015, 3, 18. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.J. The cyclopurine deoxynucleosides: DNA repair, biological effects, mechanistic insights, and unanswered questions. Free Radic. Biol. Med. 2017, 107, 90–100. [Google Scholar] [CrossRef]
- Kuraoka, I.; Bender, C.; Romieu, A.; Cadet, J.; Wood, R.D.; Lindahl, T. Removal of oxygen free-radical-induced 5’,8-purine cyclodeoxynucleosides from DNA by the nucleotide excision-repair pathway in human cells. Proc. Natl. Acad. Sci. USA 2000, 97, 3832–3837. [Google Scholar] [CrossRef] [PubMed]
- Kropachev, K.; Ding, S.; Terzidis, M.A.; Masi, A.; Liu, Z.; Cai, Y.; Kolbanovskiy, M.; Chatgilialoglu, C.; Broyde, S.; Geacintov, N.E.; et al. Structural basis for the recognition of diastereomeric 5’,8-cyclo-2’-deoxypurine lesions by the human nucleotide excision repair system. Nucleic Acids Res. 2014, 42, 5020–5032. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Kropachev, K.; Terzidis, M.A.; Masi, A.; Chatgilialoglu, C.; Shafirovich, V.; Geacintov, N.E.; Broyde, S. Differences in the access of lesions to the nucleotide excision repair machinery in nucleosomes. Biochemistry 2015, 54, 4181–4185. [Google Scholar] [CrossRef] [PubMed]
- You, C.; Swanson, L.A.; Dai, X.; Yuan, B.; Wang, J.; Wang, Y. Translesion synthesis of 8,5’-cyclopurine-2’-deoxynucleosides by DNA polymerases eta, iota, and zeta. J. Biol. Chem. 2013, 288, 28548–28556. [Google Scholar] [CrossRef] [PubMed]
- Pednekar, V.; Weerasooriya, S.; Jasti, V.P.; Basu, A.K. Mutagenicity and genotoxicity of (5’S)-8,5’-cyclo-2’-deoxyadenosine in Escherichia coli and replication of (5’S)-8,5’-cyclopurine-2’-deoxynucleosides in vitro by DNA polymerase IV, exo-free Klenow fragment, and Dpo4. Chem. Res. Toxicol. 2014, 27, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Kuraoka, I.; Robins, P.; Masutani, C.; Hanaoka, F.; Gasparutto, D.; Cadet, J.; Wood, R.D.; Lindahl, T. Oxygen Free Radical Damage to DNA. J. Biol. Chem. 2001, 276, 49283–49288. [Google Scholar] [CrossRef]
- Jiang, Z.; Xu, M.; Lai, Y.; Laverde, E.E.; Terzidis, M.A.; Masi, A.; Chatgilialoglu, C.; Liu, Y. Bypass of a 5’,8-cyclopurine-2’-deoxynucleoside by DNA polymerase β during DNA replication and base excision repair leads to nucleotide misinsertions and DNA strand breaks. DNA Repair 2015, 33, 24–34. [Google Scholar] [CrossRef]
- Xu, M.; Lai, Y.; Jiang, Z.; Terzidis, M.A.; Masi, A.; Chatgilialoglu, C.; Liu, Y. A 5’, 8-cyclo-2’-deoxypurine lesion induces trinucleotide repeat deletion via a unique lesion bypass by DNA polymerase β. Nucleic Acids Res. 2014, 42, 13749–13763. [Google Scholar] [CrossRef] [PubMed]
- Weng, P.J.; Gao, Y.; Gregory, M.T.; Wang, P.; Wang, Y.; Yang, W. Bypassing a 8,5’-cyclo-2’-deoxyadenosine lesion by human DNA polymerase η at atomic resolution. Proc. Natl. Acad. Sci. USA 2018, 115, 10660–10665. [Google Scholar] [CrossRef] [PubMed]
- Jaruga, P.; Xiao, Y.; Vartanian, V.; Lloyd, R.S.; Dizdaroglu, M. Evidence for the involvement of DNA repair enzyme NEIL1 in nucleotide excision repair of (5’R)- and (5’S)-8,5’-cyclo-2’-deoxyadenosines. Biochemistry 2010, 49, 1053–1055. [Google Scholar] [CrossRef] [PubMed]
- Arczewska, K.D.; Tomazella, G.G.; Lindvall, J.M.; Kassahun, H.; Maglioni, S.; Torgovnick, A.; Henriksson, J.; Matilainen, O.; Marquis, B.J.; Nelson, B.C.; et al. Active transcriptomic and proteomic reprogramming in the C. elegans nucleotide excision repair mutant xpa-1. Nucleic Acids Res. 2013, 41, 5368–5381. [Google Scholar] [PubMed]
- Khan, I.; Suhasini, A.N.; Banerjee, T.; Sommer, J.A.; Kaplan, D.L.; Kuper, J.; Kisker, C.; Brosh, R.M., Jr. Impact of age-associated cyclopurine lesions on DNA repair helicases. PLoS ONE 2014, 9, e113293. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Kraus, W.L. On PAR with PARP: Cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012, 26, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Swindall, A.F.; Stanley, J.A.; Yang, E.S. PARP1: Friend or foe of DNA damage and repair in tumorigenesis? Cancers 2013, 5, 943–958. [Google Scholar] [CrossRef] [PubMed]
- Robu, M.; Shah, R.G.; Petitclerc, N.; Brind’Amour, J.; Kandan-Kulangara, F.; Shah, G.M. Role of poly(ADP-ribose) polymerase-1 in the removal of UV-induced DNA lesions by nucleotide excision repair. Proc. Natl. Acad. Sci. USA 2013, 110, 1658–1663. [Google Scholar] [CrossRef] [Green Version]
- Langelier, M.F.; Planck, J.L.; Roy, S.; Pascal, J.M. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP1. Science 2012, 336, 728–732. [Google Scholar] [CrossRef]
- Eustermann, S.; Videler, H.; Yang, J.C.; Cole, P.T.; Gruszka, D.; Veprintsev, D.; Neuhaus, D. The DNA-binding domain of human PARP1 interacts with DNA single-strand breaks as a monomer through its second zinc finger. J. Mol. Biol. 2011, 407, 149–170. [Google Scholar] [CrossRef]
- Ray Chaudhui, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Pines, A.; Vrouwe, M.G.; Marteijn, J.A.; Typas, D.; Luijsterburg, M.S.; Cansoy, M.; Hensbergen, P.; Deelder, A.; de Groot, A.; Matsumoto, S.; et al. PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J. Cell Biol. 2012, 199, 235–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khodyreva, S.N.; Prasad, R.; Ilina, E.S.; Sukhanova, M.V.; Kutuzov, M.M.; Liu, Y.; Hou, E.W.; Wilson, S.H.; Lavrik, O.I. Apurinic/apyrimidinic (AP) site recognition by the 5’-dRP/AP lyase in poly(ADP-ribose) polymerase-1 (PARP-1). Proc. Natl. Acad. Sci. USA 2010, 107, 22090–22095. [Google Scholar] [CrossRef] [PubMed]
- Purohit, N.K.; Robu, M.; Shah, R.G.; Geacintov, N.E.; Shah, G.M. Characterization of the interactions of PARP-1 with UV-damaged DNA in vivo and in vitro. Sci. Rep. 2016, 6, 19020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herceg, Z.; Wang, Z.Q. Functions of poly(ADP-ribose) polymerase (PARP) in DNA repair, genomic integrity and cell death. Mutat. Res. 2001, 477, 97–110. [Google Scholar] [CrossRef]
- Bai, P.; Canto, C.; Oudart, H.; Brunyanszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD+/SIRT1 reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef]
- Thorslund, T.; von Kobbe, C.; Harrigan, J.A.; Indig, F.E.; Christiansen, M.; Stevnsner, T.; Bohr, V.A. Cooperation of the Cockayne syndrome group B protein and poly(ADP-ribose) polymerase 1 in the response to oxidative stress. Mol. Cell Biol. 2005, 25, 7625–7636. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Tseng, A.; Borch Jensen, M.; Scheibye-Alsing, K.; Fang, E.F.; Iyama, T.; Bharti, S.K.; Marosi, K.; Froetscher, L.; Kassahun, H.; et al. Cockayne syndrome group A and B proteins converge on transcription-linked resolution of non-B DNA. Proc. Natl. Acad. Sci. USA 2016, 113, 12502–12507. [Google Scholar] [CrossRef] [Green Version]
- Langelier, M.F.; Pascal, J.M. PARP-1 mechanism for coupling DNA damage detection to poly(ADP-ribose) synthesis. Curr. Opin. Struct. Biol. 2013, 23, 134–143. [Google Scholar] [CrossRef] [Green Version]
- Faraone-Mennella, M.R.; Marini, M.; Ferone, A.; Cacace, O.; Liguoro, A.; Margonato, V.; Farina, B.; Veicsteinas, A. Physical exercise activates the poly-ADPribosylation system in rat testes. J. Biol. Regul. Homeost. Agents 2010, 24, 325–334. [Google Scholar] [PubMed]
- Hooten, N.N.; Kompaniez, K.; Barnes, J.; Lohani, A.; Evans, M.K. Poly(ADP-ribose) Polymerase 1 (PARP-1) binds to 8-Oxoguanine-DNA glycosylase (OGG1). JBC 2011, 286, 44679–44690. [Google Scholar] [CrossRef] [PubMed]
- Flyunt, R.; Bazzanini, R.; Chatgilialoglu, C.; Mulazzani, Q.G. Fate of the 2’-Deoxyadenosin-5’-yl Radical under Anaerobic Conditions. J. Am. Chem. Soc. 2000, 122, 4225–4226. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Ferreri, C.; Masi, A.; Sansone, A.; Terzidis, M.A.; Tsakos, M. A problem solving approach for the diastereoselective synthesis of (5’S)- and (5’R)-5’,8-cyclopurine lesions. Org. Chem. Front. 2014, 1, 698–702. [Google Scholar] [CrossRef]
- Huang, H.; Das, R.S.; Basu, A.K.; Stone, M.P. Structure of (5’S)-8,5’-cyclo-2’-deoxyguanosine in DNA. J. Am. Chem. Soc. 2011, 133, 20357–20368. [Google Scholar] [CrossRef] [PubMed]
- Zaliznyak, T.; Lukin, M.; de los Santos, C. Structure and stability of duplex DNA containing (5’S)-5’,8-cyclo-2’-deoxyadenosine: An oxidatively generated lesion repaired by NER. Chem. Res. Toxicol. 2012, 25, 2103–2111. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Tidyman, W.E.; Le, K.U.; Kirsten, E.; Kun, E.; Ordahl, C.P. Analysis of nucleotide sequence-dependent DNA binding of poly(ADP-ribose) polymerase in a purified system. Biochemistry 2004, 43, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Kun, E.; Kirsten, E.; Mendeleyev, J.; Ordahl, C.P. Regulation of the enzymatic catalysis of poly(ADP-ribose) polymerase by dsDNA, polyamines, Mg2+, Ca2+, Histones H1 and H3, and ATP. Biochemistry 2004, 43, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2006, 1, 2876–2890. [Google Scholar] [CrossRef] [PubMed]
- Langelier, M.F.; Planck, J.L.; Roy, S.; Pascal, J.M. Crystal structures of poly(ADP-ribose) polymerase-1 (PARP1) zinc fingers bound to DNA. J. Biol. Chem. 2011, 286, 10690–10701. [Google Scholar] [CrossRef] [PubMed]
- Yelamos, J.; Farres, J.; Llacuna, L.; Ampurdanes, C.; Martin-Caballero, J. PARP-1 and PARP-2: New players in tumour development. Am. J. Cancer Res. 2011, 1, 328–346. [Google Scholar] [PubMed]
- Langelier, M.F.; Riccio, A.A.; Pascal, J.M. PARP-2 and PARP-3 are selectively activated by 5’ phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic Acids Res. 2014, 42, 127762–127775. [Google Scholar] [CrossRef] [PubMed]
- Steffen, J.D.; Brody, J.R.; Armen, R.S.; Pascal, J.M. Structural implications for selective targeting of PARPs. Front. Oncol. 2013, 3, 301. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 5′-d(GCA GAC ATA TCC TAG AGX CAT AT)-3′ 3′-d(CGT CTG TAT AGG ATC TCT GTA TA)-3′ | Tm, °C |
|---|---|
| X = dA (unmodified) | 60.0 ± 0.3 |
| X = 5’R-cdA | 59.0 ± 0.2 |
| X = 5’S-cdA | 58.0 ± 0.3 |
| Strands | Sequence (5’–3′) 1 | Mass Calcd. (Da) | Mass Found 2 (Da) |
|---|---|---|---|
| ss-N | GCA GAC ATA TCC TAG AGA CAT AT | 7040.7 | 7038.1 |
| ss-5’S | GCA GAC ATA TCC TAG AGX CAT AT | 7038.7 | 7037.2 |
| ss-5’R | GCA GAC ATA TCC TAG AGX CAT AT | 7038.7 | 7036.9 |
| CS 3 | ATA TGT CTC TAG GAT ATG TCT GC | 7044.7 | 7044.7 |
| Compounds | Θ220nm 50 nM | Θ220nm 100 nM | Θ220nm 200 nM |
|---|---|---|---|
| PARP1 | −2.84 | −2.84 | −2.84 |
| PARP1 + ds-N | −7.14 | −7.79 | −2.46 |
| PARP1 + ds-5’S | −7.96 | −5.82 | −0.53 |
| PARP1 + ds-5’R | −9.09 | −8.26 | −7.26 |
| Substrates | Saturation at (nM) | Ka * (M−1) |
|---|---|---|
| ds-N | 100 | 1.51 × 107 |
| ds-5’S | 200 | 3.19 × 109 |
| ds-5’R | 200 | 1.21 × 107 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masi, A.; Sabbia, A.; Ferreri, C.; Manoli, F.; Lai, Y.; Laverde, E.; Liu, Y.; Krokidis, M.G.; Chatgilialoglu, C.; Faraone Mennella, M.R. Diastereomeric Recognition of 5’,8-cyclo-2’-Deoxyadenosine Lesions by Human Poly(ADP-ribose) Polymerase 1 in a Biomimetic Model. Cells 2019, 8, 116. https://doi.org/10.3390/cells8020116
Masi A, Sabbia A, Ferreri C, Manoli F, Lai Y, Laverde E, Liu Y, Krokidis MG, Chatgilialoglu C, Faraone Mennella MR. Diastereomeric Recognition of 5’,8-cyclo-2’-Deoxyadenosine Lesions by Human Poly(ADP-ribose) Polymerase 1 in a Biomimetic Model. Cells. 2019; 8(2):116. https://doi.org/10.3390/cells8020116
Chicago/Turabian StyleMasi, Annalisa, Arianna Sabbia, Carla Ferreri, Francesco Manoli, Yanhao Lai, Eduardo Laverde, Yuan Liu, Marios G. Krokidis, Chryssostomos Chatgilialoglu, and Maria Rosaria Faraone Mennella. 2019. "Diastereomeric Recognition of 5’,8-cyclo-2’-Deoxyadenosine Lesions by Human Poly(ADP-ribose) Polymerase 1 in a Biomimetic Model" Cells 8, no. 2: 116. https://doi.org/10.3390/cells8020116
APA StyleMasi, A., Sabbia, A., Ferreri, C., Manoli, F., Lai, Y., Laverde, E., Liu, Y., Krokidis, M. G., Chatgilialoglu, C., & Faraone Mennella, M. R. (2019). Diastereomeric Recognition of 5’,8-cyclo-2’-Deoxyadenosine Lesions by Human Poly(ADP-ribose) Polymerase 1 in a Biomimetic Model. Cells, 8(2), 116. https://doi.org/10.3390/cells8020116