Epigenetically Altered T Cells Contribute to Lupus Flares

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Etiopathogenesis of Systemic Lupus Erythematosus (SLE)
2. DNA Methylation and T Cell Gene Expression
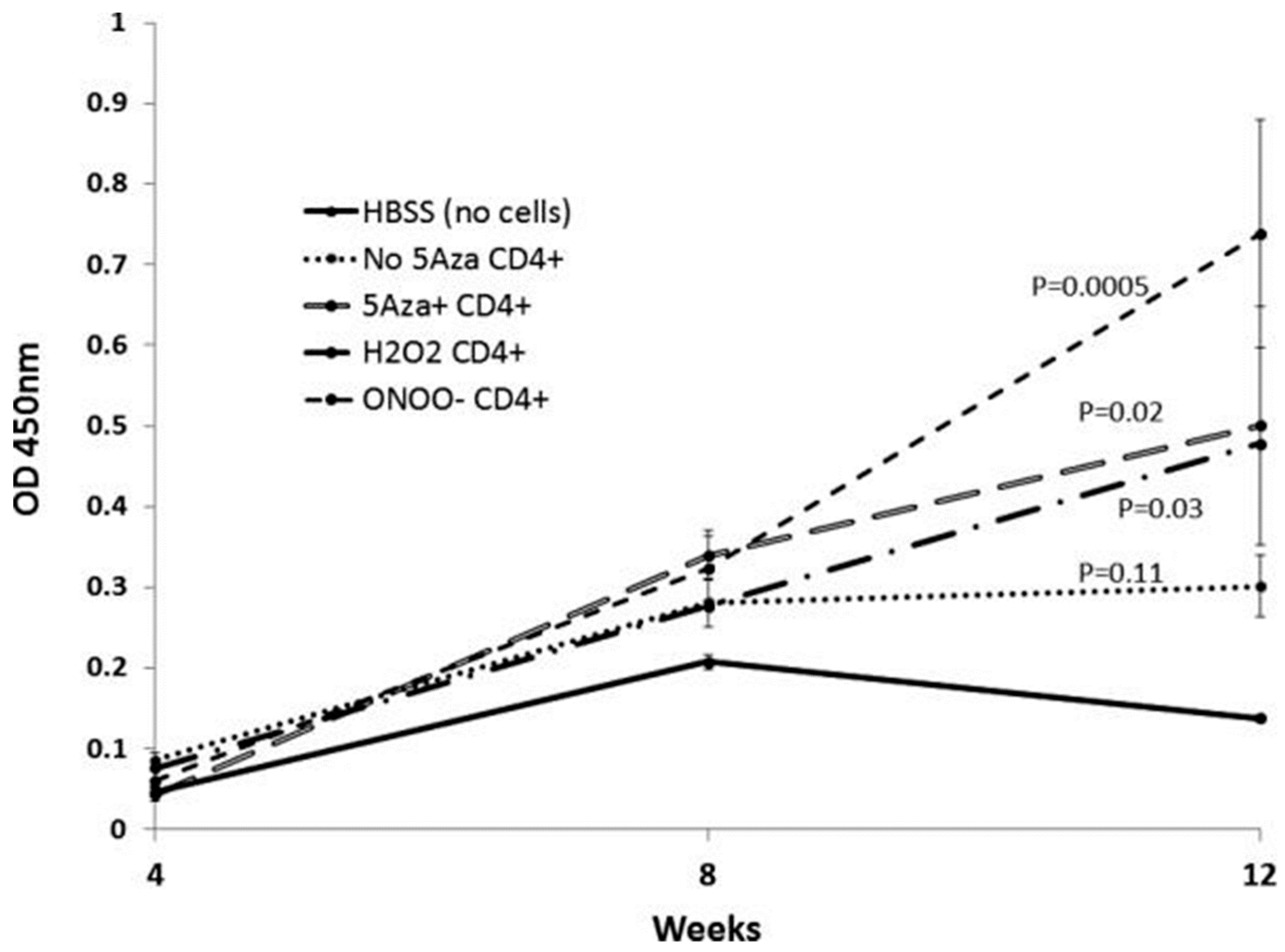
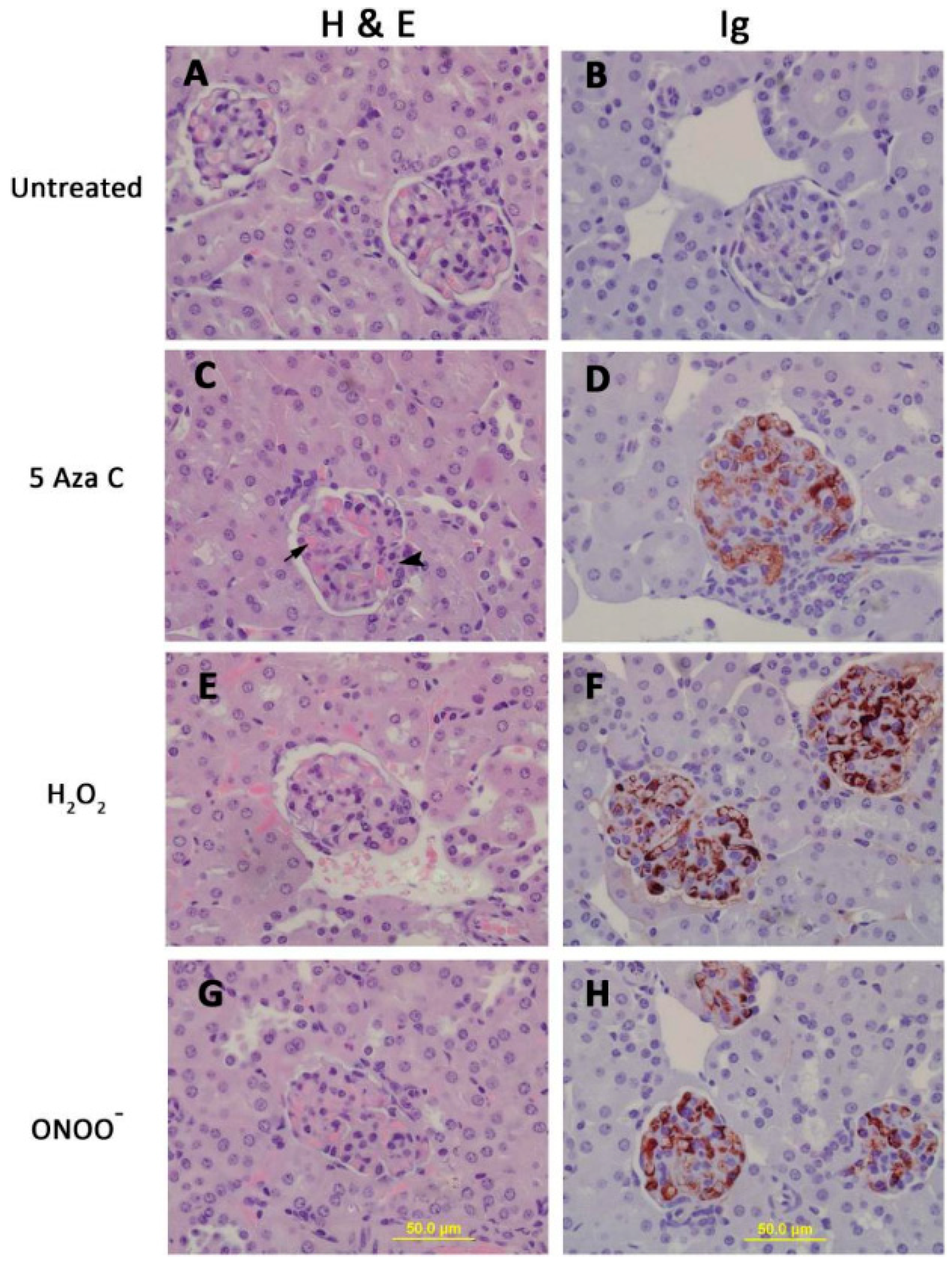
3. DNA Demethylation and T Cell Function
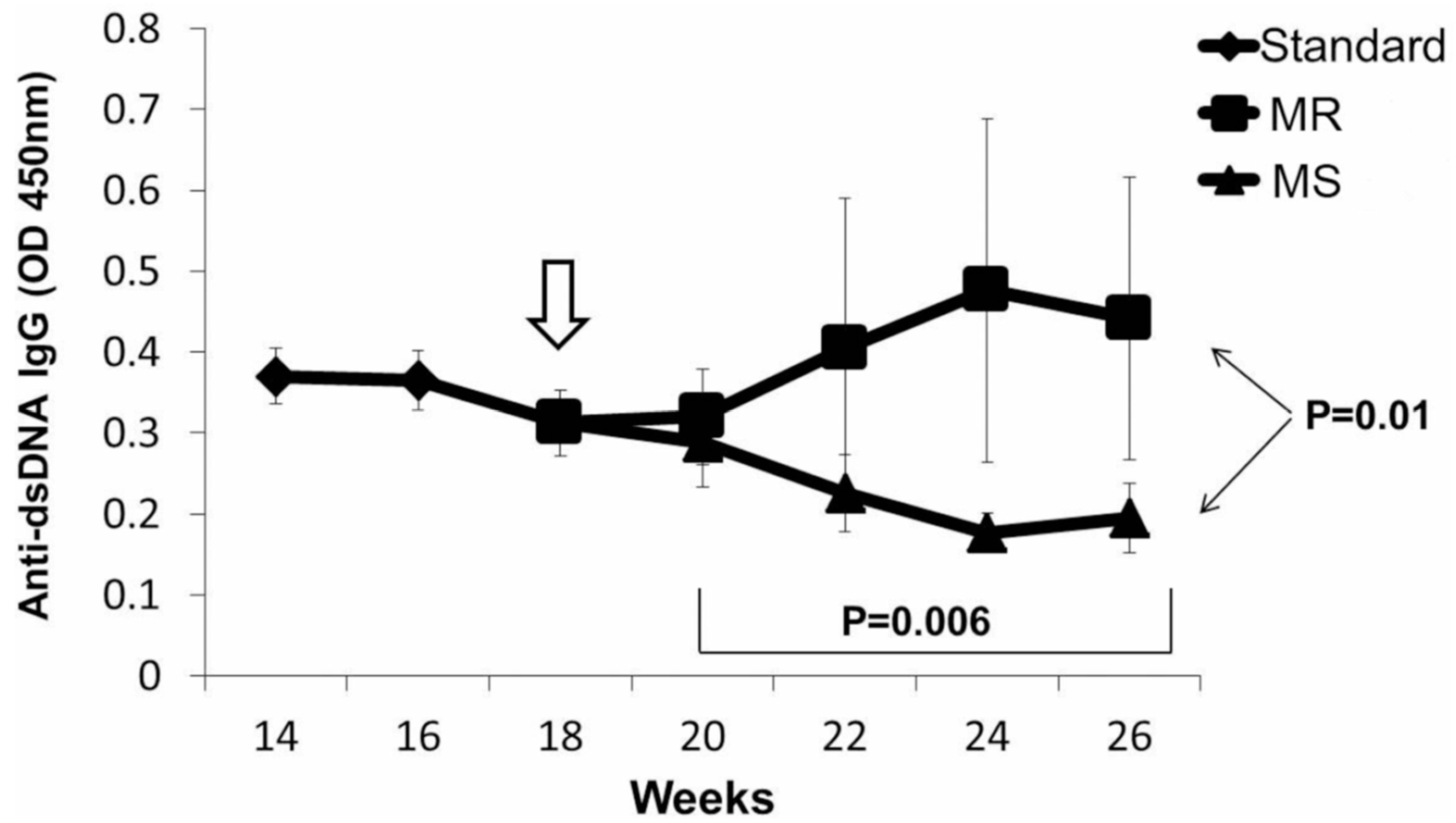
4. T Cell DNA Demethylation in Drug-Induced and Idiopathic Lupus
5. Summary
Conflicts of Interest
References
- Ghodke-Puranik, Y.; Niewold, T.B. Immunogenetics of systemic lupus erythematosus: A comprehensive review. J. Autoimmun. 2015, 64, 125–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarvinen, P.; Aho, K. Twin studies in rheumatic diseases. Semin Arthritis Rheum 1994, 24, 19–28. [Google Scholar] [CrossRef]
- Cooper, G.S.; Parks, C.G. Occupational and environmental exposures as risk factors for systemic lupus erythematosus. Curr. Rheumatol. Rep. 2004, 6, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Somers, E.C.; Richardson, B.C. Environmental exposures, epigenetic changes and the risk of lupus. Lupus 2014, 23, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Richardson, B. Primer: Epigenetics of autoimmunity. Nat. Clin. Pr. Rheumatol 2007, 3, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Ginder, G.D.; Williams, D.C., Jr. Readers of DNA methylation, the MBD family as potential therapeutic targets. Pharmacol. Pharm. 2018, 184, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Javierre, B.M.; Richardson, B. A new epigenetic challenge: Systemic lupus erythematosus. Adv. Exp. Med. Biol. 2011, 711, 117–136. [Google Scholar]
- Richardson, B.; Powers, D.; Hooper, F.; Yung, R.L.; O’Rourke, K. Lymphocyte function-associated antigen 1 overexpression and T cell autoreactivity. Arthritis Rheum 1994, 37, 1363–1372. [Google Scholar] [CrossRef]
- Lu, Q.; Wu, A.; Ray, D.; Deng, C.; Attwood, J.; Hanash, S.; Pipkin, M.; Lichtenheld, M.; Richardson, B. DNA methylation and chromatin structure regulate T cell perforin gene expression. J. Immunol. 2003, 170, 5124–5132. [Google Scholar] [CrossRef]
- Yano, S.; Ghosh, P.; Kusaba, H.; Buchholz, M.; Longo, D.L. Effect of promoter methylation on the regulation of IFN-gamma gene during in vitro differentiation of human peripheral blood T cells into a Th2 population. J. Immunol. 2003, 171, 2510–2516. [Google Scholar] [CrossRef]
- Oelke, K.; Lu, Q.; Richardson, D.; Wu, A.; Deng, C.; Hanash, S.; Richardson, B. Overexpression of CD70 and overstimulation of IgG synthesis by lupus T cells and T cells treated with DNA methylation inhibitors. Arthritis Rheum 2004, 50, 1850–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Q.; Wu, A.; Tesmer, L.; Ray, D.; Yousif, N.; Richardson, B. Demethylation of CD40LG on the inactive X in T cells from women with lupus. J. Immunol. 2007, 179, 6352–6358. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kuick, R.; Hanash, S.; Richardson, B. DNA methylation inhibition increases T cell KIR expression through effects on both promoter methylation and transcription factors. Clin. Immunol. 2009, 130, 213–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajalingam, R. Overview of the killer cell immunoglobulin-like receptor system. Methods Mol. Biol. 2012, 882, 391–414. [Google Scholar] [PubMed]
- Strickland, F.M.; Patel, D.; Khanna, D.; Somers, E.; Robida, A.M.; Pihalja, M.; Swartz, R.; Marder, W.; Richardson, B. Characterisation of an epigenetically altered CD4+ CD28+ Kir+ T cell subset in autoimmune rheumatic diseases by multiparameter flow cytometry. Lupus Sci Med. 2016, 3, e000147. [Google Scholar] [CrossRef] [PubMed]
- Gensterblum, E.; Renauer, P.; Coit, P.; Strickland, F.M.; Kilian, N.C.; Miller, S.; Ognenovski, M.; Wren, J.D.; Tsou, P.S.; Lewis, E.E.; et al. CD4+CD28+KIR+CD11ahi T cells correlate with disease activity and are characterized by a pro-inflammatory epigenetic and transcriptional profile in lupus patients. J. Autoimmun. 2018, 86, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Via, C.S. Advances in lupus stemming from the parent-into-F1 model. Trends Immunol. 2010, 31, 236–245. [Google Scholar] [PubMed] [Green Version]
- Yung, R.; Kaplan, M.; Ray, D.; Schneider, K.; Mo, R.R.; Johnson, K.; Richardson, B. Autoreactive murine Th1 and Th2 cells kill syngeneic macrophages and induce autoantibodies. Lupus 2001, 10, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.R.; Carugati, A.; Fadok, V.A.; Cook, H.T.; Andrews, M.; Carroll, M.C.; Savill, J.S.; Henson, P.M.; Botto, M.; Walport, M.J. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J. Exp. Med. 2000, 192, 359–366. [Google Scholar] [CrossRef]
- Denny, M.F.; Chandaroy, P.; Killen, P.D.; Caricchio, R.; Lewis, E.E.; Richardson, B.C.; Lee, K.D.; Gavalchin, J.; Kaplan, M.J. Accelerated macrophage apoptosis induces autoantibody formation and organ damage in systemic lupus erythematosus. J. Immunol. 2006, 176, 2095–2104. [Google Scholar] [CrossRef]
- Richardson, B.C.; Liebling, M.R.; Hudson, J.L. CD4+ cells treated with DNA methylation inhibitors induce autologous B cell differentiation. Clin. Immunol. Immunopathol. 1990, 55, 368–381. [Google Scholar] [CrossRef]
- Gorelik, G.; Sawalha, A.H.; Patel, D.; Johnson, K.; Richardson, B. T cell PKCdelta kinase inactivation induces lupus-like autoimmunity in mice. Clin. Immunol. 2015, 158, 193–203. [Google Scholar] [CrossRef]
- Yung, R.; Williams, R.; Johnson, K.; Phillips, C.; Stoolman, L.; Chang, S.; Richardson, B. Mechanisms of drug-induced lupus. III. Sex-specific differences in T cell homing may explain increased disease severity in female mice. Arthritis Rheum 1997, 40, 1334–1343. [Google Scholar] [Green Version]
- Cornacchia, E.; Golbus, J.; Maybaum, J.; Strahler, J.; Hanash, S.; Richardson, B. Hydralazine and procainamide inhibit T cell DNA methylation and induce autoreactivity. J. Immunol. 1988, 140, 2197–2200. [Google Scholar] [PubMed]
- Scheinbart, L.S.; Johnson, M.A.; Gross, L.A.; Edelstein, S.R.; Richardson, B.C. Procainamide inhibits DNA methyltransferase in a human T cell line. J. Rheumatol. 1991, 18, 530–534. [Google Scholar] [PubMed]
- Deng, C.; Lu, Q.; Zhang, Z.; Rao, T.; Attwood, J.; Yung, R.; Richardson, B. Hydralazine may induce autoimmunity by inhibiting extracellular signal-regulated kinase pathway signaling. Arthritis Rheum 2003, 48, 746–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quddus, J.; Johnson, K.J.; Gavalchin, J.; Amento, E.P.; Chrisp, C.E.; Yung, R.L.; Richardson, B.C. Treating activated CD4+ T cells with either of two distinct DNA methyltransferase inhibitors, 5-azacytidine or procainamide, is sufficient to cause a lupus-like disease in syngeneic mice. J. Clin. Investig. 1993, 92, 38–53. [Google Scholar] [CrossRef]
- Richardson, B.; Scheinbart, L.; Strahler, J.; Gross, L.; Hanash, S.; Johnson, M. Evidence for impaired T cell DNA methylation in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum 1990, 33, 1665–1673. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, M.; Sawalha, A.H.; Richardson, B.; Lu, Q. Impaired DNA methylation and its mechanisms in CD4(+)T cells of systemic lupus erythematosus. J. Autoimmun. 2013, 41, 92–99. [Google Scholar] [CrossRef]
- Gorelik, G.J.; Yarlagadda, S.; Richardson, B.C. Protein kinase Cdelta oxidation contributes to ERK inactivation in lupus T cells. Arthritis Rheum 2012, 64, 2964–2974. [Google Scholar] [CrossRef]
- Ahsan, H. 3-Nitrotyrosine: A biomarker of nitrogen free radical species modified proteins in systemic autoimmunogenic conditions. Hum. Immunol. 2013, 74, 1392–1399. [Google Scholar] [PubMed]
- Strickland, F.M.; Li, Y.; Johnson, K.; Sun, Z.; Richardson, B.C. CD4+ T cells epigenetically modified by oxidative stress cause lupus-like autoimmunity in mice. J. Autoimmun. 2015, 62, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N. Role of methionine on epigenetic modification of DNA methylation and gene expression in animals. Anim. Nutr. 2018, 4, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S. Choline, Other Methyl-Donors and Epigenetics. Nutrients 2017, 9, 445. [Google Scholar] [CrossRef] [PubMed]
- Strickland, F.M.; Hewagama, A.; Wu, A.; Sawalha, A.H.; Delaney, C.; Hoeltzel, M.F.; Yung, R.; Johnson, K.; Mickelson, B.; Richardson, B.C. Diet influences expression of autoimmune-associated genes and disease severity by epigenetic mechanisms in a transgenic mouse model of lupus. Arthritis Rheum 2013, 65, 1872–1881. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Soto, M.; Sanchez-Hidalgo, M.; Alarcon-de-la-Lastra, C. An update on diet and nutritional factors in systemic lupus erythematosus management. Nutr. Res. Rev. 2017, 30, 118–137. [Google Scholar] [CrossRef] [PubMed]
- Tomasello, G.; Mazzola, M.; Leone, A.; Sinagra, E.; Zummo, G.; Farina, F.; Damiani, P.; Cappello, F.; Geagea, G.; Jurjus, A.; et al. Nutrition, oxidative stress and intestinal dysbiosis: Influence of diet on gut microbiota in inflammatory bowel diseases. Biomed. Pap. Med. Fac Univ Palacky Olomouc Czech. Repub. 2016, 160, 461–466. [Google Scholar] [CrossRef] [Green Version]



© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Richardson, B. Epigenetically Altered T Cells Contribute to Lupus Flares. Cells 2019, 8, 127. https://doi.org/10.3390/cells8020127
Richardson B. Epigenetically Altered T Cells Contribute to Lupus Flares. Cells. 2019; 8(2):127. https://doi.org/10.3390/cells8020127
Chicago/Turabian StyleRichardson, Bruce. 2019. "Epigenetically Altered T Cells Contribute to Lupus Flares" Cells 8, no. 2: 127. https://doi.org/10.3390/cells8020127
APA StyleRichardson, B. (2019). Epigenetically Altered T Cells Contribute to Lupus Flares. Cells, 8(2), 127. https://doi.org/10.3390/cells8020127