1. Introduction
It is now well established that pluripotency of the cells is maintained as a consequence of the balance of different lineage-specifying forces. Instead, lineage specification is due to modifications in the combination of specific transcription factors and miRNAs that carry out a particular transcription program.
Differentiation of hematopoietic progenitors into distinct lineages depends on the activation of specific and tightly regulated transcription factors. First specification is between myeloid and erythroid lineages and depends on several factors playing critical roles. As an example, PU1 is an essential factor for reconstitution of the myeloid lineage [
1], and Cebp-a, -b, and -ε factors play major roles in commitment toward myeloid cells: granulocytes, monocytes and macrophages [
2,
3,
4]. Macrophages are heterogeneous and versatile cells that can acquire different functional phenotypes depending on the microenvironment and the resident cytokines. Two main subsets of macrophages are known, the classically activated or inflammatory (M1 type), and the alternatively activated anti-inflammatory (M2 type). They polarize in the presence of granulocyte macrophage (GM) or macrophage (M) colony-stimulating factor. On the other hand, osteoclasts derive from the commitment of hematopoietic stem cells (HSCs) into the monocyte/macrophage lineage through PU1 [
5]. Afterwards, macrophages develop into pre-osteoclasts, undergoing a complex differentiation process implying a series of steps—proliferation, migration and fusion—that lead to differentiated osteoclasts (i.e., multinucleated and polarized cells with bone resorptive activity) [
5]. Osteoclastogenesis depends on two main cytokines, the M-CSF and the receptor activator of nuclear factor-κB ligand (RANKL), which are essential for osteoclast lifespan and function through their receptors, the colony-stimulating factor receptor (c-fms) and receptor activator of nuclear factor-κB (RANK), respectively [
6,
7,
8]. Therefore, osteoclasts share with macrophages a trait of differentiation due to the activation of many signalling pathways and transcription factors as a consequence of the same cytokine stimulation (M-CSF). Osteoclast differentiation is a complex mechanism that involves multiple factors such as cytokines, transcription factors and miRNAs [
9]. NFATc1 is considered the master transcription factor regulating osteoclast differentiation [
10] and it is maintained in the cytosol as inactive hyper-phosphorylated protein in pre-osteoclasts. During the following differentiation, calcium signalling, in particular the calcium oscillations observed in osteoclast precursors, stimulates the phosphatase calcineurin, which dephosphorylates NFATc1 and allows its nuclear translocation [
10,
11]. In the nucleus, NFATc1 regulates the expression of many osteoclast-specific genes such as tartrate-resistant acid phosphatase (TRAP), c-Fos, cathepsin K (CtsK) and microphthalmia transcription factor (MITF) [
8].
NFATc1 is widely expressed in different cell types and is essential for the development of many tissues [
12]. Although it is considered the master regulator of osteoclastogenesis, it is not clear how the ubiquitous NFATc1 can direct an osteoclast-specific transcription program.
In this study, we tried to unravel the molecular pathway underlying pre-osteoclast differentiation by PCR array analysis, using RAW 264.7 macrophage cell line. We studied the expression profiles of untransfected and siRNA-NFATc1 transfected cells during the early steps of osteoclast differentiation after RANKL induction. Gene Ontology (GO) and Ingenuity Pathway Analysis (IPA) mechanistic analyses were performed taking into consideration the function of mRNAs, which changed their expression significantly in PCR arrays. Altogether, our results suggest that GATA2 was a new target downstream of NFATc1, whose depletion directly or indirectly caused GATA2 activation. Thus, GATA2 might be involved in the differentiation of bone marrow-derived macrophages in the absence of NFATc1, while precisely NFATc1 could downregulate it during commitment of macrophages towards the osteoclast lineage.
Overall, we hypothesize that these two transcription factors could be involved in the specification of different cell lineages.
2. Materials and Methods
2.1. Cell Culture and Osteoclastogenesis In Vitro
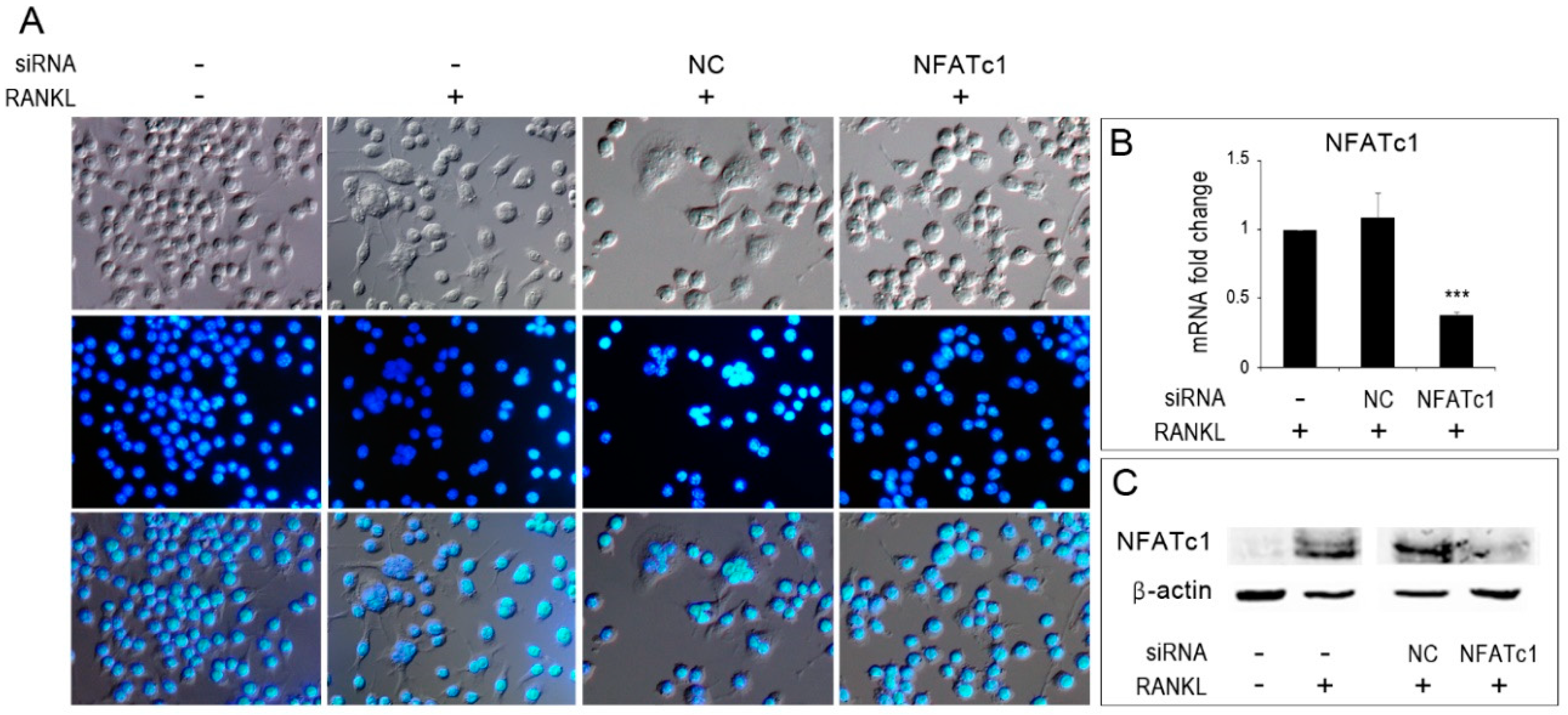
The murine RAW 264.7 macrophage cell line was purchased from the American Type Culture Collection (Manassas, VA, USA). Cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco, NY, USA) with 10% heat-inactivated fetal bovine serum (FBS, Sigma Chemical Co., St. Louis, MO, USA), 100 U/ml penicillin and 100 µg/ml streptomycin. To induce osteoclast differentiation, cells were suspended in alpha-minimal essential medium (α-MEM Gibco, NY, USA) with 10% heat-inactivated fetal bovine serum (FBS, Sigma), 100 U/ml penicillin and 100 µg/ml streptomycin with RANKL 50 ng/mL (Peprotech, USA). To identify multinucleated osteoclasts, RAW 264.7 cells (untransfected and transfected) were grown on coverslips for 24 h with or without RANKL (50 ng/mL). Cells were fixed with 4% paraformaldehyde (Sigma Chemical Co., St. Louis, MO, USA) in physiological phosphate buffered saline (PBS) for 15 min. All the coverslips were mounted on glass slides with mounting medium containing DAPI (Invitrogen, Thermo Fisher Scientific Carlsbad, CA, USA), and observed under an Axioskop-2 Plus microscope (Zeiss, Germany), equipped for epifluorescence. Images were recorded using a digital camera system, cropped and grouped, and contrast and brightness were adjusted using Photoshop CS2 (Adobe Systems, San Jose, CA, USA).
2.2. Small Interfering RNA (siRNA) Transfection
Cells were transfected with NFATc1-siRNA or non-correlated (NC) siRNA (Qiagen, Germantown, Maryland, USA) as previously reported [
13]. In brief, cells (2.5 × 10
5) were seeded onto 6-well plates in medium without antibiotics; 24 h later, the transfection of siRNAs was carried out with Lipofectamine RNAiMAX (Invitrogen, Thermo Fisher Scientific Carlsbad, CA, USA). All transfections were carried out with 20 μM duplex siRNA in medium without FBS or antibiotics. Then, 6 h later, we added RANKL (50 ng/mL) to the medium. One or two days after transfection, cells were recovered to perform further analyses. Experiments were repeated three times.
2.3. RNA Extraction and cDNA Synthesis
RAW 264.7 cells were cultured (1 × 106 cells/well) in a 6-well plate overnight. Cells were untransfected or transfected with specific siRNA and treated with RANKL 50 ng/mL for 24, 48 and 72 h. After this time of stimulation, the cells were washed once with PBS. Total RNA was isolated using the GenElute Mammalian Total RNA Miniprep Kit (Sigma Chemical Co., St. Louis, MO, USA) and quantified by using a spectrophotometer (Eppendorf S.r.l., Hamburg, Germany). Total RNA (3μg) was reverse-transcribed to cDNA using the SuperScript Vilo (Invitrogen, Thermo Fisher Scientific Carlsbad, CA, USA).
2.4. Quantitative Polymerase Chain Reaction (QPCR)
Quantification of gene expression was performed using the StepOne Plus Real-Time PCR as described in the manufacturer’s manual (Applied Biosystems Life Technologies, Carlsbad, CA, USA) with SYBR Green chemistry and the comparative threshold cycle method [
14]. The quantitative PCR (QPCR) was run as follows: 1 × cycle, denaturing at 95 °C for 10 min for DNA polymerase activation; and 38 cycles, melting at 95 °C for 15 s and annealing/extension at 60 °C for 60 s. QPCR was then performed in triplicate on each cDNA sample for each gene using primers by Qiagen: QT001676692 (
NFATc1), QT00166663 (
TRAF6), QT00108815 (
MMP9), QT00131313 (
MITF1), QT00131012 (
TRAP), QT01047032 (
DC-STAMP), QT02589489 (
CTSK 2), QT00149415 (
RelA), QT00197568 (
RhoA), QT01167257 (
OSCAR), QT00096194 (
Myc), QT00102193 (
Runx2), QT00159768 (
SMAD5), QT00103005 (
STAT4), QT00170975 (
Hnf1), QT00249781 (
Esr1), QT00265846 (
Egr1), QT00160524 (
GATA2) and AQT01658692 (
GAPDH). For
Fos we used the following primers, after validation F: 5′CACTCCAAGCGGAGACAGAT3′ and R: 5′TCGGTGGGCTGCCAAAATAA3′. The threshold cycle (CT) values were calculated against the housekeeping gene
GAPDH. At least three distinct biological samples were examined for each gene and treatment (each performed in triplicate).
2.5. RT2 Profiler PCR Array Analysis
Mouse RT
2 Profiler PCR array for transcription factors (PAMM-075ZC) and for osteoporosis (PAMM-170ZC) in 96-well plate format (Qiagen Sciences, Germantown, MD, USA) were used to analyze gene expression changes. Samples were prepared from pooled RNA extracted from RAW264.7 cells using the RNeasy MinElute Cleanup Kit (Qiagen Sciences, Germantown, MD, USA). RAW 264.7 cells were untransfected (+/- RANKL) or transfected +RANKL (siRNA-control/siRNA-NFATc1). Total RNA was reverse-transcribed in cDNA according to Qiagen’s instructions for the RT
2 First Strand Kit. QPCR was run as described above, with RT
2 SYBR Green ROX QPCR Mastermix, in a StepOne Real-Time instrument (Applied Biosystem Life Technologies, Carlsbad, CA, USA). Normalization was done on automatic HKG panel. This method uses the five genes (wells H1–H5) that have small changes in threshold cycle (CT) values across all of the included samples. Array analysis of the results was performed using the software provided by SABiosciences (
www.SABiosciences.com).
2.6. Western Blot
RAW 264.7 cells were transfected or untransfected and cultured as described above for 24 and 72 h with RANKL. Then, cells were lysed and total cellular protein were extracted using RIPA buffer (Cell Signaling Inc. Beverly, MA, USA). The protein concentration of cell lysates was determined by the Bradford method. For western blot, 30 μg of protein was separated on 10% SDS–polyacrylamide gels by electrophoresis and transferred to a nitrocellulose membrane (Millipore Temecula, CA, USA). Membranes were incubated overnight at 4 °C with the following antibodies: NFATc1 and NFATc2 (Santa Cruz, CA, USA) at 1:1000 dilution; DC-STAMP (Millipore Temecula, CA, USA) at 1:500 dilution; GATA2 (Cloud Clone Corporation USA) at 1:500 dilution; TRAF6 (Santa Cruz, CA, USA) at 1:500 dilution; RANK (Santa Cruz, CA, USA) at 1:500 dilution; MITF (Abcam, England) at 1:500 dilution; and STAT6 (Santa Cruz, CA, USA) at 1:500 dilution.
The secondary antibodies Alexa Fluor 680 Goat anti-Rabbit (1:2000) and Alexa Fluor 800 Rabbit anti-Mouse (1:5000) (Molecular Probes, Life Technologies, Carlsbad, CA, USA) were incubated for 1 h at room temperature. Proteins were visualized using an Odyssey Infrared Imaging System (LI-COR Lincoln, Nebraska USA) according to the manufacturer’s instructions. Densitometry analyses were conducted using Quantity One software (Bio-Rad Laboratories, Mississauga, Canada).
2.7. GO and Pathway Analysis
Gene Ontology Consortium (
http://geneontology.org/) database (released 9 August 2018) was used to analyze the potential functions of mRNAs differentially expressed in untransfected and siRNA-transfected RANKL-induced RAW 264.7 cells. The PANTHER database (Protein Analysis Through Evolutionary Relationships,
http://pantherdb.org) was used for the analysis as GO molecular function complete and GO biological process complete. Analysis was performed with the PANTHER Overrepresentation Test (released 5 December 2017), using a
Mus musculus reference list (all genes in database). The test that was performed is the Fisher’s exact test with FDR correction. The default output was sorted by hierarchy of the categories. By default, only the categories with
p value better than 0.05 were displayed. In the hierarchy view, the results were sorted by the fold enrichment of the most specific categories, with their parent terms (
p value better than 0.05) indented directly below. Results of all
p values have been displayed.
Protein network analysis was performed using Qiagen’s Ingenuity Pathway Analysis (IPA, Qiagen Redwood City, CA, USA) software.
2.8. Statistical Analysis
Data are expressed as mean ± S.D. of at least three independent experiments. Statistical significance between two groups was determined by a two-tailed Student’s T test. p < 0.05 was considered to indicate a statistically significant difference.
4. Discussion
Osteoclasts are derived from hematopoietic stem cell (HSCs) and later from monocyte-macrophage lineage through the action of two cytokines, the M-CSF and RANKL [
5]. The RAW 264.7 murine macrophage cell line consists of a population of pre-osteoclasts, which need only RANKL-stimulation to differentiate in osteoclasts in vitro over a four day period. As already reported, after RANKL-induction the first step is the expression of NFATc1 transcription factor. As a master regulator, NFATc1 may indeed regulate expression of many other genes [
10]. However, this transcription factor is expressed in many different cell types where it does not necessarily imply differentiation into osteoclasts. The comprehension of the mechanisms that regulate initial processes of osteoclastogenesis is of paramount importance to understanding the process of osteoclast differentiation, and in turn for managing many bone-related diseases, such as osteoporosis, osteopetrosis and rheumatoid arthritis (AR) [
9,
18,
19].
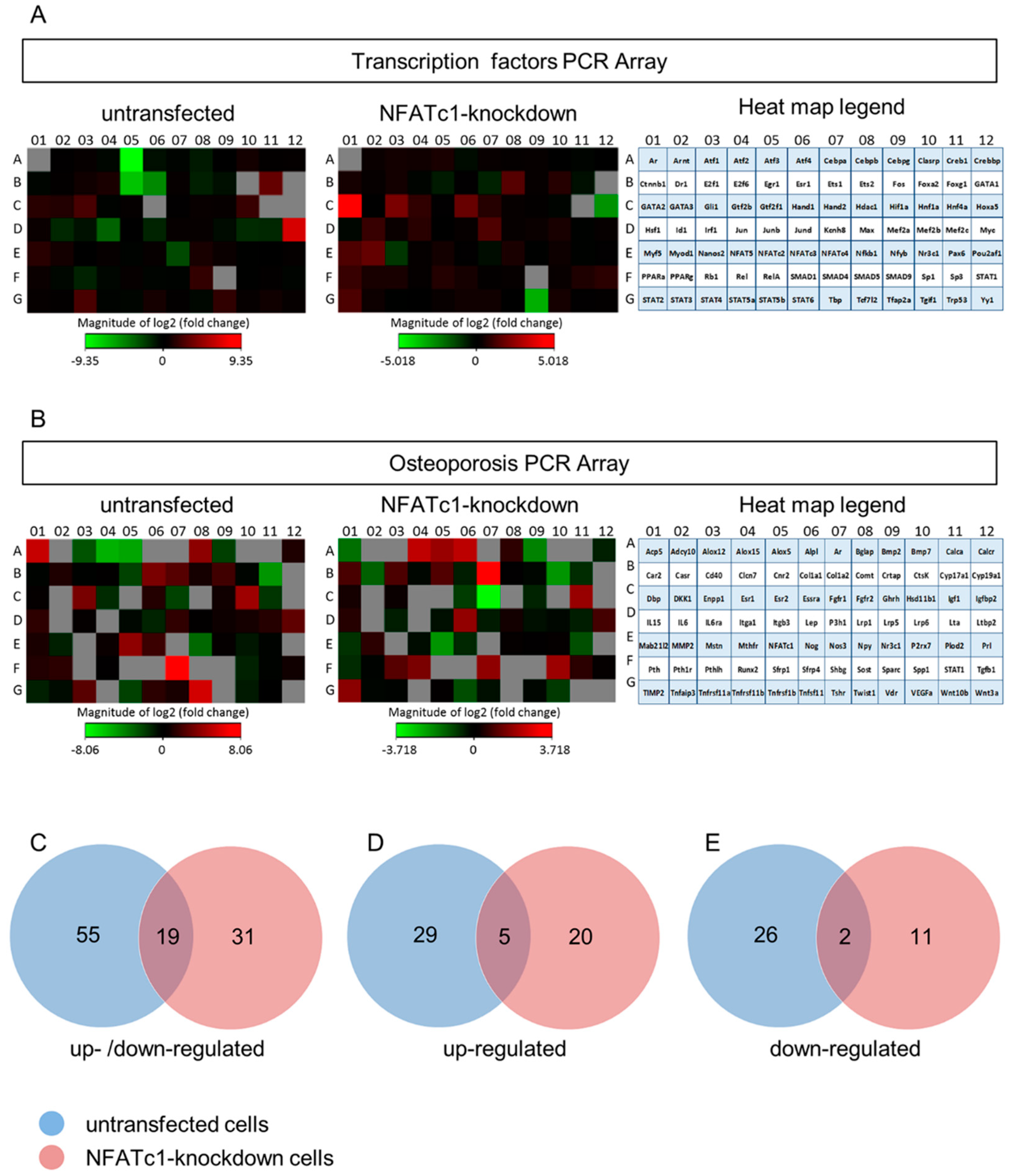
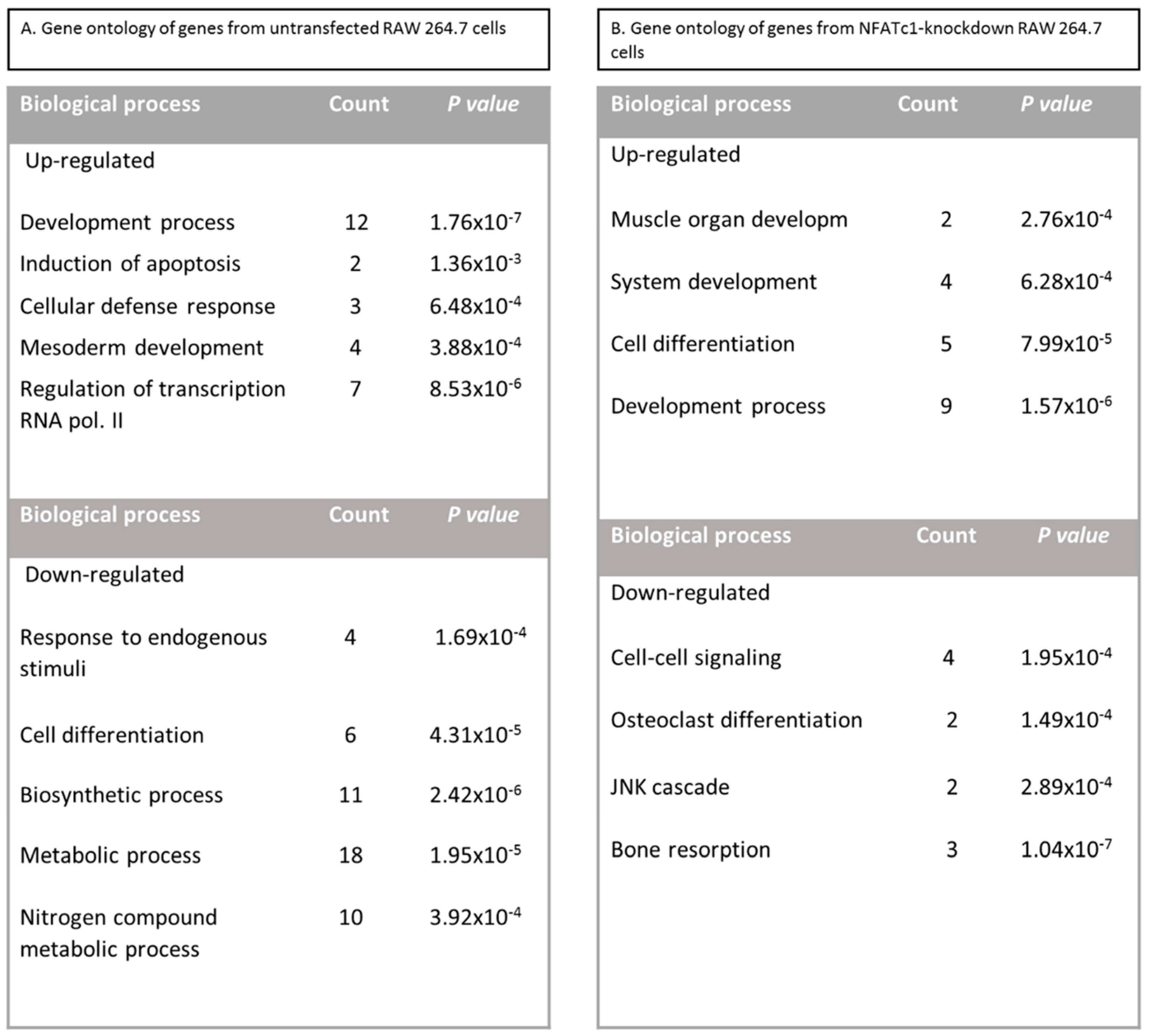
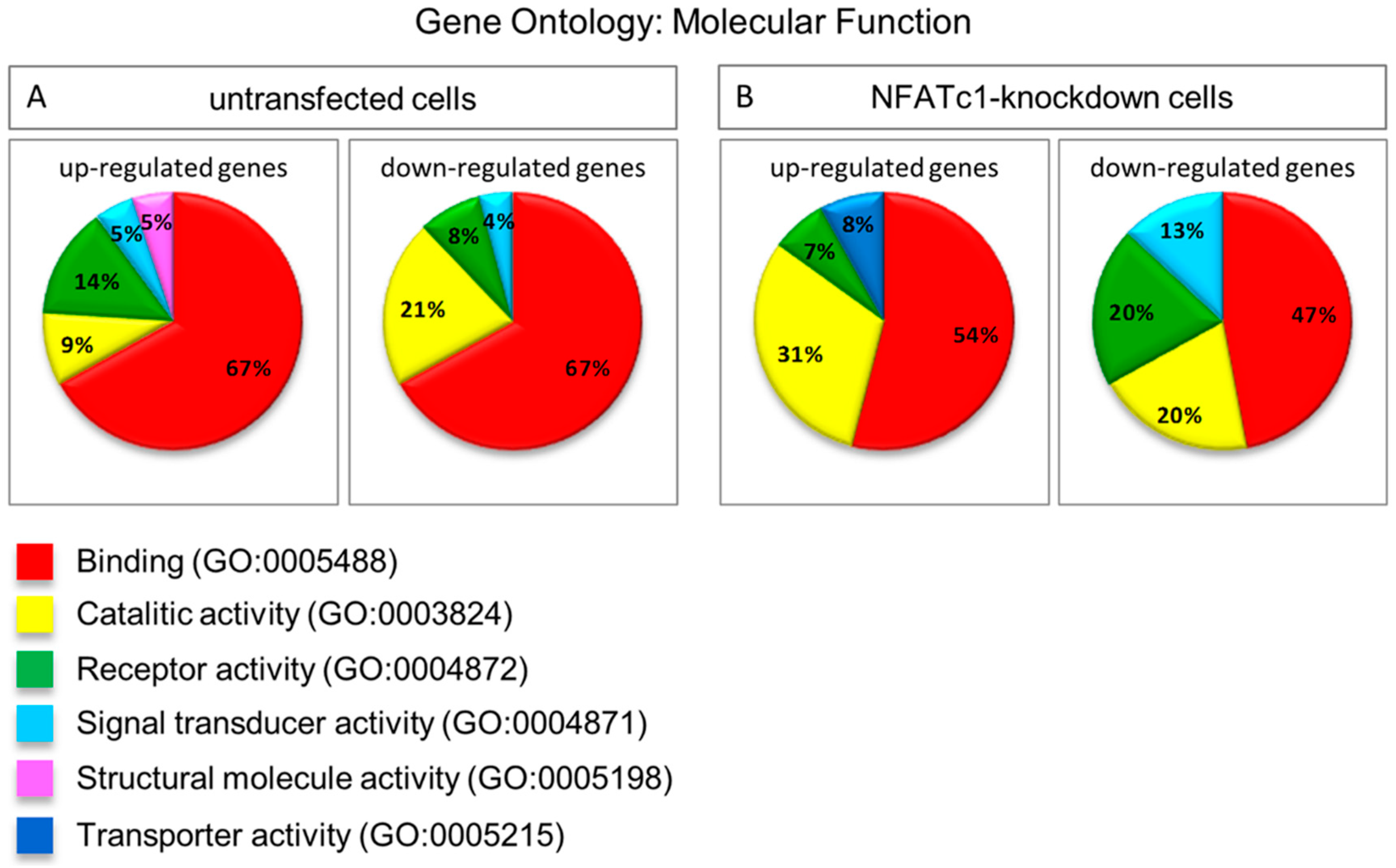
In this study, we utilized PCR array analysis to identify downstream targets of NFATc1 to clarify steps involved in pre-osteoclasts differentiation. We compared the transcriptomes of RANKL-induced pre-osteoclasts with defective or complete expression of NFATc1. We found that in untransfected cells, the cytokine modulated expression of many genes grouping in different functional categories. Gene Ontology analysis (as biological processes) grouped the up-regulated genes into “development process” (12 genes p 1.76 × 10−7) and “regulation of transcription RNA pol. II” (7 genes p 8.53 × 10−6). On the contrary, in NFATc1-knockdown cells, up-regulated genes were mainly grouped into the categories “development process” (9 genes p 1.57 × 10−6) and “cell differentiation” (5 genes p 7.99 × 10−5). Instead, the genes down-regulated in untransfected cells were grouped into “metabolic process” (18 genes p 1.95 × 10−5), “biosynthetic process” (11 genes p 2.42 × 10−6), “nitrogen compound metabolic process” (10 genes p 3.92 × 10−4) and “cell differentiation” (6 genes p 4.31 × 10−5). Most of these genes are involved in the “housekeeping” activities of many cell types. On the contrary, down-regulated genes in NFATc1-knockdown cells belong mainly to groups related to “cell signaling” (4 genes p 1.95 × 10−4), “bone resorption” (3 genes p 1.04 × 10−7) and “osteoclast differentiation” (2 genes p 1.49 × 10−4), as expected in cases of impairment of the key regulator for osteoclast transcriptional program. We focused on the “developmental processes” and “cell differentiation” related genes, since the identification of these genes could help to elucidate the molecular mechanism that underlies osteoclast differentiation.
Venn analysis revealed that 19 genes showed changes in their expression levels in both groups (untransfected and NFATc1-knockdown). In detail, we found genes that were up-regulated in one group and down-regulated in the other, and genes that were up-regulated or down-regulated in both groups. Among them, there were genes that changed expression level dramatically and genes that showed very mild changes. As an example, Acp5 was up-regulated in untransfected cells and down-regulated in NFATc1-knockdown cells (fold changes: 41.89 and −3.06, respectively). This result was expected, since Acp5 codes for TRAP, which is a hallmark of osteoclast differentiation. Another interesting gene that changed in the two conditions was the transcription factor Jun. This gene was significantly down-regulated (fold change: –11.86) in untransfected cells, while it was just under the exclusion threshold in NFATc1-knockdown cells (1.66). Our GO analysis grouped this gene in the “cell differentiation” category together with Ets2 (−2.53 and 2.76), Runx2 (1.05 and 2.88) and Myod1 (1.79 and 3.21), showing the same trend in the two groups. On the contrary, bone morphogenetic protein 2 (Bmp2) was down-regulated in the two groups (fold changes: −4.15 and −3.69), which was also expected. In fact, Bmp2 is a hallmark of osteoblast differentiation and it is likely that NFATc1 does not regulate it.
Among the up-regulated genes in untransfected cells,
Myc appeared interesting since it strongly increased in our PCR array (108-fold) and was validated by QPCR.
Myc is a proto-oncogene known for controlling proliferation, differentiation, apoptosis and cancer. However, it has been reported that
Myc is correlated with osteoclasts differentiation induced by RANKL, and its dominant negative isoform reduces the expression of osteoclast marker proteins such as CtsK and TRAP [
20]. In our bioinformatics analysis,
Myc belonged to the GO group “regulation of transcription RNA pol. II” together with NFATc1. This group collected transcription factors that could participate in osteoclasts differentiation. Accordingly, our data showed a strong increase of
Myc expression in the initial phase of differentiation in untransfected cells but not in NFATc1-knockdown cells, correlating
Myc expression to NFATc1 levels. Recently, it has been demonstrated that
Myc has a role in the induction of estrogen receptor–related receptor
α (ERR
α), a nuclear receptor that cooperates with NFATc1 to drive osteoclastogenesis [
21].
Another transcription factor up-regulated in untransfected cells and down-regulated in NFATc1-knockdown cells was
MITF, which we analyzed regardless of the PCR arrays data. MITF is a family of transcription factors expressed in a tissue- and cell lineage-specific manner; among them MITF-E is expressed in monocyte-macrophage cell lineage and osteoclasts. It has been reported that miR-155 inhibits osteoclast differentiation by targeting MITF [
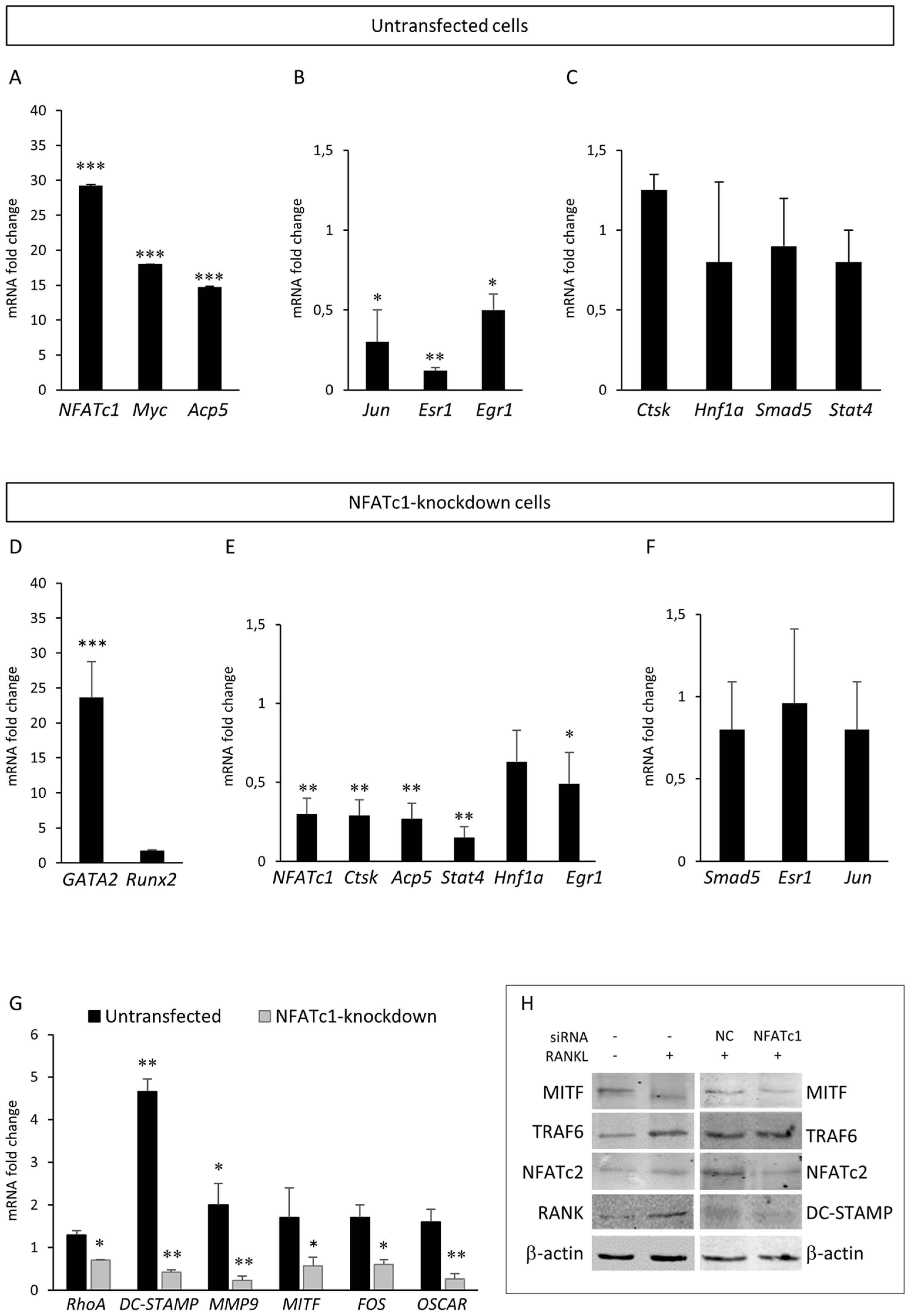
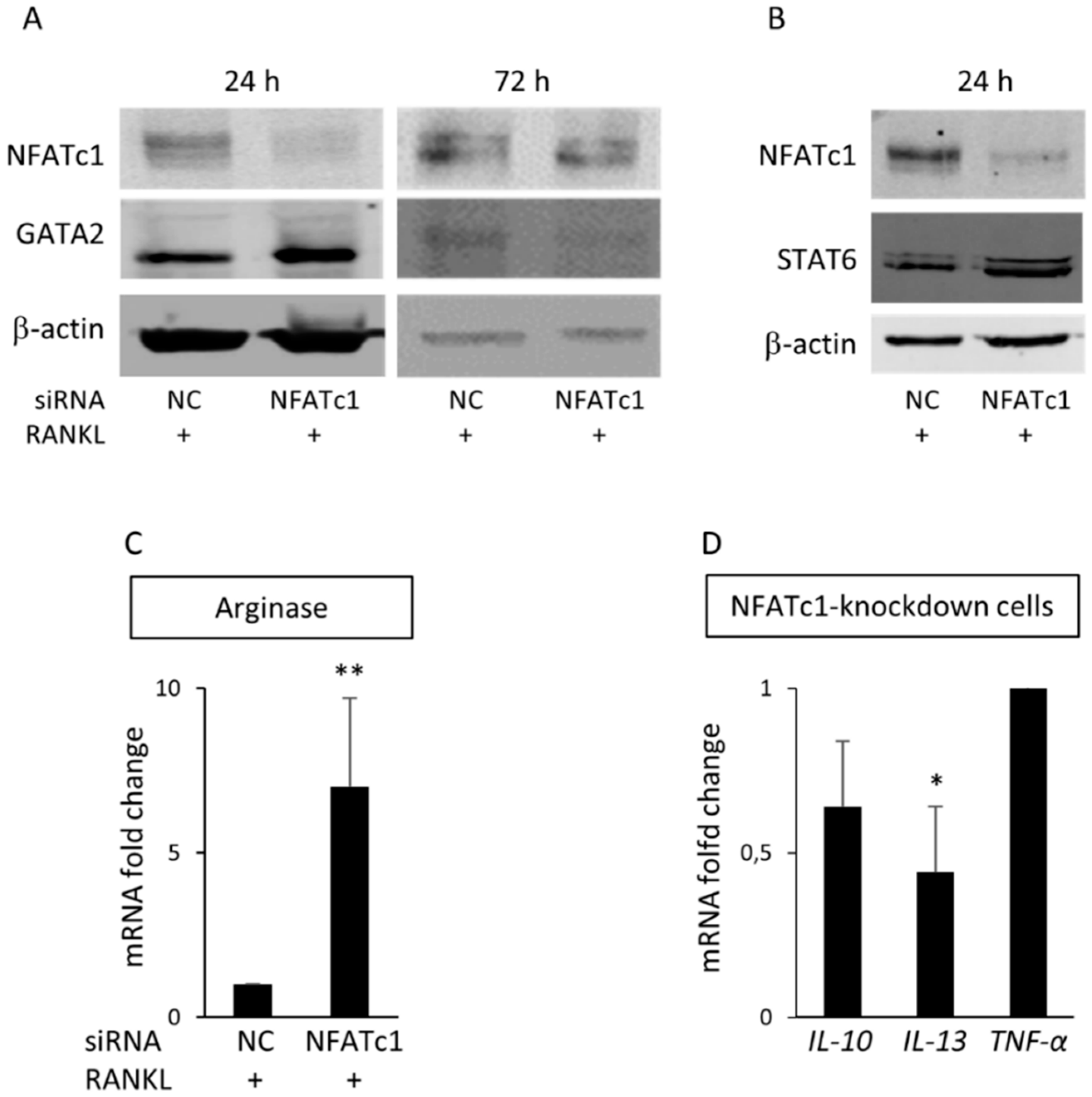
22]. Data obtained by QPCR and western blot showed that MITF-mRNA and protein decreased in NFATc1-knockdown cells (
Figure 5G–H). Lu et al. [
23] found that MITF silencing did not influence NFATc1 expression or activation, and that MITF up-regulation did not have positive influence on NFATc1 expression level. These results suggest that MITF functions downstream of NFATc1 within RANKL signaling, even if it was found that MITF up-regulation was sufficient to induce osteoclast markers such as TRAP, DC-STAMP, CtsK and ATP6V0 [
23]. Accordingly, our data point to MITF as a target of NFATc1, since NFATc1-depletion inhibits MITF expression. Ultimately, the role of MITF is to promote the amplification of the NFATc1 signaling during osteoclastogenesis [
23], whereas NFATc1 is critical for MITF expression (
Figure 5F,G). Therefore, it might be proposed that MITF is an inducer for the osteoclast differentiation acting in concert with NFATc1. In fact, as we have already discussed, NFATc1 is ubiquitous and is involved in many differentiation programs other than osteoclastogenesis, whereas MITF-E is present only in the osteoclasts.
Another interesting gene is
GATA2, which is up-regulated in the two groups of cells but with strong differences. The level of its expression is just above the exclusion threshold (2.59-fold) in untransfected cells, but is strongly up-regulated in NFATc1-knockdown cells (32.40-fold). We validated these results finding increased expression levels of both mRNA and protein in NFATc1-knockdown cells (
Figure 5D and
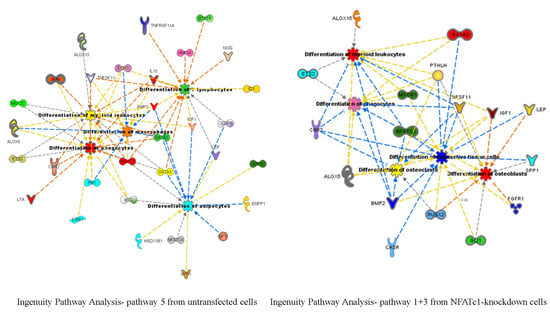
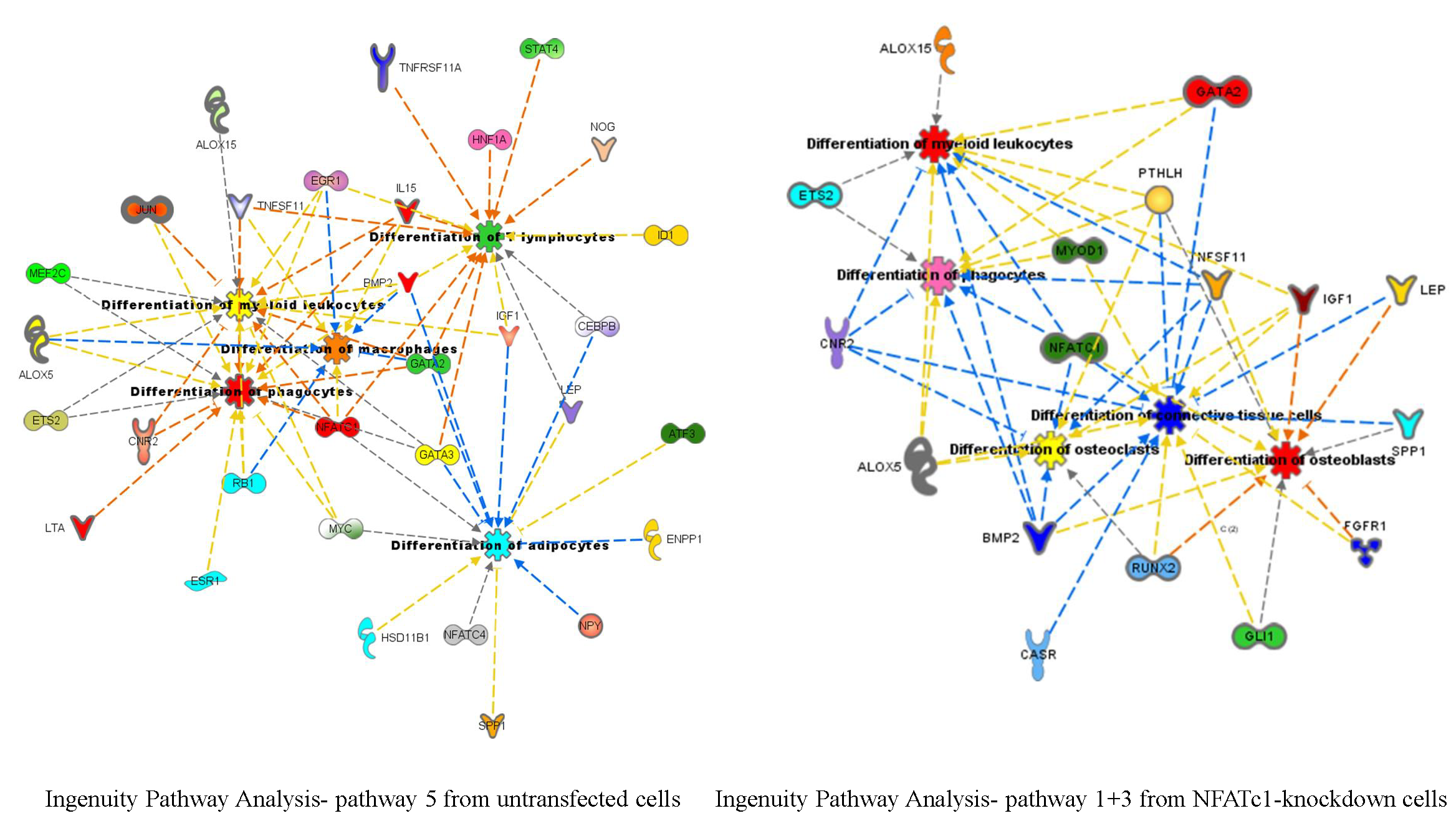
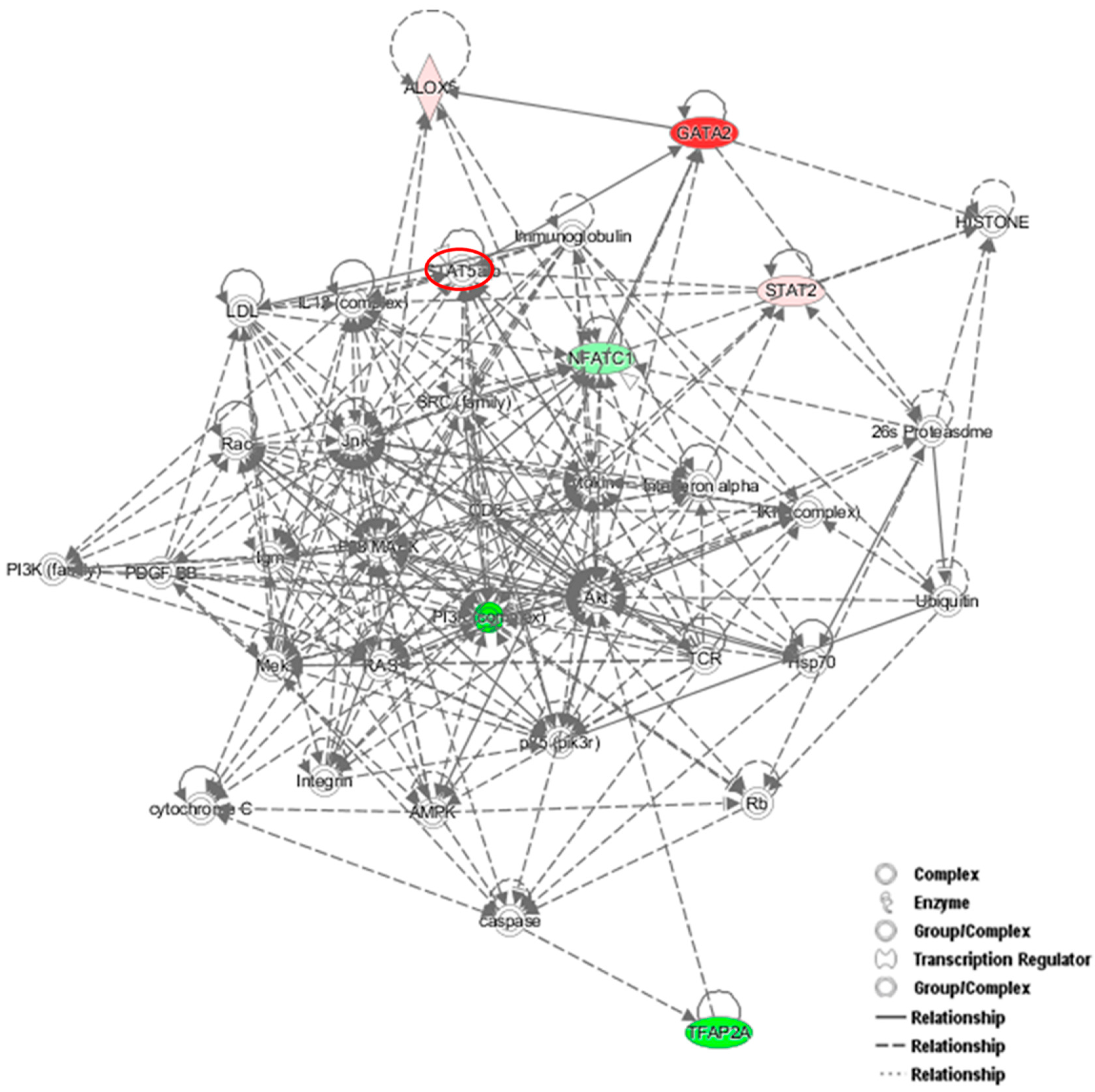
Figure 7A). Interestingly, the GATA2-mRNA levels were inversely correlated with those of NFATc1, suggesting the existence of a cross-regulation between NFATc1 and GATA2 in RAW 264.7 cells. In our bioinformatics analyses, GATA2 belonged to the GO group of “cell differentiation” and, most importantly, it was a central node in the protein network analysis by IPA (
Figure 6), closely related to NFATc1 and STAT family members. GATA2 is considered an inducer for the reprogramming of cell pluripotency and belongs to a family of six transcription factors with well-established roles in cellular differentiation. GATA1, GATA2 and GATA3 are expressed in hematopoietic cells [
24,
25,
26], whereas GATA4, GATA5 and GATA6 regulate cell differentiation in non-hematopoietic cells [
27,
28,
29]. GATA1 and GATA2 are often expressed in a mutually antagonistic manner, regulating the differentiation of hematopoietic stem cells (HSCs) at different times [
26,
30]. In fact, GATA2 acts very early in the differentiation of bone marrow–derived macrophage (BMM)–HSCs and is down-regulated during lineage commitment, whereas GATA1 interferes in the last steps of the differentiation [
30]. In addition, GATA2 is required to generate osteoclast progenitors [
31], while GATA1 is dispensable for osteoclastogenesis [
32]. Therefore, the GATA2–GATA1 ratio in hematopoietic progenitors controls lineage divergence between osteoclasts and erythrocytes/megakaryocytes. In agreement, our PCR array data showed the GATA2–GATA1 ratio was 2.59:1 in untransfected cells and 32.40:1 in NFATc1-knockdown cells, as expected in the case of lineage divergence towards osteoclasts. In addition, it should be taken into account that GATA2, but not GATA1, is critical in maintaining the differentiated state of mast cell/basophil as well as eosinophil lineages [
33]. In fact, deletion of GATA2 in bone marrow mononuclear cells (BMMCs) results in an impairment of mast cell–like phenotype with a strong expression of macrophage markers, such as CD11b- and/or Ly6G/C [
34]. GATA2 is also crucial for maintaining the expression of the hallmarks of mast cell/basophil phenotypes, for example,
Alox5, which is commonly expressed in both cell types [
35]. In agreement, our array data showed that
Alox 5 and
Alox 15 increased mRNA expression levels (5.25 and 3.83, respectively) in NFATc1-knockdown cells parallel to
GATA2 up-regulation, while, on the contrary, they were strongly down-regulated (−36.52 and −47.76, respectively) in untransfected cells. Our data strongly suggests that GATA2 is a downstream target of NFATc1, and we might speculate that NFATc1 possibly represses its expression through miRNA induction.
Many cytokines are crucial for the differentiation of pre-osteoclasts, such as IL-1, IL-6, IL-15 and TNF-α, whereas others suppress osteoclastogenesis including IL-3, IL-4, IL-10 and IFN [
36]. Accordingly, our PCR arrays data showed that the pro-osteoclastogenesis cytokine
IL-15 increased (2.50-fold) in untransfected cells because of RANKL-stimulation (
Table 1). Cytokines are mainly produced by T lymphocytes, although IL-6, IL-10, IL-13 and TNF-α are also produced by mast cells and monocyte-macrophages in which they induce the differentiation [
37]. In addition, the pro-inflammatory cytokines IL-6 and TNF-α are considered hallmarks of activated macrophage phenotype M1, whereas IL-10 and IL-13 are hallmarks of alternatively activated macrophage phenotype M2 (osteoclast-like) [
38]. Our PCR array data showed that
IL6 expression level is under the exclusion threshold (≥2) in both treatments (
Tables S1 and S2). Further, we analyzed the mRNA expression levels of
IL-10, IL-13 and
TNF-α cytokines. Our results indicated that
TNF-α and
IL-10 expression levels did not significantly change in NFATc1-knockdown cells with respect to the untransfected cells, whereas
IL-13 mRNA was significantly inhibited in NFATc1-knockdown cells (
Figure 7D). The cytokine IL-4 has anti-osteoclastogenesis activity through binding with its receptor (IL4-R) and activation of the transcription factor STAT6 [
39]. In agreement, our results indicate that STAT6 protein levels increased in NFATc1-knockdown cells, suggesting anti-osteoclastogenesis activity. Altogether, these data suggest that NFATc1 ablation caused GATA2 and STAT6 activation, but did not inhibit the expression of pro-osteoclastogenesis cytokines (TNF-α and IL-6), whereas it caused an impairment of the expression of some cytokines (IL-13) associated with macrophage differentiation of the M2 phenotype (osteoclast-like).
Ultimately, the balances and order of expression of multiple transcription factors, rather than the expression level of a single factor, such as
GATA2,
STAT6 and
NFATc1, are important for the specification of cellular fate. This is not surprising since it has been reported that exactly these factors—GATA2, STAT6 and NFATc1—are tightly controlled for the genome-wide occupancy and consequent transcriptional programs that could be activated, such as in the case of endothelial cell activation [
40].
Nevertheless, our data suggest that GATA2 could be one of the important factors that determine differentiation towards macrophages in the absence of NFATc1, even if the cellular fate in the presence of GATA2 up-regulation is presently unknown. However, the observed activation of the transcription factors STAT6 and GATA2 in the absence of NFATc1 are worthy of further studies.
Unraveling the molecular program that drives the differentiation and function of osteoclasts is important for a better understanding of many diseases linked to defective osteoclastogenesis.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}