C/EBPβ Is a Transcriptional Regulator of Wee1 at the G2/M Phase of the Cell Cycle

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. siRNA Transfection and Generation of Conditional Knockdown Cell Lines
2.3. Cell Proliferation Assay
2.4. Live/Dead Cell Staining
2.5. Cell Cycle Analysis
2.6. Live Cell Fluorescence Imaging for Analysis of Motisis
2.7. Western Blot Analysis
2.8. Quantitative Real-Time PCR
2.9. Chromatin Immunoprecipitation
2.10. Luciferase Reporter Assay
2.11. Immunoprecipitation
2.12. Xenograft Mouse Model and siRNA Delivery
2.13. Immunohistochemical Staining for Xenograft Tumor
2.14. Immunohistochemical Staining for Lung Cancer Tissue Microarray
2.15. Statistical Analysis
3. Results
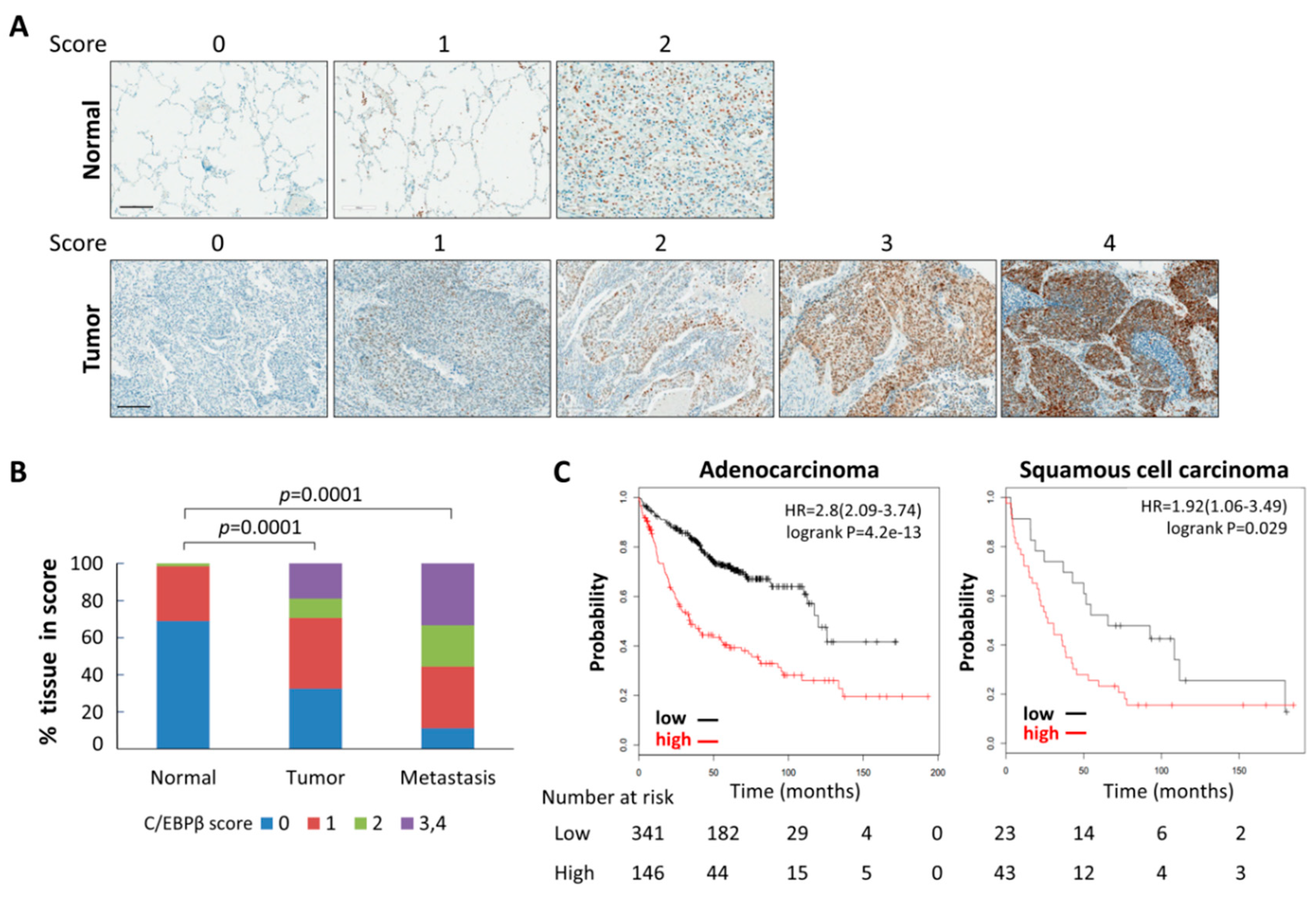
3.1. Levels of C/EBPβ Protein in Human Lung Cancer Tissues Were Elevated
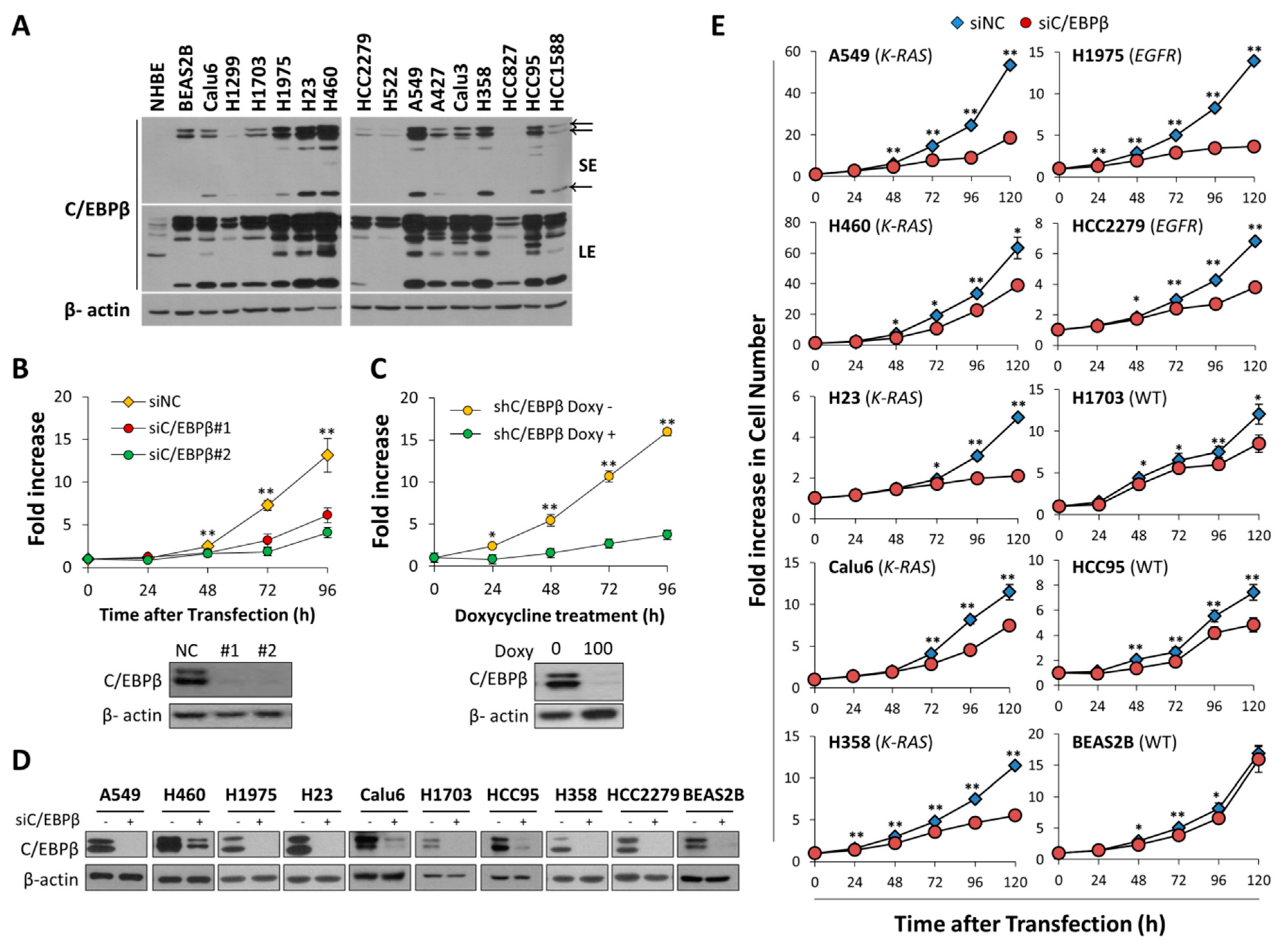
3.2. Knockdown of C/EBPβ Inhibits Cell Proliferation Rates of NSCLC Cells
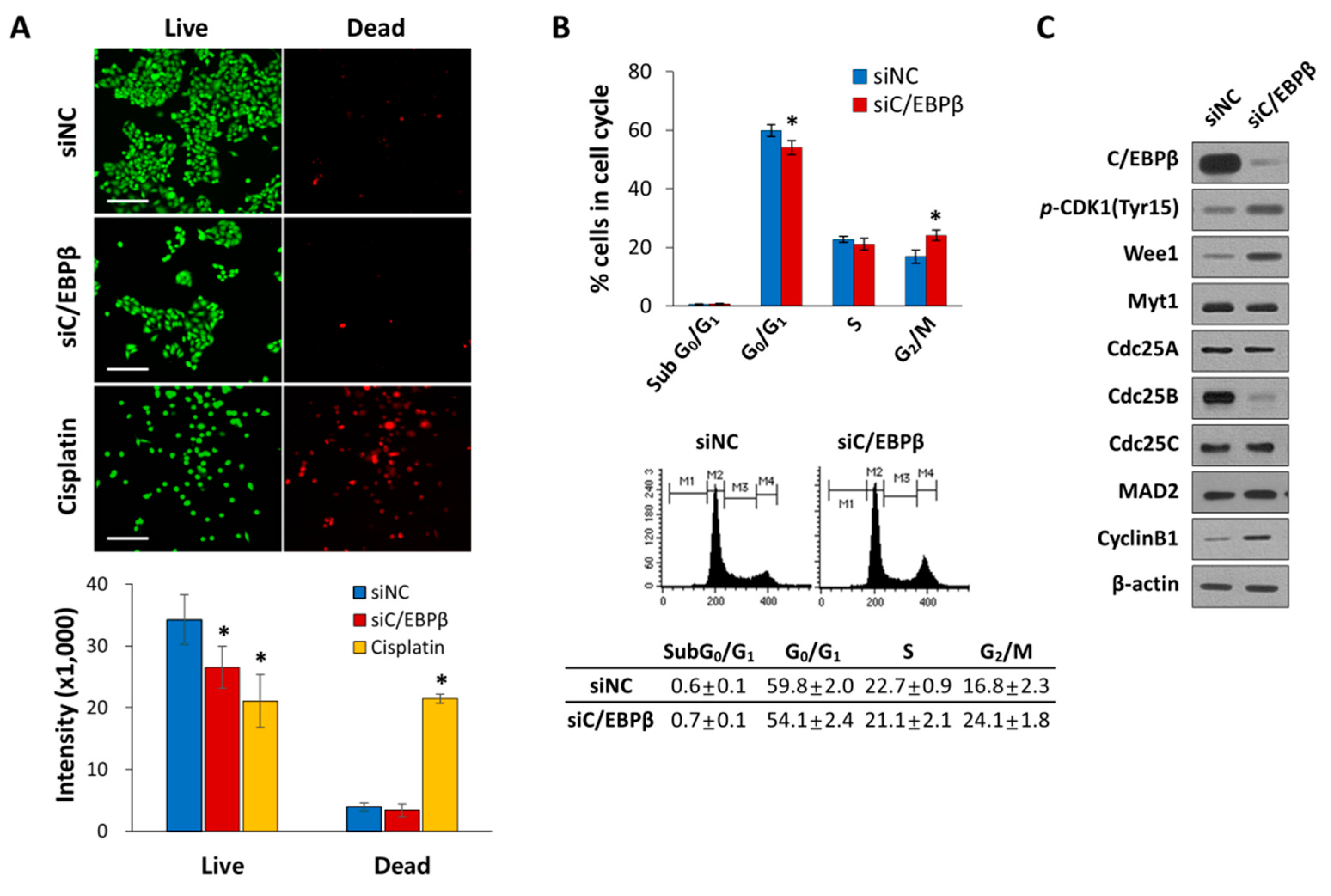
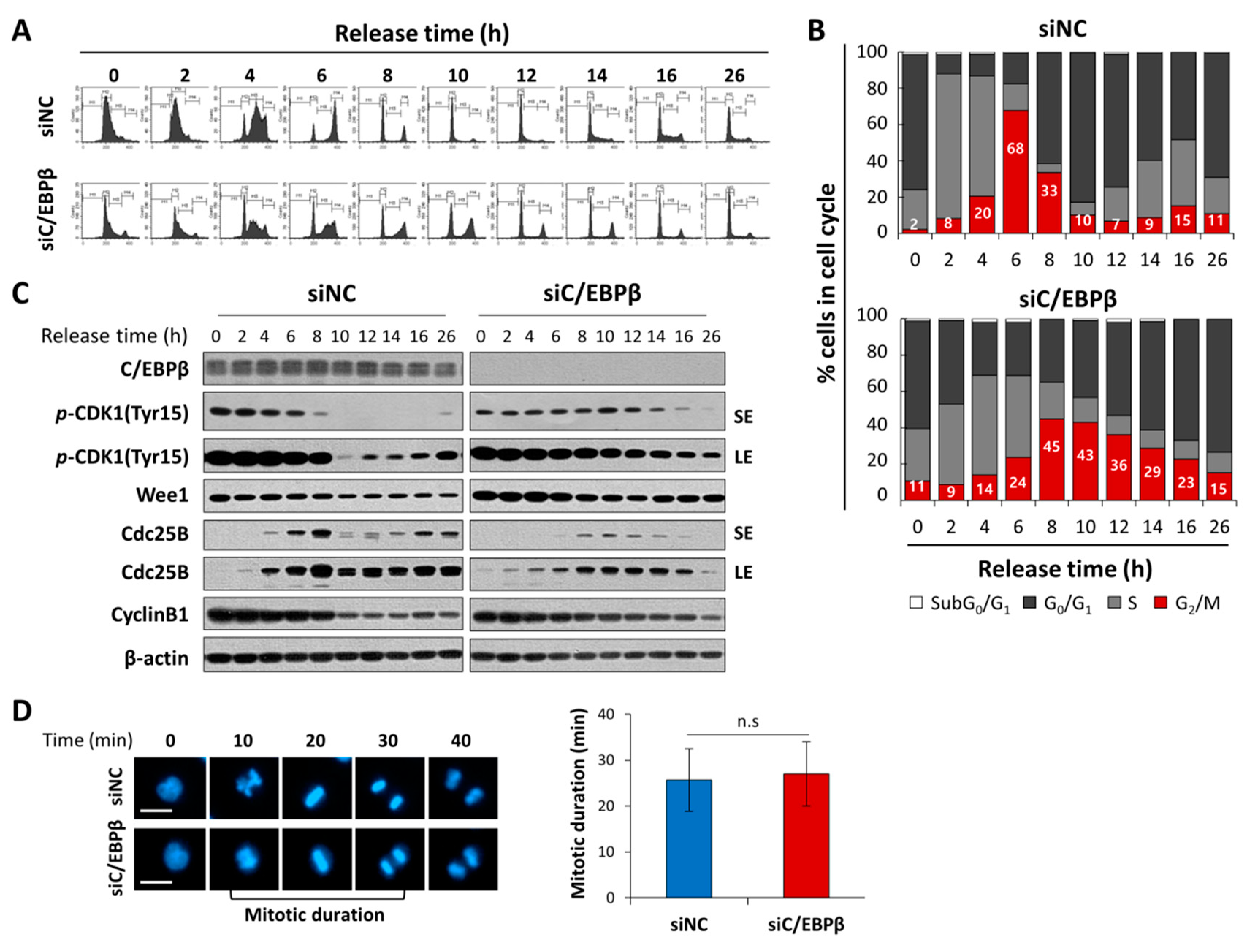
3.3. C/EBPβ-Knockdown Delayed G2/M Phase of the Cell Cycle Progression with Elevated Levels of Inhibitory Phosphorylation of CDK1
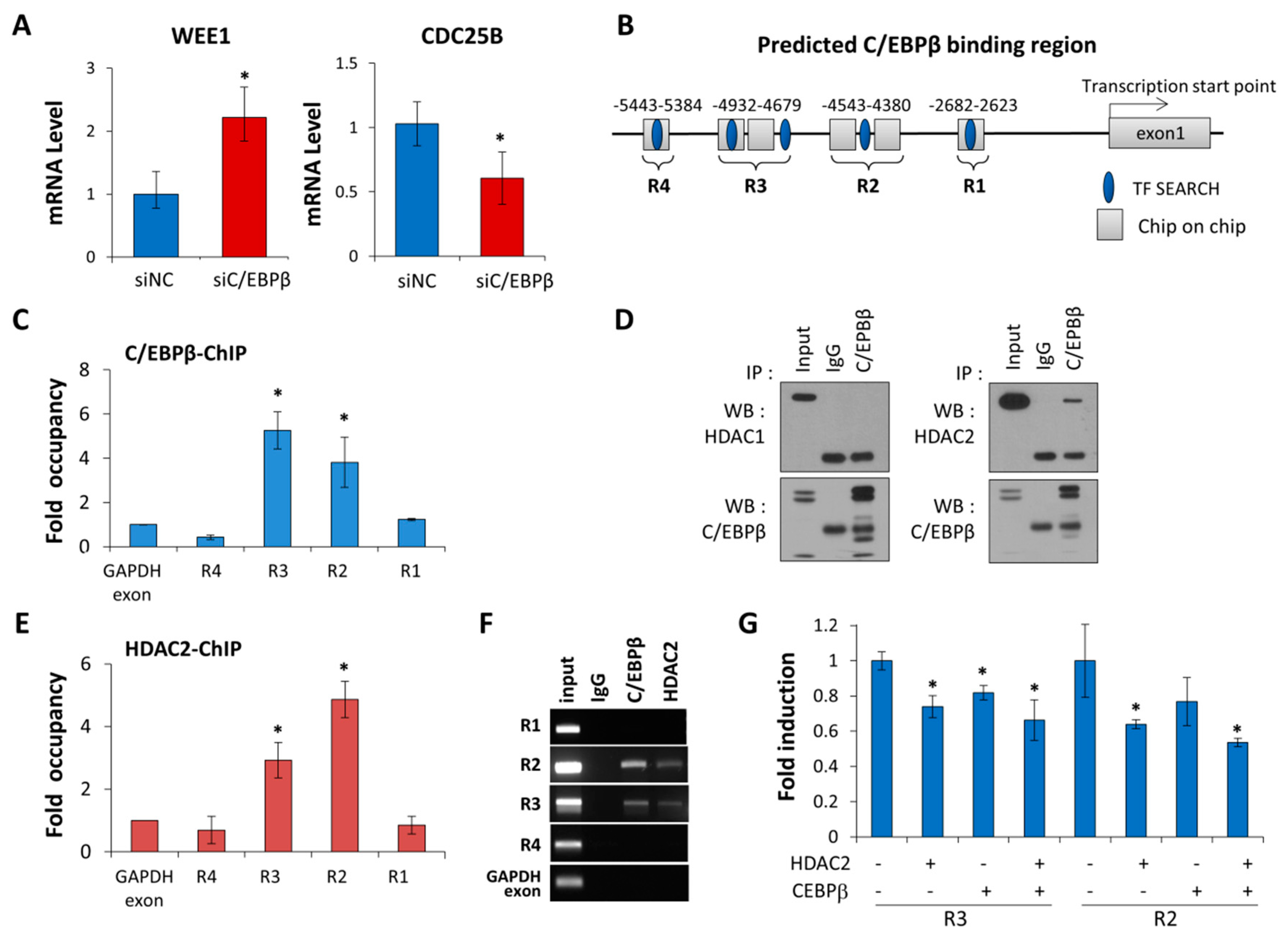
3.4. C/EBPβ Regulates Wee1 Expression at the Transcription Levels
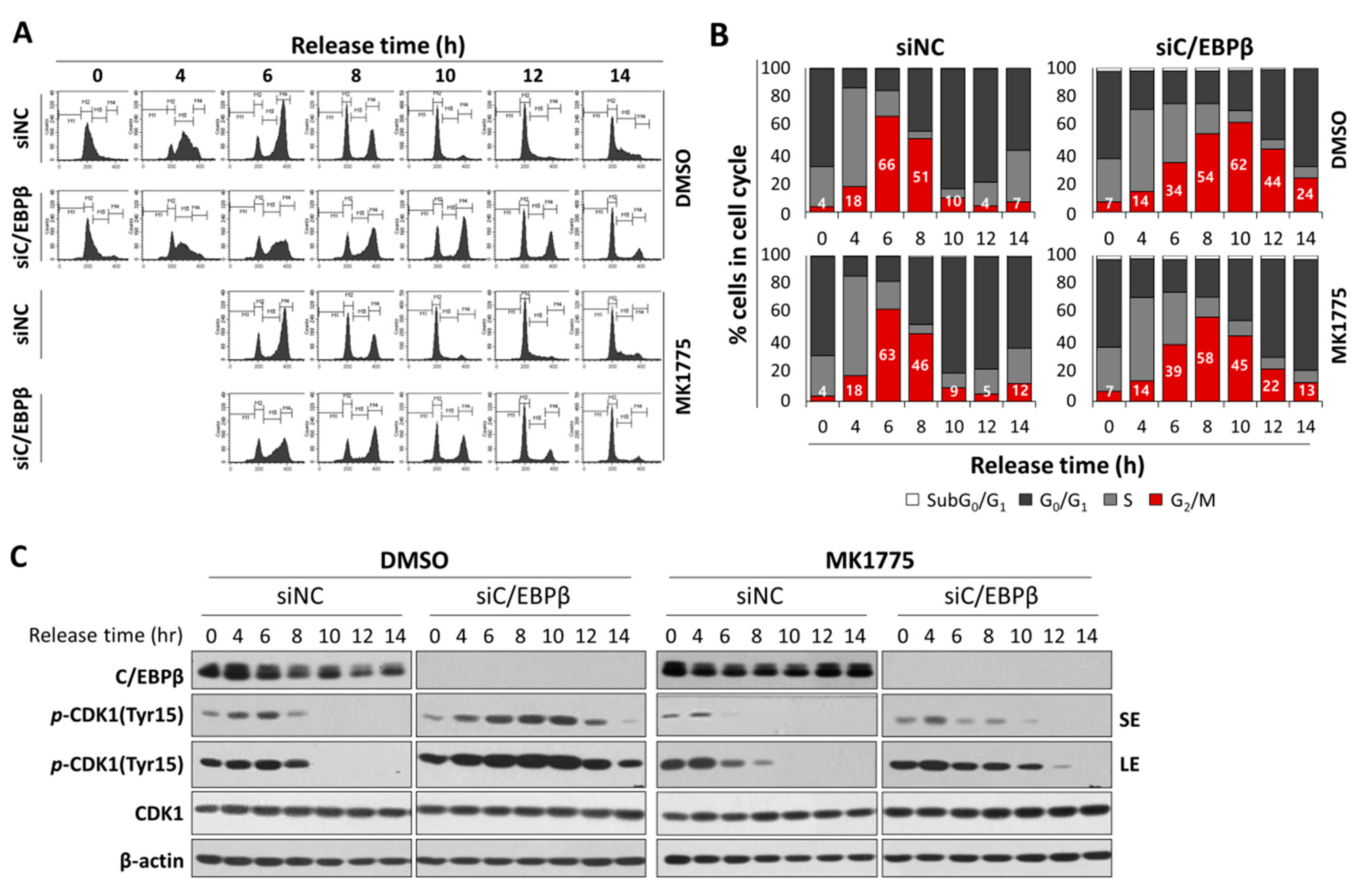
3.5. C/EBPβ-Knockdown Cells Treated with MK1775 Were Recovered from G2/M Delay
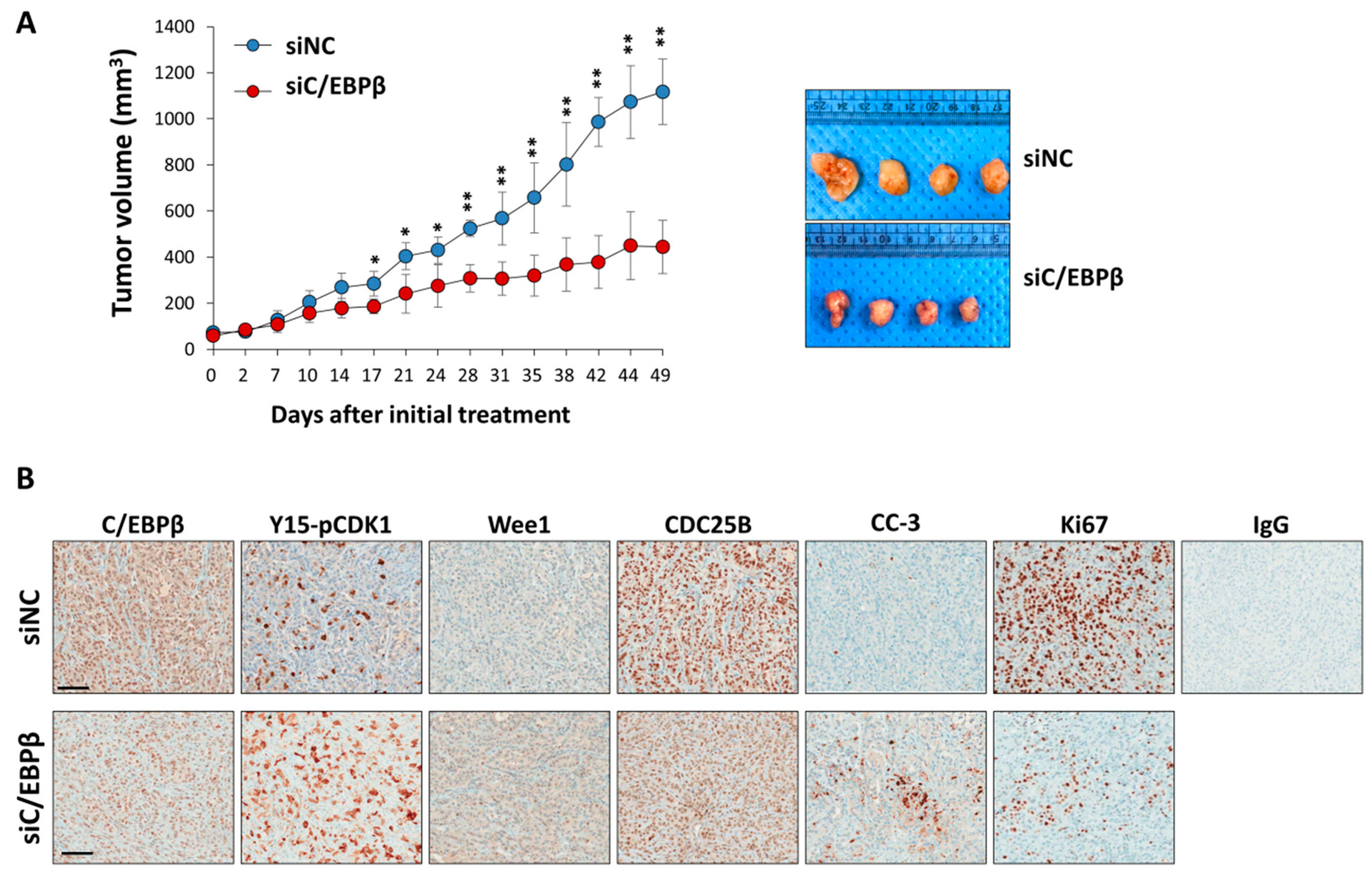
3.6. C/EBPβ-Knockdown Inhibits Tumor Growth In Vivo
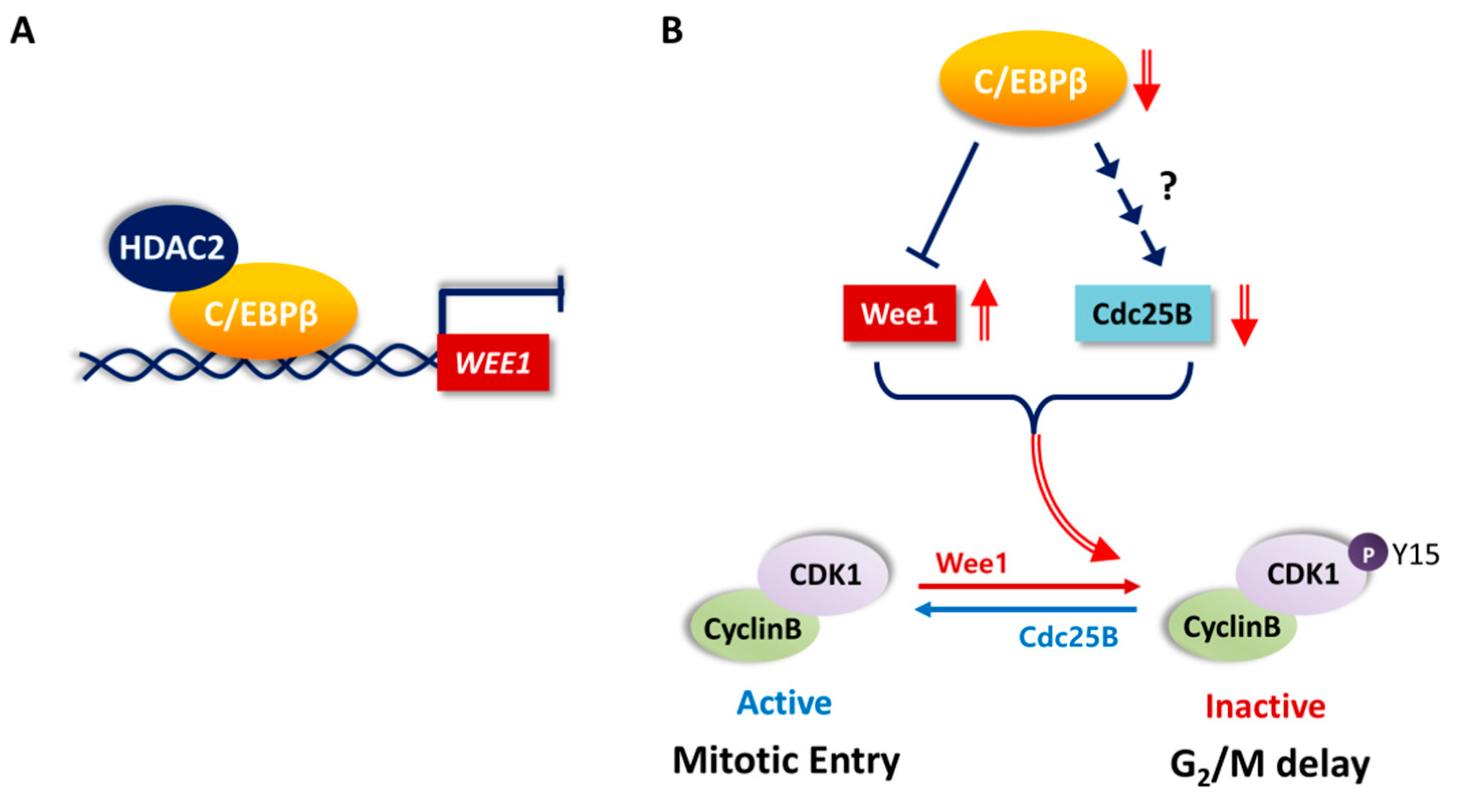
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sebastian, T.; Johnson, P.F. Stop and go: Anti-proliferative and mitogenic functions of the transcription factor C/EBPbeta. Cell Cycle 2006, 5, 953–957. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Umek, R.M.; McKnight, S.L. Regulated expression of three C/EBP isoforms during adipose conversion of 3T3-L1 cells. Genes Dev. 1991, 5, 1538–1552. [Google Scholar] [CrossRef] [PubMed]
- Poli, V. The role of C/EBP isoforms in the control of inflammatory and native immunity functions. J. Biol. Chem. 1998, 273, 29279–29282. [Google Scholar] [CrossRef] [PubMed]
- Zahnow, C.A. CCAAT/enhancer binding proteins in normal mammary development and breast cancer. Breast Cancer Res. 2002, 4, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Ramji, D.P.; Foka, P. CCAAT/enhancer-binding proteins: Structure, function and regulation. Biochem.J. 2002, 365, 561–575. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Oh, H.S.; Shim, M.; Sterneck, E.; Johnson, P.F.; Smart, R.C. C/EBPbeta modulates the early events of keratinocyte differentiation involving growth arrest and keratin 1 and keratin 10 expression. Mol. Cell. Biol. 1999, 19, 7181–7190. [Google Scholar] [CrossRef] [PubMed]
- Guerzoni, C.; Bardini, M.; Mariani, S.A.; Ferrari-Amorotti, G.; Neviani, P.; Panno, M.L.; Zhang, Y.; Martinez, R.; Perrotti, D.; Calabretta, B. Inducible activation of CEBPB, a gene negatively regulated by BCR/ABL, inhibits proliferation and promotes differentiation of BCR/ABL-expressing cells. Blood 2006, 107, 4080–4089. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, W.; Ambrosino, C.; Ruocco, M.R.; Poli, V.; Romani, L.; Quinto, I.; Barbieri, S.; Holmes, K.L.; Venuta, S.; et al. Impaired generation of bone marrow B lymphocytes in mice deficient in C/EBPbeta. Blood 1997, 90, 156–164. [Google Scholar] [PubMed]
- Regalo, G.; Forster, S.; Resende, C.; Bauer, B.; Fleige, B.; Kemmner, W.; Schlag, P.M.; Meyer, T.F.; Machado, J.C.; Leutz, A. C/EBPbeta regulates homeostatic and oncogenic gastric cell proliferation. J. Mol. Med. 2016, 94, 1385–1395. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, L.E.; Li, W.; Cressman, D.E.; Peng, Y.; Ciliberto, G.; Poli, V.; Taub, R. CCAAT enhancer- binding protein beta is required for normal hepatocyte proliferation in mice after partial hepatectomy. J. Clin. Invest. 1998, 102, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Morante, D.; Cortes-Canteli, M.; Sanz-Sancristobal, M.; Santos, A.; Perez-Castillo, A. Decreased CCAAT/enhancer binding protein beta expression inhibits the growth of glioblastoma cells. Neuroscience 2011, 176, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Yoon, K.; Sterneck, E.; Johnson, P.F.; Smart, R.C. CCAAT/enhancer binding protein-beta is a mediator of keratinocyte survival and skin tumorigenesis involving oncogenic Ras signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.; Zhu, S.; Ewing, S.J.; Smart, R.C. Decreased survival of C/EBP beta-deficient keratinocytes is due to aberrant regulation of p53 levels and function. Oncogene 2007, 26, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J.; Ramaswamy, S.; Ford, H.L.; Contreras, B.; Martinez, R.V.; Kittrell, F.S.; Zahnow, C.A.; Patterson, N.; Golub, T.R.; Ewen, M.E. A mechanism of cyclin D1 action encoded in the patterns of gene expression in human cancer. Cell 2003, 114, 323–334. [Google Scholar] [CrossRef]
- Zahnow, C.A.; Younes, P.; Laucirica, R.; Rosen, J.M. Overexpression of C/EBPbeta-LIP, a naturally occurring, dominant-negative transcription factor, in human breast cancer. J. Natl. Cancer Inst. 1997, 89, 1887–1891. [Google Scholar] [PubMed]
- Rask, K.; Thorn, M.; Ponten, F.; Kraaz, W.; Sundfeldt, K.; Hedin, L.; Enerback, S. Increased expression of the transcription factors CCAAT-enhancer binding protein-beta (C/EBBeta) and C/EBzeta (CHOP) correlate with invasiveness of human colorectal cancer. Int. J. Cancer 2000, 86, 337–343. [Google Scholar] [CrossRef]
- Sundfeldt, K.; Ivarsson, K.; Carlsson, M.; Enerback, S.; Janson, P.O.; Brannstrom, M.; Hedin, L. The expression of CCAAT/enhancer binding protein (C/EBP) in the human ovary in vivo: Specific increase in C/EBPbeta during epithelial tumour progression. Br. J. Cancer 1999, 79, 1240–1248. [Google Scholar] [CrossRef] [PubMed]
- Sankpal, N.V.; Moskaluk, C.A.; Hampton, G.M.; Powell, S.M. Overexpression of CEBPbeta correlates with decreased TFF1 in gastric cancer. Oncogene 2006, 25, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Oya, M.; Horiguchi, A.; Mizuno, R.; Marumo, K.; Murai, M. Increased activation of CCAAT/enhancer binding protein-beta correlates with the invasiveness of renal cell carcinoma. Clin. Cancer Res. 2003, 9, 1021–1027. [Google Scholar] [PubMed]
- Regalo, G.; Canedo, P.; Suriano, G.; Resende, C.; Campos, M.L.; Oliveira, M.J.; Figueiredo, C.; Rodrigues-Pereira, P.; Blin, N.; Seruca, R.; et al. C/EBPbeta is over-expressed in gastric carcinogenesis and is associated with COX-2 expression. J. Pathol. 2006, 210, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, K.; Van Bockstaele, D.R.; Berneman, Z.N. The cell cycle: A review of regulation, deregulation and therapeutic targets in cancer. Cell Proliferat. 2003, 36, 131–149. [Google Scholar] [CrossRef]
- Sherr, C.J. G1 phase progression: Cycling on cue. Cell 1994, 79, 551–555. [Google Scholar] [CrossRef]
- Girard, F.; Strausfeld, U.; Fernandez, A.; Lamb, N.J. Cyclin A is required for the onset of DNA replication in mammalian fibroblasts. Cell 1991, 67, 1169–1179. [Google Scholar] [CrossRef]
- Ohtsubo, M.; Theodoras, A.M.; Schumacher, J.; Roberts, J.M.; Pagano, M. Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol. Cell. Biol. 1995, 15, 2612–2624. [Google Scholar] [CrossRef] [PubMed]
- King, R.W.; Jackson, P.K.; Kirschner, M.W. Mitosis in transition. Cell 1994, 79, 563–571. [Google Scholar] [CrossRef]
- Jeffrey, P.D.; Russo, A.A.; Polyak, K.; Gibbs, E.; Hurwitz, J.; Massague, J.; Pavletich, N.P. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature 1995, 376, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Lew, D.J.; Kornbluth, S. Regulatory roles of cyclin dependent kinase phosphorylation in cell cycle control. Curr. Opin. Cell Biol. 1996, 8, 795–804. [Google Scholar] [CrossRef]
- Carnero, A.; Hannon, G.J. The INK4 family of CDK inhibitors. Curr. Top. Microbiol. Immunol. 1998, 227, 43–55. [Google Scholar] [PubMed]
- Lee, M.H.; Reynisdottir, I.; Massague, J. Cloning of p57KIP2, a cyclin-dependent kinase inhibitor with unique domain structure and tissue distribution. Genes Dev. 1995, 9, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Lee, M.H.; Erdjument-Bromage, H.; Koff, A.; Roberts, J.M.; Tempst, P.; Massague, J. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 1994, 78, 59–66. [Google Scholar] [CrossRef]
- Harper, J.W.; Elledge, S.J.; Keyomarsi, K.; Dynlacht, B.; Tsai, L.H.; Zhang, P.; Dobrowolski, S.; Bai, C.; Connell-Crowley, L.; Swindell, E.; et al. Inhibition of cyclin-dependent kinases by p21. Mol. Biol. Cell 1995, 6, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [PubMed]
- Singhal, S.; Vachani, A.; Antin-Ozerkis, D.; Kaiser, L.R.; Albelda, S.M. Prognostic implications of cell cycle, apoptosis, and angiogenesis biomarkers in non-small cell lung cancer: A review. Clin. Cancer Res. 2005, 11, 3974–3986. [Google Scholar] [CrossRef] [PubMed]
- Selagea, L.; Mishra, A.; Anand, M.; Ross, J.; Tucker-Burden, C.; Kong, J.; Brat, D.J. EGFR and C/EBP-beta oncogenic signaling is bidirectional in human glioma and varies with the C/EBP-beta isoform. FASEB J. 2016, 30, 4098–4108. [Google Scholar] [CrossRef] [PubMed]
- Gavvovidis, I.; Rost, I.; Trimborn, M.; Kaiser, F.J.; Purps, J.; Wiek, C.; Hanenberg, H.; Neitzel, H.; Schindler, D. A novel MCPH1 isoform complements the defective chromosome condensation of human MCPH1-deficient cells. PloS ONE 2012, 7, e40387. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Lee, H.K.; Kim, H.S.; Yang, H.K.; Kim, W.H. Tumour suppressor gene expression correlates with gastric cancer prognosis. J. Pathol. 2003, 200, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Gyorffy, B.; Surowiak, P.; Budczies, J.; Lanczky, A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PloS ONE 2013, 8, e82241. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, K.D.; Berns, A. Cell of origin of lung cancer. Mol. Oncol. 2010, 4, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Blanco, R.; Iwakawa, R.; Tang, M.; Kohno, T.; Angulo, B.; Pio, R.; Montuenga, L.M.; Minna, J.D.; Yokota, J.; Sanchez-Cespedes, M. A gene-alteration profile of human lung cancer cell lines. Hum. Mutat. 2009, 30, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Den Dunnen, J.T.; Antonarakis, S.E. Mutation nomenclature extensions and suggestions to describe complex mutations: A discussion. Hum. Mutat. 2000, 15, 7–12. [Google Scholar] [CrossRef]
- Forbes, S.; Clements, J.; Dawson, E.; Bamford, S.; Webb, T.; Dogan, A.; Flanagan, A.; Teague, J.; Wooster, R.; Futreal, P.A.; et al. COSMIC 2005. Br. J. Cancer 2006, 94, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Wistuba, II.; Bryant, D.; Behrens, C.; Milchgrub, S.; Virmani, A.K.; Ashfaq, R.; Minna, J.D.; Gazdar, A.F. Comparison of features of human lung cancer cell lines and their corresponding tumors. Clin. Cancer Res. 1999, 5, 991–1000. [Google Scholar] [PubMed]
- Yamamoto, H.; Shigematsu, H.; Nomura, M.; Lockwood, W.W.; Sato, M.; Okumura, N.; Soh, J.; Suzuki, M.; Wistuba, II.; Fong, K.M.; Lee, H.; et al. PIK3CA mutations and copy number gains in human lung cancers. Cancer Res. 2008, 68, 6913–6921. [Google Scholar] [CrossRef] [PubMed]
- Obaya, A.J.; Sedivy, J.M. Regulation of cyclin-Cdk activity in mammalian cells. Cell. Mol. Life Sci. 2002, 59, 126–142. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, P.H. Triggering the all-or-nothing switch into mitosis. Trends Cell Biol. 2001, 11, 512–519. [Google Scholar] [CrossRef]
- Parker, L.L.; Piwnica-Worms, H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science 1992, 257, 1955–1957. [Google Scholar] [CrossRef] [PubMed]
- Mueller, P.R.; Coleman, T.R.; Kumagai, A.; Dunphy, W.G. Myt1: A membrane-associated inhibitory kinase that phosphorylates Cdc2 on both threonine-14 and tyrosine-15. Science 1995, 270, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Donzelli, M.; Draetta, G.F. Regulating mammalian checkpoints through Cdc25 inactivation. EMBO Rep. 2003, 4, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Dunphy, W.G.; Kumagai, A. The cdc25 protein contains an intrinsic phosphatase activity. Cell 1991, 67, 189–196. [Google Scholar] [CrossRef]
- Glotzer, M. Mitosis: Don’t get mad, get even. Curr. Biol. 1996, 6, 1592–1594. [Google Scholar] [CrossRef]
- Meraldi, P.; Draviam, V.M.; Sorger, P.K. Timing and checkpoints in the regulation of mitotic progression. Dev. Cell 2004, 7, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Hendzel, M.J.; Wei, Y.; Mancini, M.A.; Van Hooser, A.; Ranalli, T.; Brinkley, B.R.; Bazett-Jones, D.P.; Allis, C.D. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma 1997, 106, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Salisbury, E.; Shi, X.; Timchenko, L.; Medrano, E.E.; Timchenko, N.A. HDAC1 promotes liver proliferation in young mice via interactions with C/EBPbeta. J. Biol. Chem. 2008, 283, 26179–26187. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Perfettini, J.L.; Roumier, T.; Kroemer, G. Cyclin-dependent kinase-1: Linking apoptosis to cell cycle and mitotic catastrophe. Cell Death Differ. 2002, 9, 1287–1293. [Google Scholar] [CrossRef] [PubMed]
- McGowan, C.H.; Russell, P. Cell cycle regulation of human WEE1. EMBO J. 1995, 14, 2166–2175. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.A.; Kornbluth, S. Cdc25 and Wee1: Analogous opposites? Cell Div. 2007, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Katayama, K.; Fujita, N.; Tsuruo, T. Akt/protein kinase B-dependent phosphorylation and inactivation of WEE1Hu promote cell cycle progression at G2/M transition. Mol. Cell. Biol. 2005, 25, 5725–5737. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Arai, H.; Nishihara, Y.; Taniguchi, M.; Watanabe, N.; Hunter, T.; Osada, H. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proc. Natl. Acad. Sci. USA 2004, 101, 4419–4424. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhu, Y.; Huang, Y.; McAvoy, S.; Johnson, W.B.; Cheung, T.H.; Chung, T.K.; Lo, K.W.; Yim, S.F.; Yu, M.M.; et al. Transcriptional repression of WEE1 by Kruppel-like factor 2 is involved in DNA damage-induced apoptosis. Oncogene 2005, 24, 3875–3885. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Komai, K.; Ouyang, Z.; Murata, M.; Hikasa, M.; Ohgiri, M.; Shiozawa, S. c-Fos/activator protein-1 transactivates wee1 kinase at G(1)/S to inhibit premature mitosis in antigen-specific Th1 cells. EMBO J. 2001, 20, 4618–4627. [Google Scholar] [CrossRef] [PubMed]
- Quan, J.; Adelmant, G.; Marto, J.A.; Look, A.T.; Yusufzai, T. The chromatin remodeling factor CHD5 is a transcriptional repressor of WEE1. PloS ONE 2014, 9, e108066. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Taylor, R.N.; Bagchi, I.C.; Bagchi, M.K. Regulation of human endometrial stromal proliferation and differentiation by C/EBPbeta involves cyclin E-cdk2 and STAT3. Mol. Endocrinol. 2012, 26, 2016–2030. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Larris, B.; Peiris, T.H.; Zhang, L.; Le Lay, J.; Gao, Y.; Greenbaum, L.E. C/EBPbeta activates E2F-regulated genes in vivo via recruitment of the coactivator CREB-binding protein/P300. J. Biol. Chem. 2007, 282, 24679–24688. [Google Scholar] [CrossRef] [PubMed]
- Cassel, T.N.; Nord, M. C/EBP transcription factors in the lung epithelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, L773–L781. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.; Iyama, K.I.; Kimura, T.; Sano, K.; Darlington, G.J.; Akiba, T.; Takiguchi, M. Mice lacking CCAAt/enhancer-binding protein-alpha show hyperproliferation of alveolar type II cells and increased surfactant protein mRNAs. Cell Tissue Res. 2001, 306, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Flodby, P.; Barlow, C.; Kylefjord, H.; Ahrlund-Richter, L.; Xanthopoulos, K.G. Increased hepatic cell proliferation and lung abnormalities in mice deficient in CCAAT/enhancer binding protein alpha. J. Biol.l Chem. 1996, 271, 24753–24760. [Google Scholar] [CrossRef]
- Park, Y.; Kemper, B. The CYP2B1 proximal promoter contains a functional C/EBP regulatory element. DNA Cell Biol. 1996, 15, 693–701. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Crouch, E. Surfactant protein D gene regulation. Interactions among the conserved CCAAT/enhancer-binding protein elements. J. Biol. Chem. 2002, 277, 19530–19537. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.S.; Chen, J.F.; Johnson, P.F.; Muppala, V.; Lee, Y.H. C/EBPbeta, when expressed from the C/ebpalpha gene locus, can functionally replace C/EBPalpha in liver but not in adipose tissue. Mol. Cell. Biol. 2000, 20, 7292–7299. [Google Scholar] [CrossRef] [PubMed]
- House, J.S.; Zhu, S.; Ranjan, R.; Linder, K.; Smart, R.C. C/EBPalpha and C/EBPbeta are required for Sebocyte differentiation and stratified squamous differentiation in adult mouse skin. PloS ONE 2010, 5, e9837. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.; Sadohara, T.; Sugita, M.; Iyama, K.; Takiguchi, M. Differential expression of CCAAT enhancer binding protein family in rat alveolar epithelial cell proliferation and in acute lung injury. Cell Tissue Res. 1999, 297, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Gombart, A.F.; Hofmann, W.K.; Kawano, S.; Takeuchi, S.; Krug, U.; Kwok, S.H.; Larsen, R.J.; Asou, H.; Miller, C.W.; Hoelzer, D.; et al. Mutations in the gene encoding the transcription factor CCAAT/enhancer binding protein alpha in myelodysplastic syndromes and acute myeloid leukemias. Blood 2002, 99, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Halmos, B.; Huettner, C.S.; Kocher, O.; Ferenczi, K.; Karp, D.D.; Tenen, D.G. Down-regulation and antiproliferative role of C/EBPalpha in lung cancer. Cancer Res. 2002, 62, 528–534. [Google Scholar] [PubMed]
- Tada, Y.; Brena, R.M.; Hackanson, B.; Morrison, C.; Otterson, G.A.; Plass, C. Epigenetic modulation of tumor suppressor CCAAT/enhancer binding protein alpha activity in lung cancer. J. Natl. Cancer Inst. 2006, 98, 396–406. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Score | Normal n (%) | Tumor n (%) | Metastasis n (%) |
|---|---|---|---|
| 0 | 47 (69.1) | 31 (32.6) | 1 (11.1) |
| 1 | 20 (29.4) | 36 (37.9) | 3 (33.3) |
| 2 | 1 (1.5) | 10 (10.5) | 2 (22.2) |
| 3 | 0 (0) | 13 (13.7) | 3 (33.3) |
| 4 | 0 (0) | 5 (5.3) | 0 (0) |
| Total | 68 (100) | 95 (100) | 9 (100) |
| C/EBPβ Expression | |||||
|---|---|---|---|---|---|
| Negative n (%) | Positive n (%) | Total n (%) | p Value | ||
| Sex | |||||
| Male | 23 (31) | 52 (69) | 75 (100) | 0.4343 | |
| Female | 8 (40) | 12 (60) | 20 (100) | ||
| Age | |||||
| ≤57 | 11 (31) | 24 (69) | 35 (100) | 0.8505 | |
| >57 | 20 (33) | 40 (67) | 60 (100) | ||
| Clinical Stage | |||||
| I | 10 (27) | 27 (73) | 37 (100) | 0.2305 | |
| II | 12 (32) | 25 (68) | 37 (100) | ||
| III | 9 (43) | 12 (57) | 21 (100) | ||
| N classification | |||||
| 0 | 15 (28) | 38 (72) | 53 (100) | 0.2204 | |
| 1 | 8 (33) | 16 (67) | 24 (100) | ||
| 2 | 8 (44) | 10 (66) | 18 (100) | ||
| T classification | |||||
| 1 | 1 (13) | 7 (87) | 8 (100) | 0.3039 | |
| 2 | 23 (33) | 47 (67) | 70 (100) | ||
| 3 | 5 (46) | 6 (54) | 11 (100) | ||
| 4 | 2 (33) | 4 (67) | 17 (100) | ||
| Histology | |||||
| Adenocarcinoma | 13 (46) | 15 (54) | 28 (100) | 0.0180 * | 0.0256 † |
| Squamous carcinoma | 15 (31) | 34 (69) | 49 (100) | 0.2941 # | |
| Other NSCLC | 3 (17) | 15 (83) | 18 (100) | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.H.; Sung, J.Y.; Choi, E.K.; Yoon, H.-K.; Kang, B.R.; Hong, E.K.; Park, B.-K.; Kim, Y.-N.; Rho, S.B.; Yoon, K. C/EBPβ Is a Transcriptional Regulator of Wee1 at the G2/M Phase of the Cell Cycle. Cells 2019, 8, 145. https://doi.org/10.3390/cells8020145
Lee JH, Sung JY, Choi EK, Yoon H-K, Kang BR, Hong EK, Park B-K, Kim Y-N, Rho SB, Yoon K. C/EBPβ Is a Transcriptional Regulator of Wee1 at the G2/M Phase of the Cell Cycle. Cells. 2019; 8(2):145. https://doi.org/10.3390/cells8020145
Chicago/Turabian StyleLee, Ji Hae, Jee Young Sung, Eun Kyung Choi, Hyun-Kyoung Yoon, Bo Ram Kang, Eun Kyung Hong, Byung-Kiu Park, Yong-Nyun Kim, Seung Bae Rho, and Kyungsil Yoon. 2019. "C/EBPβ Is a Transcriptional Regulator of Wee1 at the G2/M Phase of the Cell Cycle" Cells 8, no. 2: 145. https://doi.org/10.3390/cells8020145
APA StyleLee, J. H., Sung, J. Y., Choi, E. K., Yoon, H. -K., Kang, B. R., Hong, E. K., Park, B. -K., Kim, Y. -N., Rho, S. B., & Yoon, K. (2019). C/EBPβ Is a Transcriptional Regulator of Wee1 at the G2/M Phase of the Cell Cycle. Cells, 8(2), 145. https://doi.org/10.3390/cells8020145