Essential Roles for the Non-Canonical IκB Kinases in Linking Inflammation to Cancer, Obesity, and Diabetes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
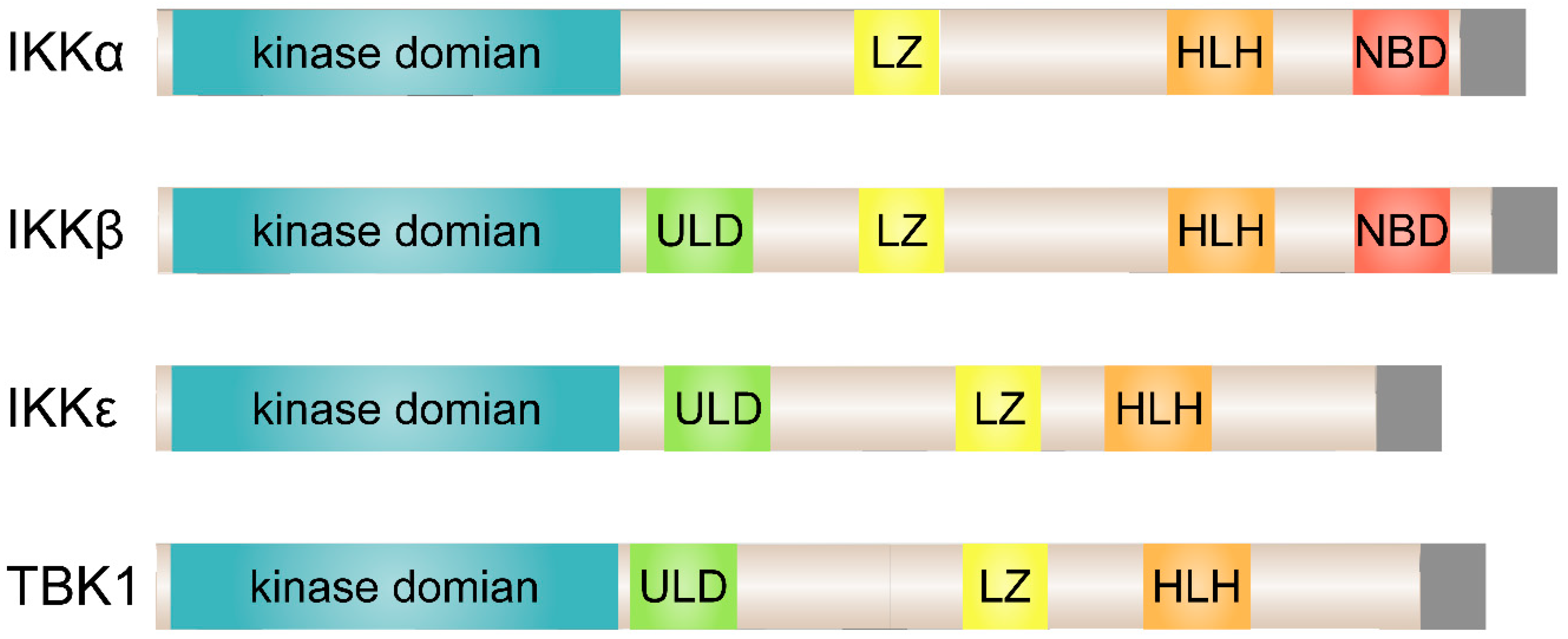
2. TBK1 and IKKε in NF-κB Signaling
3. TBK1 and IKKε in Interferon Signal Transduction
4. TBK1 and IKKε in Cancer
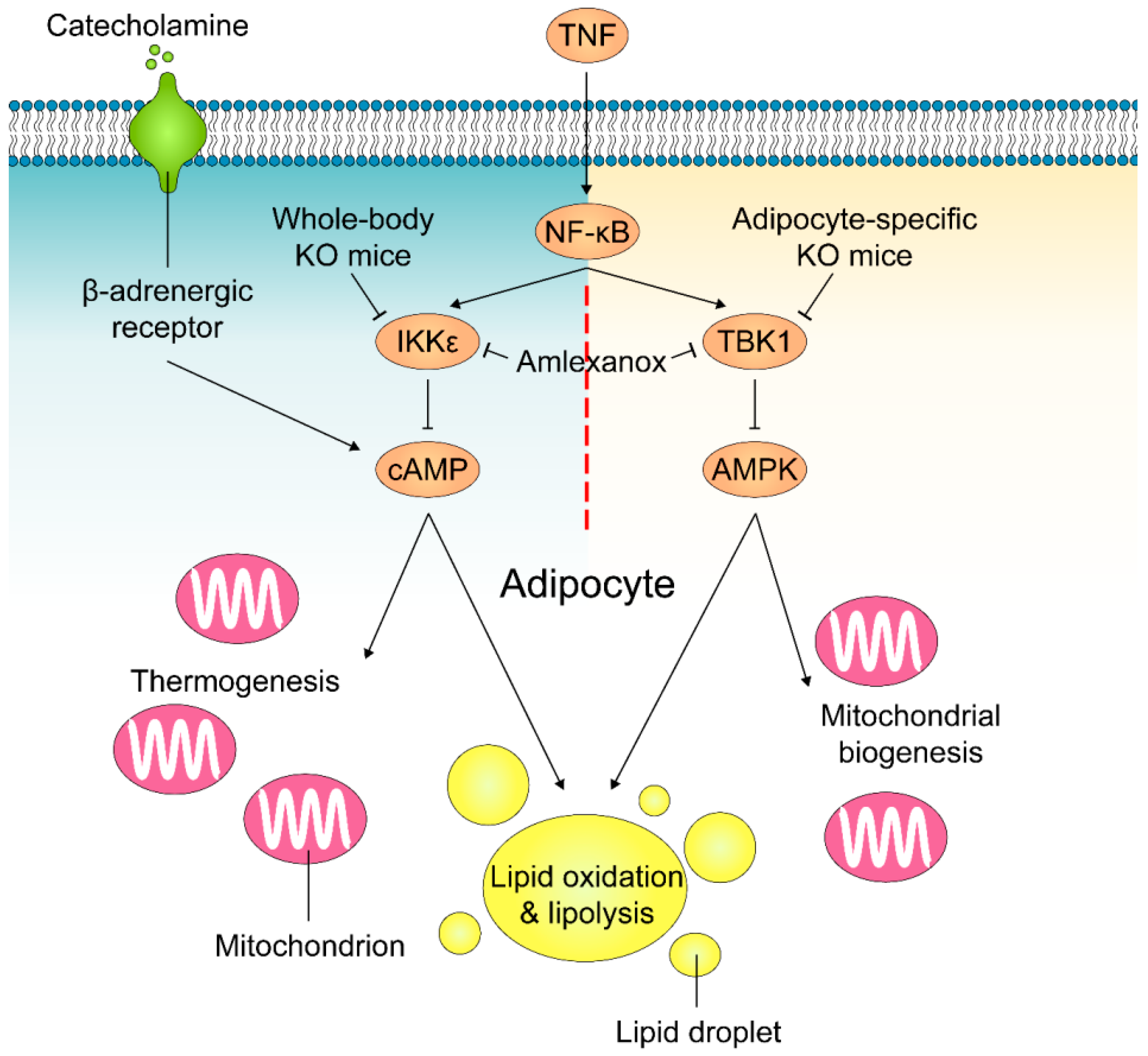
5. TBK1 and IKKε in Obesity
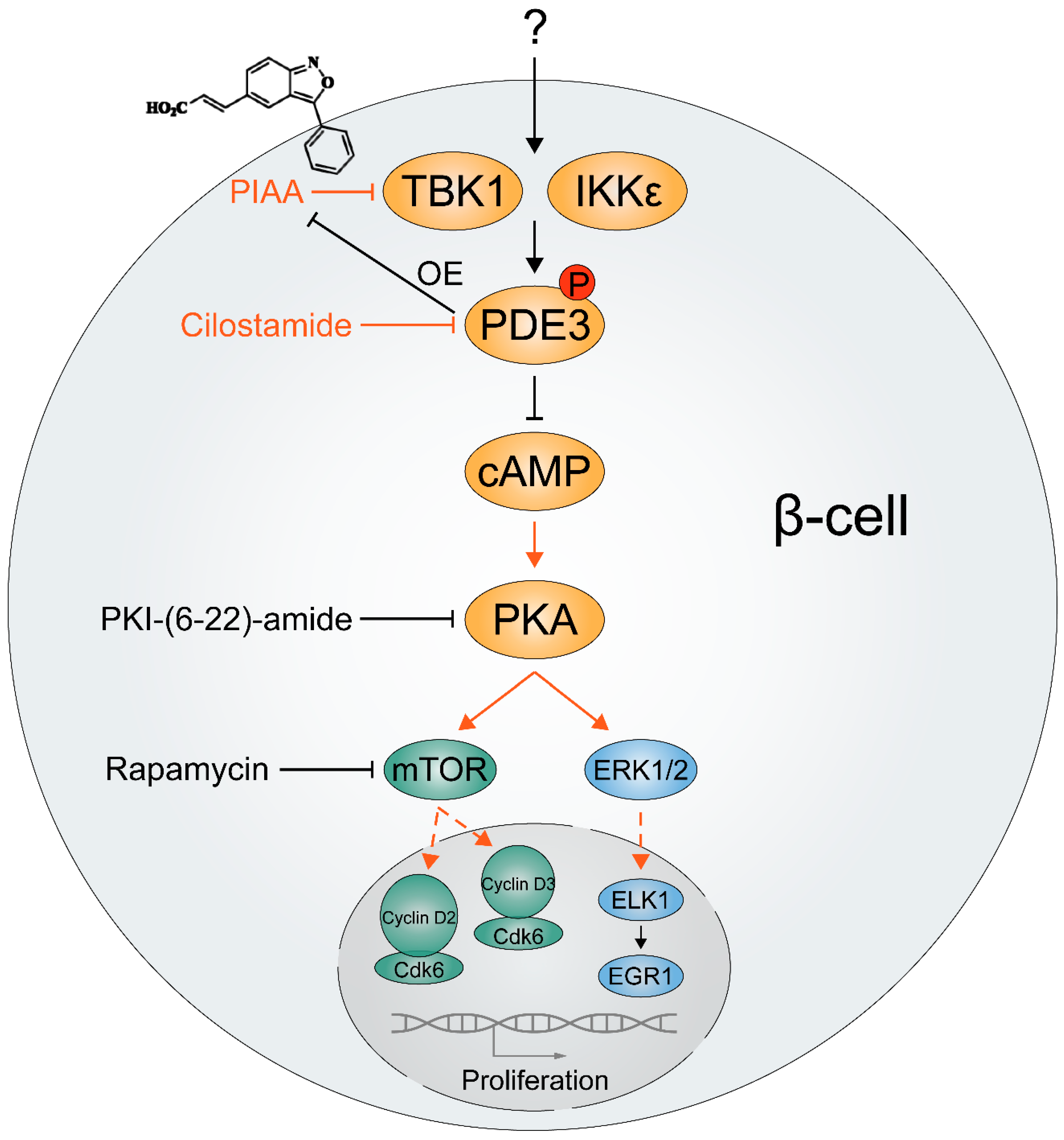
6. TBK1 and IKKε in Diabetes
7. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jing, H.; Lee, S. NF-kappaB in cellular senescence and cancer treatment. Mol. Cells 2014, 37, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, C.M.; Davis, R.E.; Demchenko, Y.; Bellamy, W.; Gabrea, A.; Zhan, F.; Lenz, G.; Hanamura, I.; Wright, G.; Xiao, W.; et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 2007, 12, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Compagno, M.; Lim, W.K.; Grunn, A.; Nandula, S.V.; Brahmachary, M.; Shen, Q.; Bertoni, F.; Ponzoni, M.; Scandurra, M.; Califano, A.; et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature 2009, 459, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Novak, U.; Rinaldi, A.; Kwee, I.; Nandula, S.V.; Rancoita, P.M.; Compagno, M.; Cerri, M.; Rossi, D.; Murty, V.V.; Zucca, E.; et al. The NF-{kappa}B negative regulator TNFAIP3 (A20) is inactivated by somatic mutations and genomic deletions in marginal zone lymphomas. Blood 2009, 113, 4918–4921. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, F.T.; Luedde, T.; Singer, S.; Schmidt-Supprian, M.; Baumgartl, J.; Schirmacher, P.; Pasparakis, M.; Bruning, J.C. Hepatic NF-kappa B essential modulator deficiency prevents obesity-induced insulin resistance but synergizes with high-fat feeding in tumorigenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 1297–1302. [Google Scholar] [CrossRef] [PubMed]
- Arkan, M.C.; Hevener, A.L.; Greten, F.R.; Maeda, S.; Li, Z.W.; Long, J.M.; Wynshaw-Boris, A.; Poli, G.; Olefsky, J.; Karin, M. IKK-beta links inflammation to obesity-induced insulin resistance. Nat. Med. 2005, 11, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Goldfine, A.B.; Fonseca, V.; Jablonski, K.A.; Pyle, L.; Staten, M.A.; Shoelson, S.E. The effects of salsalate on glycemic control in patients with type 2 diabetes: A randomized trial. Ann. Intern. Med. 2010, 152, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.H.; Bazuine, M.; Lumeng, C.N.; Geletka, L.M.; Mowers, J.; White, N.M.; Ma, J.T.; Zhou, J.; Qi, N.; Westcott, D.; et al. The protein kinase IKKepsilon regulates energy balance in obese mice. Cell 2009, 138, 961–975. [Google Scholar] [CrossRef]
- Reilly, S.M.; Chiang, S.H.; Decker, S.J.; Chang, L.; Uhm, M.; Larsen, M.J.; Rubin, J.R.; Mowers, J.; White, N.M.; Hochberg, I.; et al. An inhibitor of the protein kinases TBK1 and IKK-varepsilon improves obesity-related metabolic dysfunctions in mice. Nat. Med. 2013, 19, 313–321. [Google Scholar] [CrossRef]
- Eldor, R.; Yeffet, A.; Baum, K.; Doviner, V.; Amar, D.; Ben-Neriah, Y.; Christofori, G.; Peled, A.; Carel, J.C.; Boitard, C.; et al. Conditional and specific NF-kappaB blockade protects pancreatic beta cells from diabetogenic agents. Proc. Natl. Acad. Sci. USA 2006, 103, 5072–5077. [Google Scholar] [CrossRef]
- Giannoukakis, N.; Rudert, W.A.; Trucco, M.; Robbins, P.D. Protection of human islets from the effects of interleukin-1beta by adenoviral gene transfer of an Ikappa B repressor. J. Biol. Chem. 2000, 275, 36509–36513. [Google Scholar] [CrossRef]
- Heimberg, H.; Heremans, Y.; Jobin, C.; Leemans, R.; Cardozo, A.K.; Darville, M.; Eizirik, D.L. Inhibition of cytokine-induced NF-kappaB activation by adenovirus-mediated expression of a NF-kappaB super-repressor prevents beta-cell apoptosis. Diabetes 2001, 50, 2219–2224. [Google Scholar] [CrossRef]
- Hofmann, M.A.; Schiekofer, S.; Kanitz, M.; Klevesath, M.S.; Joswig, M.; Lee, V.; Morcos, M.; Tritschler, H.; Ziegler, R.; Wahl, P.; et al. Insufficient glycemic control increases nuclear factor-kappa B binding activity in peripheral blood mononuclear cells isolated from patients with type 1 diabetes. Diabetes Care 1998, 21, 1310–1316. [Google Scholar] [CrossRef]
- Mabley, J.G.; Hasko, G.; Liaudet, L.; Soriano, F.G.; Southan, G.J.; Salzman, A.L.; Szabo, C. NFkappaB1 (p50)-deficient mice are not susceptible to multiple low-dose streptozotocin-induced diabetes. J. Endocrinol. 2002, 173, 457–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Listrat, A.; Lebret, B.; Louveau, I.; Astruc, T.; Bonnet, M.; Lefaucheur, L.; Picard, B.; Bugeon, J. How Muscle Structure and Composition Influence Meat and Flesh Quality. Sci. World J. 2016, 2016, 3182746. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-kappaB, inflammation, and metabolic disease. Cell Metab. 2011, 13, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.J.; Yamamoto, Y.; Gaynor, R.B. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 1998, 396, 77–80. [Google Scholar] [CrossRef]
- Kim, J.K.; Kim, Y.J.; Fillmore, J.J.; Chen, Y.; Moore, I.; Lee, J.; Yuan, M.; Li, Z.W.; Karin, M.; Perret, P.; et al. Prevention of fat-induced insulin resistance by salicylate. J. Clin. Investig. 2001, 108, 437–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, M.; Konstantopoulos, N.; Lee, J.; Hansen, L.; Li, Z.W.; Karin, M.; Shoelson, S.E. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science 2001, 293, 1673–1677. [Google Scholar] [CrossRef] [PubMed]
- Rohl, M.; Pasparakis, M.; Baudler, S.; Baumgartl, J.; Gautam, D.; Huth, M.; De Lorenzi, R.; Krone, W.; Rajewsky, K.; Bruning, J.C. Conditional disruption of IkappaB kinase 2 fails to prevent obesity-induced insulin resistance. J. Clin. Investig. 2004, 113, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Polkinghorne, E.; Lau, Q.; Cooney, G.J.; Kraegen, E.W.; Cleasby, M.E. Local activation of the IkappaK-NF-kappaB pathway in muscle does not cause insulin resistance. Am. J. Physiol. 2008, 294, E316–E325. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harbor Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C.; Chang, J.H.; Jin, J. Regulation of nuclear factor-kappaB in autoimmunity. Trends Immunol. 2013, 34, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. Non-canonical NF-kappaB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Beinke, S.; Ley, S.C. Functions of NF-kappaB1 and NF-kappaB2 in immune cell biology. Biochem. J. 2004, 382, 393–409. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Signaling to NF-kappaB. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef] [PubMed]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef]
- Karin, M.; Delhase, M. The I kappa B kinase (IKK) and NF-kappa B: Key elements of proinflammatory signalling. Semin. Immunol. 2000, 12, 85–98. [Google Scholar] [CrossRef]
- Sun, S.C.; Ley, S.C. New insights into NF-kappaB regulation and function. Trends Immunol. 2008, 29, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Israel, A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harbor Perspect. Biol. 2010, 2, a000158. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. The noncanonical NF-kappaB pathway. Immunol. Rev. 2012, 246, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C.; Liu, Z.G. A special issue on NF-kappaB signaling and function. Cell Res. 2011, 21, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sun, S.C. NF-kappaB in inflammation and renal diseases. Cell Biosci. 2015, 5, 63. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Harhaj, E.W.; Sun, S.C. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol. Cell 2001, 7, 401–409. [Google Scholar] [CrossRef]
- Senftleben, U.; Cao, Y.; Xiao, G.; Greten, F.R.; Krahn, G.; Bonizzi, G.; Chen, Y.; Hu, Y.; Fong, A.; Sun, S.C.; et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science 2001, 293, 1495–1499. [Google Scholar] [CrossRef]
- Perkins, N.D. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat. Rev. Mol. Cell Biol. 2007, 8, 49–62. [Google Scholar] [CrossRef]
- Peters, R.T.; Liao, S.M.; Maniatis, T. IKKepsilon is part of a novel PMA-inducible IkappaB kinase complex. Mol. Cell 2000, 5, 513–522. [Google Scholar] [CrossRef]
- Shimada, T.; Kawai, T.; Takeda, K.; Matsumoto, M.; Inoue, J.; Tatsumi, Y.; Kanamaru, A.; Akira, S. IKK-i, a novel lipopolysaccharide-inducible kinase that is related to IkappaB kinases. Int. Immunol. 1999, 11, 1357–1362. [Google Scholar] [CrossRef] [PubMed]
- Pomerantz, J.L.; Baltimore, D. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 1999, 18, 6694–6704. [Google Scholar] [CrossRef] [PubMed]
- Tojima, Y.; Fujimoto, A.; Delhase, M.; Chen, Y.; Hatakeyama, S.; Nakayama, K.; Kaneko, Y.; Nimura, Y.; Motoyama, N.; Ikeda, K.; et al. NAK is an IkappaB kinase-activating kinase. Nature 2000, 404, 778–782. [Google Scholar] [CrossRef] [PubMed]
- Bonnard, M.; Mirtsos, C.; Suzuki, S.; Graham, K.; Huang, J.; Ng, M.; Itie, A.; Wakeham, A.; Shahinian, A.; Henzel, W.J.; et al. Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription. EMBO J. 2000, 19, 4976–4985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kravchenko, V.V.; Mathison, J.C.; Schwamborn, K.; Mercurio, F.; Ulevitch, R.J. IKKi/IKKepsilon plays a key role in integrating signals induced by pro-inflammatory stimuli. J. Biol. Chem. 2003, 278, 26612–26619. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; tenOever, B.R.; Grandvaux, N.; Zhou, G.P.; Lin, R.; Hiscott, J. Triggering the interferon antiviral response through an IKK-related pathway. Science 2003, 300, 1148–1151. [Google Scholar] [CrossRef] [PubMed]
- McWhirter, S.M.; Fitzgerald, K.A.; Rosains, J.; Rowe, D.C.; Golenbock, D.T.; Maniatis, T. IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc. Natl. Acad. Sci. USA 2004, 101, 233–238. [Google Scholar] [CrossRef]
- Xu, J.; Jia, Y.F.; Tapadar, S.; Weaver, J.D.; Raji, I.O.; Pithadia, D.J.; Javeed, N.; Garcia, A.J.; Choi, D.S.; Matveyenko, A.V.; et al. Inhibition of TBK1/IKKepsilon Promotes Regeneration of Pancreatic beta-cells. Sci. Rep. 2018, 8, 15587. [Google Scholar] [CrossRef]
- Peters, R.T.; Maniatis, T. A new family of IKK-related kinases may function as I kappa B kinase kinases. BBA Mol. Cell. Biol. Lipids 2001, 1471, M57–M62. [Google Scholar]
- Buss, H.; Dorrie, A.; Schmitz, M.L.; Hoffmann, E.; Resch, K.; Kracht, M. Constitutive and interleukin-1-inducible phosphorylation of p65 NF-{kappa}B at serine 536 is mediated by multiple protein kinases including I{kappa}B kinase (IKK)-{alpha}, IKK{beta}, IKK{epsilon}, TRAF family member-associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-binding protein-associated factor II31-mediated interleukin-8 transcription. J. Biol. Chem. 2004, 279, 55633–55643. [Google Scholar] [CrossRef]
- Fujita, F.; Taniguchi, Y.; Kato, T.; Narita, Y.; Furuya, A.; Ogawa, T.; Sakurai, H.; Joh, T.; Itoh, M.; Delhase, M.; et al. Identification of NAP1, a regulatory subunit of IkappaB kinase-related kinases that potentiates NF-kappaB signaling. Mol. Cell. Biol. 2003, 23, 7780–7793. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.; Oliere, S.; Sharma, S.; Sun, Q.; Lin, R.; Hiscott, J.; Grandvaux, N. Nuclear accumulation of cRel following C-terminal phosphorylation by TBK1/IKK epsilon. J. Immunol. 2006, 177, 2527–2535. [Google Scholar] [CrossRef] [PubMed]
- Mattioli, I.; Geng, H.; Sebald, A.; Hodel, M.; Bucher, C.; Kracht, M.; Schmitz, M.L. Inducible phosphorylation of NF-kappa B p65 at serine 468 by T cell costimulation is mediated by IKK epsilon. J. Biol. Chem. 2006, 281, 6175–6183. [Google Scholar] [CrossRef]
- Wietek, C.; Cleaver, C.S.; Ludbrook, V.; Wilde, J.; White, J.; Bell, D.J.; Lee, M.; Dickson, M.; Ray, K.P.; O’Neill, L.A. IkappaB kinase epsilon interacts with p52 and promotes transactivation via p65. J. Biol. Chem. 2006, 281, 34973–34981. [Google Scholar] [CrossRef] [PubMed]
- Hemmi, H.; Takeuchi, O.; Sato, S.; Yamamoto, M.; Kaisho, T.; Sanjo, H.; Kawai, T.; Hoshino, K.; Takeda, K.; Akira, S. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J. Exp. Med. 2004, 199, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.K.; Chow, E.K.; Goodnough, J.B.; Yeh, W.C.; Cheng, G. Differential requirement for TANK-binding kinase-1 in type I interferon responses to toll-like receptor activation and viral infection. J. Exp. Med. 2004, 199, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- Chau, T.L.; Gioia, R.; Gatot, J.S.; Patrascu, F.; Carpentier, I.; Chapelle, J.P.; O’Neill, L.; Beyaert, R.; Piette, J.; Chariot, A. Are the IKKs and IKK-related kinases TBK1 and IKK-epsilon similarly activated? Trends Biochem. Sci. 2008, 33, 171–180. [Google Scholar] [CrossRef]
- Clement, J.F.; Meloche, S.; Servant, M.J. The IKK-related kinases: From innate immunity to oncogenesis. Cell Res. 2008, 18, 889–899. [Google Scholar] [CrossRef]
- Hacker, H.; Redecke, V.; Blagoev, B.; Kratchmarova, I.; Hsu, L.C.; Wang, G.G.; Kamps, M.P.; Raz, E.; Wagner, H.; Hacker, G.; et al. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 2006, 439, 204–207. [Google Scholar] [CrossRef]
- Newton, K.; Dixit, V.M. Signaling in innate immunity and inflammation. Cold Spring Harbor Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Karin, M. Missing pieces in the NF-kappaB puzzle. Cell 2002, 109, S81–S96. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. NF-kappaB in immunobiology. Cell Res. 2011, 21, 223–244. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Clement, J.F.; Bibeau-Poirier, A.; Gravel, S.P.; Grandvaux, N.; Bonneil, E.; Thibault, P.; Meloche, S.; Servant, M.J. Phosphorylation of IRF-3 on Ser 339 generates a hyperactive form of IRF-3 through regulation of dimerization and CBP association. J. Virol. 2008, 82, 3984–3996. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Yoneyama, M.; Ito, T.; Takahashi, K.; Inagaki, F.; Fujita, T. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. J. Biol. Chem. 2004, 279, 9698–9702. [Google Scholar] [CrossRef]
- Panne, D.; McWhirter, S.M.; Maniatis, T.; Harrison, S.C. Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. J. Biol. Chem. 2007, 282, 22816–22822. [Google Scholar] [CrossRef]
- Ten Oever, B.R.; Sharma, S.; Zou, W.; Sun, Q.; Grandvaux, N.; Julkunen, I.; Hemmi, H.; Yamamoto, M.; Akira, S.; Yeh, W.C.; et al. Activation of TBK1 and IKKvarepsilon kinases by vesicular stomatitis virus infection and the role of viral ribonucleoprotein in the development of interferon antiviral immunity. J. Virol. 2004, 78, 10636–10649. [Google Scholar] [CrossRef]
- Caillaud, A.; Hovanessian, A.G.; Levy, D.E.; Marie, I.J. Regulatory serine residues mediate phosphorylation-dependent and phosphorylation-independent activation of interferon regulatory factor 7. J. Biol. Chem. 2005, 280, 17671–17677. [Google Scholar] [CrossRef]
- Nomura, F.; Kawai, T.; Nakanishi, K.; Akira, S. NF-kappaB activation through IKK-i-dependent I-TRAF/TANK phosphorylation. Genes Cells 2000, 5, 191–202. [Google Scholar] [Green Version]
- Gatot, J.S.; Gioia, R.; Chau, T.L.; Patrascu, F.; Warnier, M.; Close, P.; Chapelle, J.P.; Muraille, E.; Brown, K.; Siebenlist, U.; et al. Lipopolysaccharide-mediated interferon regulatory factor activation involves TBK1-IKKepsilon-dependent Lys(63)-linked polyubiquitination and phosphorylation of TANK/I-TRAF. J. Biol. Chem. 2007, 282, 31131–31146. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Cheng, G. Modulation of the interferon antiviral response by the TBK1/IKKi adaptor protein TANK. J. Biol. Chem. 2007, 282, 11817–11826. [Google Scholar] [CrossRef]
- Rothe, M.; Xiong, J.; Shu, H.B.; Williamson, K.; Goddard, A.; Goeddel, D.V. I-TRAF is a novel TRAF-interacting protein that regulates TRAF-mediated signal transduction. Proc. Natl. Acad. Sci. USA 1996, 93, 8241–8246. [Google Scholar] [CrossRef] [PubMed]
- Sasai, M.; Oshiumi, H.; Matsumoto, M.; Inoue, N.; Fujita, F.; Nakanishi, M.; Seya, T. Cutting Edge: NF-kappaB-activating kinase-associated protein 1 participates in TLR3/Toll-IL-1 homology domain-containing adapter molecule-1-mediated IFN regulatory factor 3 activation. J. Immunol. 2005, 174, 27–30. [Google Scholar] [CrossRef]
- Ryzhakov, G.; Randow, F. SINTBAD, a novel component of innate antiviral immunity, shares a TBK1-binding domain with NAP1 and TANK. EMBO J. 2007, 26, 3180–3190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef]
- Andrejeva, J.; Childs, K.S.; Young, D.F.; Carlos, T.S.; Stock, N.; Goodbourn, S.; Randall, R.E. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc. Natl. Acad. Sci. USA 2004, 101, 17264–17269. [Google Scholar] [CrossRef]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Tenoever, B.R.; Ng, S.L.; Chua, M.A.; McWhirter, S.M.; Garcia-Sastre, A.; Maniatis, T. Multiple functions of the IKK-related kinase IKKepsilon in interferon-mediated antiviral immunity. Science 2007, 315, 1274–1278. [Google Scholar] [CrossRef] [PubMed]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, Y.; Kim, S.; Bumeister, R.; Loo, Y.M.; Kwon, S.W.; Johnson, C.L.; Balakireva, M.G.; Romeo, Y.; Kopelovich, L.; Gale, M., Jr.; et al. RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell 2006, 127, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Eskiocak, B.; McMillan, E.A.; Mendiratta, S.; Kollipara, R.K.; Zhang, H.; Humphries, C.G.; Wang, C.; Garcia-Rodriguez, J.; Ding, M.; Zaman, A.; et al. Biomarker Accessible and Chemically Addressable Mechanistic Subtypes of BRAF Melanoma. Cancer Discov. 2017, 7, 832–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, J.M.; Ou, Y.H.; McMillan, E.A.; Vaden, R.M.; Zaman, A.; Bodemann, B.O.; Makkar, G.; Posner, B.A.; White, M.A. TBK1 Provides Context-Selective Support of the Activated AKT/mTOR Pathway in Lung Cancer. Cancer Res. 2017, 77, 5077–5094. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, L.; Cai, S.H.; Cheng, H. Identification of TBK1 and IKKepsilon, the non-canonical IkappaB kinases, as crucial pro-survival factors in HTLV-1-transformed T lymphocytes. Leuk. Res. 2016, 46, 37–44. [Google Scholar] [CrossRef]
- Wei, C.; Cao, Y.; Yang, X.; Zheng, Z.; Guan, K.; Wang, Q.; Tai, Y.; Zhang, Y.; Ma, S.; Cao, Y.; et al. Elevated expression of TANK-binding kinase 1 enhances tamoxifen resistance in breast cancer. Proc. Natl. Acad. Sci. USA 2014, 111, E601–E610. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, A.S., Jr. Series introduction: The transcription factor NF-kappaB and human disease. J. Clin. Investig. 2001, 107, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef]
- Hutti, J.E.; Shen, R.R.; Abbott, D.W.; Zhou, A.Y.; Sprott, K.M.; Asara, J.M.; Hahn, W.C.; Cantley, L.C. Phosphorylation of the tumor suppressor CYLD by the breast cancer oncogene IKKepsilon promotes cell transformation. Mol. Cell 2009, 34, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Brummelkamp, T.R.; Nijman, S.M.; Dirac, A.M.; Bernards, R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature 2003, 424, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Kovalenko, A.; Chable-Bessia, C.; Cantarella, G.; Israel, A.; Wallach, D.; Courtois, G. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature 2003, 424, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Trompouki, E.; Hatzivassiliou, E.; Tsichritzis, T.; Farmer, H.; Ashworth, A.; Mosialos, G. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature 2003, 424, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.F.; Guo, S.; Demicco, E.G.; Romieu-Mourez, R.; Landesman-Bollag, E.; Seldin, D.C.; Sonenshein, G.E. Inducible IkappaB kinase/IkappaB kinase epsilon expression is induced by CK2 and promotes aberrant nuclear factor-kappaB activation in breast cancer cells. Cancer Res. 2005, 65, 11375–11383. [Google Scholar] [CrossRef]
- Barbie, T.U.; Alexe, G.; Aref, A.R.; Li, S.; Zhu, Z.; Zhang, X.; Imamura, Y.; Thai, T.C.; Huang, Y.; Bowden, M.; et al. Targeting an IKBKE cytokine network impairs triple-negative breast cancer growth. J. Clin. Investig. 2014, 124, 5411–5423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, H.; Zhang, H.; Cai, J.; Wu, J.; Yuan, J.; Li, J.; Huang, Z.; Li, M. IKBKE is over-expressed in glioma and contributes to resistance of glioma cells to apoptosis via activating NF-kappaB. J. Pathol. 2011, 223, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Guo, J.; Henderson-Jackson, E.; Kim, D.; Malafa, M.; Coppola, D. IkappaB Kinase epsilon expression in pancreatic ductal adenocarcinoma. Am. J. Clin. Pathol. 2011, 136, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Rajurkar, M.; Dang, K.; Fernandez-Barrena, M.G.; Liu, X.; Fernandez-Zapico, M.E.; Lewis, B.C.; Mao, J. IKBKE Is Required during KRAS-Induced Pancreatic Tumorigenesis. Cancer Res. 2017, 77, 320–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.P.; Shu, S.K.; He, L.; Lee, Y.C.; Kruk, P.A.; Grenman, S.; Nicosia, S.V.; Mor, G.; Schell, M.J.; Coppola, D.; et al. Deregulation of IKBKE is associated with tumor progression, poor prognosis, and cisplatin resistance in ovarian cancer. Am. J. Pathol. 2009, 175, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.; Kim, M.; Hernandez, L.; Grajales, V.; Noonan, A.; Anver, M.; Davidson, B.; Annunziata, C.M. IKK-epsilon coordinates invasion and metastasis of ovarian cancer. Cancer Res. 2012, 72, 5494–5504. [Google Scholar] [CrossRef] [PubMed]
- Michalaki, V.; Syrigos, K.; Charles, P.; Waxman, J. Serum levels of IL-6 and TNF-alpha correlate with clinicopathological features and patient survival in patients with prostate cancer. Br. J. Cancer 2004, 90, 2312–2316. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, J.; Tachibana, M.; Horiguchi, Y.; Oya, M.; Ohigashi, T.; Asakura, H.; Murai, M. Serum interleukin 6 as a prognostic factor in patients with prostate cancer. Clin. Cancer Res. 2000, 6, 2702–2706. [Google Scholar] [PubMed]
- Peant, B.; Gilbert, S.; Le Page, C.; Poisson, A.; L’Ecuyer, E.; Boudhraa, Z.; Bienz, M.N.; Delvoye, N.; Saad, F.; Mes-Masson, A.M. IkappaB-Kinase-epsilon (IKKepsilon) over-expression promotes the growth of prostate cancer through the C/EBP-beta dependent activation of IL-6 gene expression. Oncotarget 2017, 8, 14487–14501. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef] [Green Version]
- Olefsky, J.M.; Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Reilly, S.M.; Saltiel, A.R. Adapting to obesity with adipose tissue inflammation. Nat. Rev. Endocrinol. 2017, 13, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.J.; Santos, A.; Prada, P.O. Linking Gut Microbiota and Inflammation to Obesity and Insulin Resistance. Physiology 2016, 31, 283–293. [Google Scholar] [CrossRef] [Green Version]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat. Med. 2005, 11, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, G.; Zhang, H.; Karin, M.; Bai, H.; Cai, D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 2008, 135, 61–73. [Google Scholar] [CrossRef]
- Oral, E.A.; Reilly, S.M.; Gomez, A.V.; Meral, R.; Butz, L.; Ajluni, N.; Chenevert, T.L.; Korytnaya, E.; Neidert, A.H.; Hench, R.; et al. Inhibition of IKKvarepsilon and TBK1 Improves Glucose Control in a Subset of Patients with Type 2 Diabetes. Cell Metab. 2017, 26, 157–170. [Google Scholar] [CrossRef]
- Mowers, J.; Uhm, M.; Reilly, S.M.; Simon, J.; Leto, D.; Chiang, S.H.; Chang, L.; Saltiel, A.R. Inflammation produces catecholamine resistance in obesity via activation of PDE3B by the protein kinases IKKepsilon and TBK1. Elife 2013, 2, e01119. [Google Scholar] [CrossRef]
- Zmuda-Trzebiatowska, E.; Oknianska, A.; Manganiello, V.; Degerman, E. Role of PDE3B in insulin-induced glucose uptake, GLUT-4 translocation and lipogenesis in primary rat adipocytes. Cell. Signal. 2006, 18, 382–390. [Google Scholar] [CrossRef] [Green Version]
- Reilly, S.M.; Ahmadian, M.; Zamarron, B.F.; Chang, L.; Uhm, M.; Poirier, B.; Peng, X.; Krause, D.M.; Korytnaya, E.; Neidert, A.; et al. A subcutaneous adipose tissue-liver signalling axis controls hepatic gluconeogenesis. Nat. Commun. 2015, 6, 6047. [Google Scholar] [CrossRef]
- Ramadoss, P.; Unger-Smith, N.E.; Lam, F.S.; Hollenberg, A.N. STAT3 targets the regulatory regions of gluconeogenic genes in vivo. Mol. Endocrinol. 2009, 23, 827–837. [Google Scholar] [CrossRef]
- Inoue, H.; Ogawa, W.; Ozaki, M.; Haga, S.; Matsumoto, M.; Furukawa, K.; Hashimoto, N.; Kido, Y.; Mori, T.; Sakaue, H.; et al. Role of STAT-3 in regulation of hepatic gluconeogenic genes and carbohydrate metabolism in vivo. Nat. Med. 2004, 10, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Ogawa, W.; Asakawa, A.; Okamoto, Y.; Nishizawa, A.; Matsumoto, M.; Teshigawara, K.; Matsuki, Y.; Watanabe, E.; Hiramatsu, R.; et al. Role of hepatic STAT3 in brain-insulin action on hepatic glucose production. Cell Metab. 2006, 3, 267–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, P.; Wong, K.I.; Sun, X.; Reilly, S.M.; Uhm, M.; Liao, Z.; Skorobogatko, Y.; Saltiel, A.R. TBK1 at the Crossroads of Inflammation and Energy Homeostasis in Adipose Tissue. Cell 2018, 172, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Daval, M.; Foufelle, F.; Ferre, P. Functions of AMP-activated protein kinase in adipose tissue. J. Physiol. 2006, 574, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G. AMP-activated protein kinase: An energy sensor that regulates all aspects of cell function. Genes Dev. 2011, 25, 1895–1908. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.J.; Bhushan, A.; Butler, A.E.; Rizza, R.A.; Butler, P.C. Sustained beta cell apoptosis in patients with long-standing type 1 diabetes: Indirect evidence for islet regeneration? Diabetologia 2005, 48, 2221–2228. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Fiaschi-Taesch, N.M.; Vasavada, R.C.; Scott, D.K.; Garcia-Ocana, A.; Stewart, A.F. Diabetes mellitus—Advances and challenges in human beta-cell proliferation. Nat. Rev. Endocrinol. 2015, 11, 201–212. [Google Scholar] [CrossRef]
- Campbell-Thompson, M.; Fu, A.; Kaddis, J.S.; Wasserfall, C.; Schatz, D.A.; Pugliese, A.; Atkinson, M.A. Insulitis and beta-Cell Mass in the Natural History of Type 1 Diabetes. Diabetes 2016, 65, 719–731. [Google Scholar] [CrossRef]
- Chen, C.; Cohrs, C.M.; Stertmann, J.; Bozsak, R.; Speier, S. Human beta cell mass and function in diabetes: Recent advances in knowledge and technologies to understand disease pathogenesis. Mol. Metab. 2017, 6, 943–957. [Google Scholar] [CrossRef] [PubMed]
- Mallone, R.; Roep, B.O. Biomarkers for immune intervention trials in type 1 diabetes. Clin. Immunol. 2013, 149, 286–296. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, M.I. Genomics, type 2 diabetes, and obesity. N. Engl. J. Med. 2010, 363, 2339–2350. [Google Scholar] [CrossRef] [PubMed]
- Flannick, J.; Florez, J.C. Type 2 diabetes: Genetic data sharing to advance complex disease research. Nat. Rev. 2016, 17, 535–549. [Google Scholar] [CrossRef] [PubMed]
- Rahier, J.; Guiot, Y.; Goebbels, R.M.; Sempoux, C.; Henquin, J.C. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes. Metab. 2008, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Dobrian, A.D.; Morris, M.A.; Nadler, J.L. Islet inflammation: A unifying target for diabetes treatment? Trends Endocrinol. Metab. 2013, 24, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Trends Endocrinol. Metab. 2014, 105, 141–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eguchi, K.; Nagai, R. Islet inflammation in type 2 diabetes and physiology. J. Clin. Investig. 2017, 127, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Benthuysen, J.R.; Carrano, A.C.; Sander, M. Advances in beta cell replacement and regeneration strategies for treating diabetes. J. Clin. Investig. 2016, 126, 3651–3660. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Melton, D.A. Pancreas regeneration. Nature 2018, 557, 351–358. [Google Scholar] [CrossRef]
- Dor, Y.; Brown, J.; Martinez, O.I.; Melton, D.A. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004, 429, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; D’Hoker, J.; Stange, G.; Bonne, S.; De Leu, N.; Xiao, X.; Van de Casteele, M.; Mellitzer, G.; Ling, Z.; Pipeleers, D.; et al. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell 2008, 132, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Thorel, F.; Nepote, V.; Avril, I.; Kohno, K.; Desgraz, R.; Chera, S.; Herrera, P.L. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature 2010, 464, 1149–1154. [Google Scholar] [CrossRef] [PubMed]
- Baeyens, L.; Lemper, M.; Leuckx, G.; De Groef, S.; Bonfanti, P.; Stange, G.; Shemer, R.; Nord, C.; Scheel, D.W.; Pan, F.C.; et al. Transient cytokine treatment induces acinar cell reprogramming and regenerates functional beta cell mass in diabetic mice. Nat. Biotechnol. 2014, 32, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Gregg, B.E.; Moore, P.C.; Demozay, D.; Hall, B.A.; Li, M.; Husain, A.; Wright, A.J.; Atkinson, M.A.; Rhodes, C.J. Formation of a human beta-cell population within pancreatic islets is set early in life. J. Clin. Endocrinol. Metab. 2012, 97, 3197–3206. [Google Scholar] [CrossRef] [PubMed]
- Perl, S.; Kushner, J.A.; Buchholz, B.A.; Meeker, A.K.; Stein, G.M.; Hsieh, M.; Kirby, M.; Pechhold, S.; Liu, E.H.; Harlan, D.M.; et al. Significant human beta-cell turnover is limited to the first three decades of life as determined by in vivo thymidine analog incorporation and radiocarbon dating. J. Clin. Endocrinol. Metab. 2010, 95, E234–E239. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Sudo, T.; Tachibana, K.; Toga, K.; Tochizawa, S.; Inoue, Y.; Kimura, Y.; Hidaka, H. Potent effects of novel anti-platelet aggregatory cilostamide analogues on recombinant cyclic nucleotide phosphodiesterase isozyme activity. Biochem. Pharmacol. 2000, 59, 347–356. [Google Scholar] [CrossRef]
- Dang, C.V. Links between metabolism and cancer. Genes Dev. 2012, 26, 877–890. [Google Scholar] [CrossRef] [Green Version]
- Uhm, M.; Bazuine, M.; Zhao, P.; Chiang, S.H.; Xiong, T.; Karunanithi, S.; Chang, L.; Saltiel, A.R. Phosphorylation of the exocyst protein Exo84 by TBK1 promotes insulin-stimulated GLUT4 trafficking. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef]
- Munoz, M.C.; Giani, J.F.; Mayer, M.A.; Toblli, J.E.; Turyn, D.; Dominici, F.P. TANK-binding kinase 1 mediates phosphorylation of insulin receptor at serine residue 994: A potential link between inflammation and insulin resistance. J. Endocrinol. 2009, 201, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Hu, D.; Chen, H.; Shi, G.; Fetahu, I.S.; Wu, F.; Rabidou, K.; Fang, R.; Tan, L.; Xu, S.; et al. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature 2018, 559, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Low, Y.S.; Armstrong, N.A.; Ryu, J.H.; Sun, S.A.; Arvanites, A.C.; Hollister-Lock, J.; Shah, N.H.; Weir, G.C.; Annes, J.P. Repurposing cAMP-modulating medications to promote beta-cell replication. Mol. Endocrinol. 2014, 28, 1682–1697. [Google Scholar] [CrossRef] [PubMed]
- Inada, A.; Hamamoto, Y.; Tsuura, Y.; Miyazaki, J.; Toyokuni, S.; Ihara, Y.; Nagai, K.; Yamada, Y.; Bonner-Weir, S.; Seino, Y. Overexpression of inducible cyclic AMP early repressor inhibits transactivation of genes and cell proliferation in pancreatic beta cells. Mol. Cell. Biol. 2004, 24, 2831–2841. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Chen, M.; Zhang, Q.H.; Ma, Z.; Weinstein, L.S. Beta cell-specific deficiency of the stimulatory G protein alpha-subunit Gsalpha leads to reduced beta cell mass and insulin-deficient diabetes. Proc. Natl. Acad. Sci. USA 2007, 104, 19601–19606. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; Park, S.; Hockman, S.; Zmuda-Trzebiatowska, E.; Svennelid, F.; Haluzik, M.; Gavrilova, O.; Ahmad, F.; Pepin, L.; Napolitano, M.; et al. Alterations in regulation of energy homeostasis in cyclic nucleotide phosphodiesterase 3B-null mice. J. Clin. Investig. 2006, 116, 3240–3251. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Block, M.A.; Cowen, S.; Davies, A.M.; Devereaux, E.; Gingipalli, L.; Johannes, J.; Larsen, N.A.; Su, Q.; Tucker, J.A.; et al. Discovery of azabenzimidazole derivatives as potent, selective inhibitors of TBK1/IKKepsilon kinases. Bioorg. Med. Chem. Lett. 2012, 22, 2063–2069. [Google Scholar] [CrossRef] [PubMed]
- Vu, H.L.; Aplin, A.E. Targeting TBK1 inhibits migration and resistance to MEK inhibitors in mutant NRAS melanoma. Mol. Cancer Res. 2014, 12, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Makino, H.; Saijo, T.; Ashida, Y.; Kuriki, H.; Maki, Y. Mechanism of action of an antiallergic agent, amlexanox (AA-673), in inhibiting histamine release from mast cells. Acceleration of cAMP generation and inhibition of phosphodiesterase. Int. Arch. Allergy Appl. Immunol. 1987, 82, 66–71. [Google Scholar] [CrossRef]
- Bell, J. Amlexanox for the treatment of recurrent aphthous ulcers. Clin. Drug Invest. 2005, 25, 555–566. [Google Scholar] [CrossRef]
- Beyett, T.S.; Gan, X.; Reilly, S.M.; Chang, L.; Gomez, A.V.; Saltiel, A.R.; Showalter, H.D.; Tesmer, J.J.G. Carboxylic Acid Derivatives of Amlexanox Display Enhanced Potency toward TBK1 and IKKepsilon and Reveal Mechanisms for Selective Inhibition. Mol. Pharmacol. 2018, 94, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- Beyett, T.S.; Gan, X.; Reilly, S.M.; Gomez, A.V.; Chang, L.; Tesmer, J.J.G.; Saltiel, A.R.; Showalter, H.D. Design, synthesis, and biological activity of substituted 2-amino-5-oxo-5H-chromeno[2,3-b]pyridine-3-carboxylic acid derivatives as inhibitors of the inflammatory kinases TBK1 and IKKepsilon for the treatment of obesity. Bioorg. Med. Chem. 2018, 26, 5443–5461. [Google Scholar] [CrossRef] [PubMed]
- Crew, A.P.; Raina, K.; Dong, H.; Qian, Y.; Wang, J.; Vigil, D.; Serebrenik, Y.V.; Hamman, B.D.; Morgan, A.; Ferraro, C.; et al. Identification and Characterization of Von Hippel-Lindau-Recruiting Proteolysis Targeting Chimeras (PROTACs) of TANK-Binding Kinase 1. J. Med. Chem. 2018, 61, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wild, P.; Farhan, H.; McEwan, D.G.; Wagner, S.; Rogov, V.V.; Brady, N.R.; Richter, B.; Korac, J.; Waidmann, O.; Choudhary, C.; et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011, 333, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef]
- Pilli, M.; Arko-Mensah, J.; Ponpuak, M.; Roberts, E.; Master, S.; Mandell, M.A.; Dupont, N.; Ornatowski, W.; Jiang, S.; Bradfute, S.B.; et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 2012, 37, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, G.; Shimogori, T.; Hattori, N.; Nukina, N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum. Mol. Genet. 2015, 24, 4429–4442. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Jin, T.; Zhu, H.; Chen, H.; Ofengeim, D.; Zou, C.; Mifflin, L.; Pan, L.; Amin, P.; Li, W.; et al. TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging. Cell 2018, 174, 1477–1491. [Google Scholar] [CrossRef]
- Freischmidt, A.; Muller, K.; Ludolph, A.C.; Weishaupt, J.H.; Andersen, P.M. Association of Mutations in TBK1 With Sporadic and Familial Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. JAMA Neurol. 2017, 74, 110–113. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, C.H.; Choi, D.-S. Essential Roles for the Non-Canonical IκB Kinases in Linking Inflammation to Cancer, Obesity, and Diabetes. Cells 2019, 8, 178. https://doi.org/10.3390/cells8020178
Shin CH, Choi D-S. Essential Roles for the Non-Canonical IκB Kinases in Linking Inflammation to Cancer, Obesity, and Diabetes. Cells. 2019; 8(2):178. https://doi.org/10.3390/cells8020178
Chicago/Turabian StyleShin, Chong Hyun, and Doo-Sup Choi. 2019. "Essential Roles for the Non-Canonical IκB Kinases in Linking Inflammation to Cancer, Obesity, and Diabetes" Cells 8, no. 2: 178. https://doi.org/10.3390/cells8020178
APA StyleShin, C. H., & Choi, D. -S. (2019). Essential Roles for the Non-Canonical IκB Kinases in Linking Inflammation to Cancer, Obesity, and Diabetes. Cells, 8(2), 178. https://doi.org/10.3390/cells8020178