Tankyrase (PARP5) Inhibition Induces Bone Loss through Accumulation of Its Substrate SH3BP2

Abstract
:1. Introduction
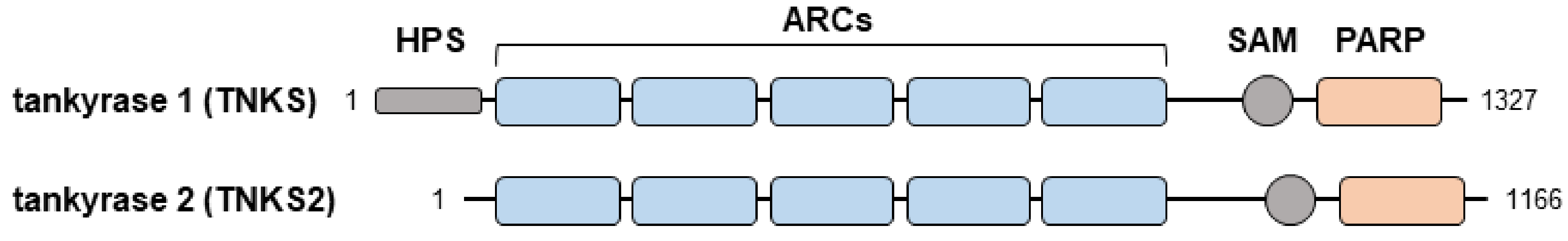
1.1. Overview of Tankyrase
1.2. Preclinical Application of Tankyrase Inhibitors in Cancer
1.3. Preclinical Application of Tankyrase Inhibitors in Fibrotic Diseases
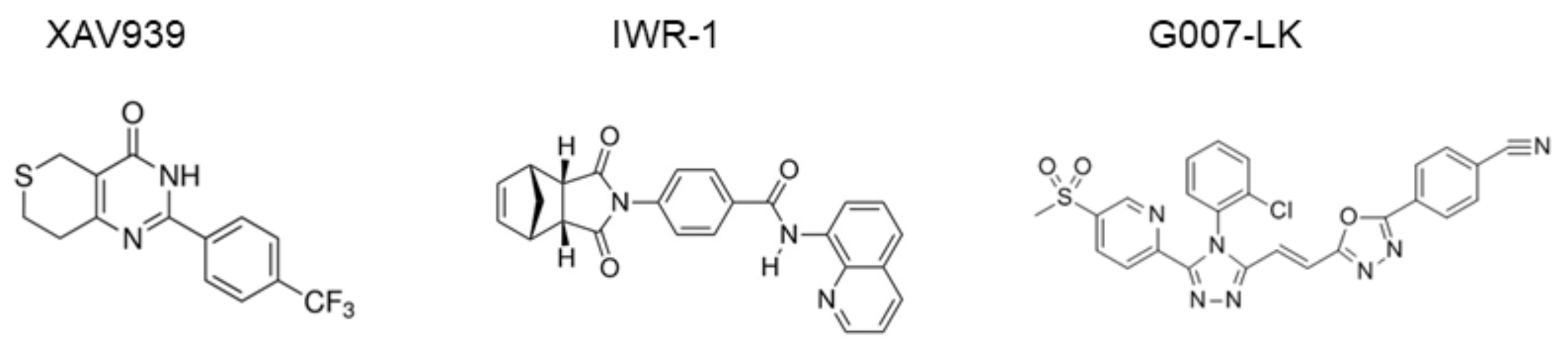
1.4. Tankyrase-Specific Inhibitors
2. Effects of Tankyrase Inhibition on Bone
2.1. SH3BP2, an Unappreciated Substrate for Tankyrase, Regulates Osteoclastogenesis
2.2. Increased Osteoclastogenesis Induced by Tankyrase Inhibitors
2.3. Increased Osteoblast Maturation Induced by Tankyrase Inhibitors
2.4. In Vivo Bone Loss Due to Tankyrase Inhibitors
2.5. Effects of PARP1 Inhibition on Bone
3. Other Possible Effects of Tankyrase Inhibition and Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Palazzo, L.; Ahel, I. PARPs in genome stability and signal transduction: Implications for cancer therapy. Biochem. Soc. Trans. 2018, 46, 1681–1695. [Google Scholar] [CrossRef] [PubMed]
- Lakshmi, T.V.; Bale, S.; Khurana, A.; Godugu, C. Tankyrase as a novel molecular target in cancer and fibrotic diseases. Curr. Drug Targets 2017, 18, 1214–1224. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.; Giriat, I.; Schmitt, A.; de Lange, T. Tankyrase, a poly(ADP-ribose) polymerase at human telomeres. Science 1998, 282, 1484–1487. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K. Novel insight into the function of tankyrase. Oncol. Lett. 2018, 16, 6895–6902. [Google Scholar] [CrossRef] [PubMed]
- Guettler, S.; La Rose, J.; Petsalaki, E.; Gish, G.; Scotter, A.; Pawson, T.; Rottapel, R.; Sicheri, F. Structural basis and sequence rules for substrate recognition by tankyrase explain the basis for cherubism disease. Cell 2011, 147, 1340–1354. [Google Scholar] [CrossRef] [PubMed]
- Levaot, N.; Voytyuk, O.; Dimitriou, I.; Sircoulomb, F.; Chandrakumar, A.; Deckert, M.; Krzyzanowski, P.M.; Scotter, A.; Gu, S.; Janmohamed, S.; et al. Loss of tankyrase-mediated destruction of 3BP2 is the underlying pathogenic mechanism of cherubism. Cell 2011, 147, 1324–1339. [Google Scholar] [CrossRef] [PubMed]
- Chiang, Y.J.; Nguyen, M.L.; Gurunathan, S.; Kaminker, P.; Tessarollo, L.; Campisi, J.; Hodes, R.J. Generation and characterization of telomere length maintenance in tankyrase 2-deficient mice. Mol. Cell Biol. 2006, 26, 2037–2043. [Google Scholar] [CrossRef] [PubMed]
- Chiang, Y.J.; Hsiao, S.J.; Yver, D.; Cushman, S.W.; Tessarollo, L.; Smith, S.; Hodes, R.J. Tankyrase 1 and tankyrase 2 are essential but redundant for mouse embryonic development. PLoS ONE 2008, 3, e2639. [Google Scholar] [CrossRef] [PubMed]
- Seimiya, H.; Smith, S. The telomeric poly (ADP-ribose) polymerase, tankyrase 1, contains multiple binding sites for telomeric repeat binding factor 1 (TRF1) and a novel acceptor, 182-kDa tankyrase-binding protein (TAB182). J. Biol. Chem. 2002, 277, 14116–14126. [Google Scholar] [CrossRef] [PubMed]
- Sbodio, J.I.; Chi, N.W. Identification of a tankyrase-binding motif shared by IRAP, TAB182, and human TRF1 but not mouse TRF1. NuMA contains this RXXPDG motif and is a novel tankyrase partner. J. Biol. Chem. 2002, 277, 31887–31892. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.A.; Mishina, Y.M.; Liu, S.M.; Cheung, A.; Stegmeier, F.; Michaud, G.A.; Charlat, O.; Wiellette, E.; Zhang, Y.; Wiessner, S.; et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009, 461, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, U.; Rehkamp, G.F.; Slany, R.; Follo, M.; Borkhardt, A. The formin-binding protein 17, FBP17, binds via a TNKS binding motif to tankyrase, a protein involved in telomere maintenance. FEBS Lett. 2003, 554, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Krastev, D.B.; Pettitt, S.J.; Campbell, J.; Song, F.; Tanos, B.E.; Stoynov, S.S.; Ashworth, A.; Lord, C.J. Coupling bimolecular PARylation biosensors with genetic screens to identify PARylation targets. Nat. Commun. 2018, 9, 2016. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.H.; Lee, D.J.; Lee, S.; Lee, S.Y.; Jun, Y.; Kim, Y.; Lee, J.S.; Lee, D.K.; Lee, S.; et al. Interaction of tankyrase and peroxiredoxin II is indispensable for the survival of colorectal cancer cells. Nat. Commun. 2017, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Lehtio, L.; Chi, N.W.; Krauss, S. Tankyrases as drug targets. FEBS J. 2013, 280, 3576–3593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizutani, A.; Yashiroda, Y.; Muramatsu, Y.; Yoshida, H.; Chikada, T.; Tsumura, T.; Okue, M.; Shirai, F.; Fukami, T.; Yoshida, M.; et al. RK-287107, a potent and specific tankyrase inhibitor, blocks colorectal cancer cell growth in a preclinical model. Cancer Sci. 2018, 109, 4003–4014. [Google Scholar] [CrossRef] [PubMed]
- Waaler, J.; Machon, O.; Tumova, L.; Dinh, H.; Korinek, V.; Wilson, S.R.; Paulsen, J.E.; Pedersen, N.M.; Eide, T.J.; Machonova, O.; et al. A novel tankyrase inhibitor decreases canonical Wnt signaling in colon carcinoma cells and reduces tumor growth in conditional APC mutant mice. Cancer Res. 2012, 72, 2822–2832. [Google Scholar] [CrossRef] [PubMed]
- Ha, G.H.; Kim, D.Y.; Breuer, E.K.; Kim, C.K. Combination treatment of polo-like kinase 1 and tankyrase-1 inhibitors enhances anticancer effect in triple-negative breast cancer cells. Anticancer Res. 2018, 38, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Bao, R.; Christova, T.; Song, S.; Angers, S.; Yan, X.; Attisano, L. Inhibition of tankyrases induces Axin stabilization and blocks Wnt signalling in breast cancer cells. PLoS ONE 2012, 7, e48670. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zheng, X.; Han, Y.; Lv, Y.; Lan, F.; Zhao, J. XAV939 inhibits the proliferation and migration of lung adenocarcinoma A549 cells through the WNT pathway. Oncol. Lett. 2018, 15, 8973–8982. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Li, X.; Wang, C.; Chen, Y.; Li, S.; Tan, J.; Tan, B.; He, Y. Inhibition of tankyrase by a novel small molecule significantly attenuates prostate cancer cell proliferation. Cancer Lett. 2018, 443, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Jin, Y.; Feng, S.; Zou, Y.; Xu, S.; Qiu, S.; Li, L.; Zheng, J. Role of Wnt/β -catenin, Wnt/c-Jun N-terminal kinase and Wnt/Ca (2+) pathways in cisplatin-induced chemoresistance in ovarian cancer. Exp. Ther. Med. 2016, 12, 3851–3858. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Qiao, Y.; Pilo, M.G.; Cigliano, A.; Liu, X.; Shao, Z.; Calvisi, D.F.; Chen, X. Tankyrase inhibitors suppress hepatocellular carcinoma cell growth via modulating the Hippo cascade. PLoS ONE 2017, 12, e0184068. [Google Scholar] [CrossRef] [PubMed]
- Martins-Neves, S.R.; Paiva-Oliveira, D.I.; Fontes-Ribeiro, C.; Bovee, J.; Cleton-Jansen, A.M.; Gomes, C.M.F. IWR-1, a tankyrase inhibitor, attenuates Wnt/β -catenin signaling in cancer stem-like cells and inhibits in vivo the growth of a subcutaneous human osteosarcoma xenograft. Cancer Lett. 2018, 414, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Stratford, E.W.; Daffinrud, J.; Munthe, E.; Castro, R.; Waaler, J.; Krauss, S.; Myklebost, O. The tankyrase-specific inhibitor JW74 affects cell cycle progression and induces apoptosis and differentiation in osteosarcoma cell lines. Cancer Med. 2014, 3, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Segditsas, S.; Tomlinson, I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene 2006, 25, 7531–7537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najdi, R.; Holcombe, R.F.; Waterman, M.L. Wnt signaling and colon carcinogenesis: Beyond APC. J. Carcinog. 2011, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni, S.M.; Fearon, E.R. AXIN1 and AXIN2 variants in gastrointestinal cancers. Cancer Lett. 2014, 355, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Li, S. Inhibition of poly (ADP-ribose) polymerase in BRCA mutation carriers. New Engl. J. Med. 2009, 361, 1707–1708. [Google Scholar] [CrossRef] [PubMed]
- Audeh, M.W.; Carmichael, J.; Penson, R.T.; Friedlander, M.; Powell, B.; Bell-McGuinn, K.M.; Scott, C.; Weitzel, J.N.; Oaknin, A.; Loman, N.; et al. Oral poly (ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: A proof-of-concept trial. Lancet 2010, 376, 245–251. [Google Scholar] [CrossRef]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly (ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Nandi, S. DNA damage response and cancer therapeutics through the lens of the Fanconi Anemia DNA repair pathway. Cell Commun. Signal. 2017, 15, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharjee, S.; Nandi, S. Rare genetic diseases with defects in DNA repair: opportunities and challenges in orphan drug development for targeted cancer therapy. Cancers 2018, 10, 298. [Google Scholar] [CrossRef] [PubMed]
- Solberg, N.T.; Waaler, J.; Lund, K.; Mygland, L.; Olsen, P.A.; Krauss, S. Tankyrase inhibition enhances the antiproliferative effect of PI3K and EGFR Inhibition, mutually affecting β-CATENIN and AKT Signaling in Colorectal Cancer. Mol. Cancer Res. 2018, 16, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Seifert, A.M.; Zhang, J.Q.; Cavnar, M.J.; Kim, T.S.; Balachandran, V.P.; Santamaria-Barria, J.A.; Cohen, N.A.; Beckman, M.J.; Medina, B.D.; et al. Wnt/β -catenin Signaling Contributes to tumor malignancy and is targetable in gastrointestinal stromal tumor. Mol. Cancer Ther. 2017, 16, 1954–1966. [Google Scholar] [CrossRef] [PubMed]
- Scarborough, H.A.; Helfrich, B.A.; Casas-Selves, M.; Schuller, A.G.; Grosskurth, S.E.; Kim, J.; Tan, A.C.; Chan, D.C.; Zhang, Z.; Zaberezhnyy, V.; et al. AZ1366: An inhibitor of tankyrase and the canonical wnt pathway that limits the persistence of non-small cell lung cancer cells following egfr inhibition. Clin. Cancer Res. 2017, 23, 1531–1541. [Google Scholar] [CrossRef] [PubMed]
- Quackenbush, K.S.; Bagby, S.; Tai, W.M.; Messersmith, W.A.; Schreiber, A.; Greene, J.; Kim, J.; Wang, G.; Purkey, A.; Pitts, T.M.; et al. The novel tankyrase inhibitor (AZ1366) enhances irinotecan activity in tumors that exhibit elevated tankyrase and irinotecan resistance. Oncotarget 2016, 7, 28273–28285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arques, O.; Chicote, I.; Puig, I.; Tenbaum, S.P.; Argiles, G.; Dienstmann, R.; Fernandez, N.; Caratu, G.; Matito, J.; Silberschmidt, D.; et al. Tankyrase inhibition blocks Wnt/β -catenin pathway and reverts resistance to PI3K and AKT inhibitors in the treatment of colorectal cancer. Clin. Cancer Res. 2016, 22, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Lau, T.; Chan, E.; Callow, M.; Waaler, J.; Boggs, J.; Blake, R.A.; Magnuson, S.; Sambrone, A.; Schutten, M.; Firestein, R.; et al. A novel tankyrase small-molecule inhibitor suppresses APC mutation-driven colorectal tumor growth. Cancer Res. 2013, 73, 3132–3144. [Google Scholar] [CrossRef] [PubMed]
- Chilosi, M.; Poletti, V.; Zamo, A.; Lestani, M.; Montagna, L.; Piccoli, P.; Pedron, S.; Bertaso, M.; Scarpa, A.; Murer, B.; et al. Aberrant Wnt/β-catenin pathway activation in idiopathic pulmonary fibrosis. Am. J. Pathol. 2003, 162, 1495–1502. [Google Scholar] [CrossRef]
- Brack, A.S.; Conboy, M.J.; Roy, S.; Lee, M.; Kuo, C.J.; Keller, C.; Rando, T.A. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science 2007, 317, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Distler, A.; Deloch, L.; Huang, J.; Dees, C.; Lin, N.Y.; Palumbo-Zerr, K.; Beyer, C.; Weidemann, A.; Distler, O.; Schett, G.; et al. Inactivation of tankyrases reduces experimental fibrosis by inhibiting canonical Wnt signalling. Ann. Rheum. Dis. 2013, 72, 1575–1580. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhu, H.; Sun, Z.; Xiang, Z.; Ge, Y.; Ni, C.; Luo, Z.; Qian, W.; Han, X. Inhibition of Wnt/β-catenin signaling promotes epithelial differentiation of mesenchymal stem cells and repairs bleomycin-induced lung injury. Am. J. Physiol. Cell Physiol. 2014, 307, C234–C244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.W.; Yin, W.T.; Jiang, R.H.; Lee, J.H.; Kim, C.D.; Lee, J.H.; Zhu, M.J.; Yoon, T.J. Inhibition of collagen synthesis by IWR-1 in normal and keloid-derived skin fibroblasts. Life Sci. 2017, 173, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Ulsamer, A.; Wei, Y.; Kim, K.K.; Tan, K.; Wheeler, S.; Xi, Y.; Thies, R.S.; Chapman, H.A. Axin pathway activity regulates in vivo pY654-β -catenin accumulation and pulmonary fibrosis. J. Biol. Chem. 2012, 287, 5164–5172. [Google Scholar] [CrossRef] [PubMed]
- Gunaydin, H.; Gu, Y.; Huang, X. Novel binding mode of a potent and selective tankyrase inhibitor. PLoS ONE 2012, 7, e33740. [Google Scholar] [CrossRef] [PubMed]
- Thorsell, A.G.; Ekblad, T.; Karlberg, T.; Low, M.; Pinto, A.F.; Tresaugues, L.; Moche, M.; Cohen, M.S.; Schuler, H. Structural basis for potency and promiscuity in poly (ADP-ribose) polymerase (PARP) and tankyrase inhibitors. J. Med. Chem. 2017, 60, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Voronkov, A.; Holsworth, D.D.; Waaler, J.; Wilson, S.R.; Ekblad, B.; Perdreau-Dahl, H.; Dinh, H.; Drewes, G.; Hopf, C.; Morth, J.P.; et al. Structural basis and SAR for G007-LK, a lead stage 1,2,4-triazole based specific tankyrase 1/2 inhibitor. J. Med. Chem. 2013, 56, 3012–3023. [Google Scholar] [CrossRef] [PubMed]
- Shultz, M.D.; Cheung, A.K.; Kirby, C.A.; Firestone, B.; Fan, J.; Chen, C.H.; Chen, Z.; Chin, D.N.; Dipietro, L.; Fazal, A.; et al. Identification of NVP-TNKS656: The use of structure-efficiency relationships to generate a highly potent, selective, and orally active tankyrase inhibitor. J. Med. Chem. 2013, 56, 6495–6511. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, L.; Pollock, K.; Guettler, S. Regulation of Wnt/β -catenin signalling by tankyrase-dependent poly(ADP-ribosyl)ation and scaffolding. Br. J. Pharmacol. 2017, 174, 4611–4636. [Google Scholar] [CrossRef] [PubMed]
- Hatani, T.; Sada, K. Adaptor protein 3BP2 and cherubism. Curr. Med. Chem. 2008, 15, 549–554. [Google Scholar] [PubMed]
- Ueki, Y.; Lin, C.Y.; Senoo, M.; Ebihara, T.; Agata, N.; Onji, M.; Saheki, Y.; Kawai, T.; Mukherjee, P.M.; Reichenberger, E.; et al. Increased myeloid cell responses to M-CSF and RANKL cause bone loss and inflammation in SH3BP2 “cherubism” mice. Cell 2007, 128, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.J.; Chen, I.P.; Koczon-Jaremko, B.; Boskey, A.L.; Ueki, Y.; Kuhn, L.; Reichenberger, E.J. Pro416Arg cherubism mutation in Sh3bp2 knock-in mice affects osteoblasts and alters bone mineral and matrix properties. Bone 2010, 46, 1306–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Sharma, T.; Chen, I.P.; Reichenberger, E.; Ueki, Y.; Arif, Y.; Parisi, D.; Maye, P. Rescue of a cherubism bone marrow stromal culture phenotype by reducing TGFβ signaling. Bone 2018, 111, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Deckert, M.; Tartare-Deckert, S.; Hernandez, J.; Rottapel, R.; Altman, A. Adaptor function for the Syk kinases-interacting protein 3BP2 in IL-2 gene activation. Immunity 1998, 9, 595–605. [Google Scholar] [CrossRef]
- Lietman, S.A.; Yin, L.; Levine, M.A. SH3BP2 mutations potentiate osteoclastogenesis via PLCγ. J. Orthop. Res. 2010, 28, 1425–1430. [Google Scholar] [CrossRef] [PubMed]
- Mukai, T.; Ishida, S.; Ishikawa, R.; Yoshitaka, T.; Kittaka, M.; Gallant, R.; Lin, Y.L.; Rottapel, R.; Brotto, M.; Reichenberger, E.J.; et al. SH3BP2 cherubism mutation potentiates TNF-α -induced osteoclastogenesis via NFATc1 and TNF-α-mediated inflammatory bone loss. J. Bone Miner. Res. 2014, 29, 2618–2635. [Google Scholar] [CrossRef] [PubMed]
- Foucault, I.; Le Bras, S.; Charvet, C.; Moon, C.; Altman, A.; Deckert, M. The adaptor protein 3BP2 associates with VAV guanine nucleotide exchange factors to regulate NFAT activation by the B-cell antigen receptor. Blood 2005, 105, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Levaot, N.; Simoncic, P.D.; Dimitriou, I.D.; Scotter, A.; La Rose, J.; Ng, A.H.; Willett, T.L.; Wang, C.J.; Janmohamed, S.; Grynpas, M.; et al. 3BP2-deficient mice are osteoporotic with impaired osteoblast and osteoclast functions. J. Clin. Investig. 2011, 121, 3244–3257. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; La Rose, J.; Kent, O.A.; Wagner, M.J.; Narimatsu, M.; Levy, A.D.; Omar, M.H.; Tong, J.; Krieger, J.R.; Riggs, E.; et al. Reciprocal stabilization of ABL and TAZ regulates osteoblastogenesis through transcription factor RUNX2. J. Clin. Investig. 2016, 126, 4482–4496. [Google Scholar] [CrossRef] [PubMed]
- Ueki, Y.; Tiziani, V.; Santanna, C.; Fukai, N.; Maulik, C.; Garfinkle, J.; Ninomiya, C.; do Amaral, C.; Peters, H.; Habal, M.; et al. Mutations in the gene encoding c-Abl-binding protein SH3BP2 cause cherubism. Nat. Genet. 2001, 28, 125–126. [Google Scholar] [CrossRef] [PubMed]
- Papadaki, M.E.; Lietman, S.A.; Levine, M.A.; Olsen, B.R.; Kaban, L.B.; Reichenberger, E.J. Cherubism: Best clinical practice. Orphanet J. Rare Dis. 2012, 7, S6. [Google Scholar] [CrossRef] [PubMed]
- Da Rosa, P.A.; Wang, Z.; Jiang, X.; Pruneda, J.N.; Cong, F.; Klevit, R.E.; Xu, W. Allosteric activation of the RNF146 ubiquitin ligase by a poly(ADP-ribosyl)ation signal. Nature 2015, 517, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, A.D.; Olsen, B.R. Tankyrase loses its grip on SH3BP2 in cherubism. Cell 2011, 147, 1222–1223. [Google Scholar] [CrossRef] [PubMed]
- Mukai, T.; Gallant, R.; Ishida, S.; Yoshitaka, T.; Kittaka, M.; Nishida, K.; Fox, D.A.; Morita, Y.; Ueki, Y. SH3BP2 gain-of-function mutation exacerbates inflammation and bone loss in a murine collagen-induced arthritis model. PLoS ONE 2014, 9, e105518. [Google Scholar] [CrossRef] [PubMed]
- Mukai, T.; Gallant, R.; Ishida, S.; Kittaka, M.; Yoshitaka, T.; Fox, D.A.; Morita, Y.; Nishida, K.; Rottapel, R.; Ueki, Y. Loss of SH3 domain-binding protein 2 function suppresses bone destruction in tumor necrosis factor-driven and collagen-induced arthritis in mice. Arthritis Rheumatol. 2015, 67, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Kittaka, M.; Mayahara, K.; Mukai, T.; Yoshimoto, T.; Yoshitaka, T.; Gorski, J.P.; Ueki, Y. Cherubism mice also deficient in c-Fos exhibit inflammatory bone destruction executed by macrophages that express MMP14 despite the absence of TRAP+ osteoclasts. J. Bone Miner. Res. 2018, 33, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Yoshitaka, T.; Mukai, T.; Kittaka, M.; Alford, L.M.; Masrani, S.; Ishida, S.; Yamaguchi, K.; Yamada, M.; Mizuno, N.; Olsen, B.R.; et al. Enhanced TLR-MYD88 signaling stimulates autoinflammation in SH3BP2 cherubism mice and defines the etiology of cherubism. Cell Rep. 2014, 8, 1752–1766. [Google Scholar] [CrossRef] [PubMed]
- Yoshitaka, T.; Ishida, S.; Mukai, T.; Kittaka, M.; Reichenberger, E.J.; Ueki, Y. Etanercept administration to neonatal SH3BP2 knock-in cherubism mice prevents TNF-α-induced inflammation and bone loss. J. Bone Miner. Res. 2014, 29, 1170–1182. [Google Scholar] [CrossRef] [PubMed]
- Yoshitaka, T.; Kittaka, M.; Ishida, S.; Mizuno, N.; Mukai, T.; Ueki, Y. Bone marrow transplantation improves autoinflammation and inflammatory bone loss in SH3BP2 knock-in cherubism mice. Bone 2015, 71, 201–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichenberger, E.J.; Levine, M.A.; Olsen, B.R.; Papadaki, M.E.; Lietman, S.A. The role of SH3BP2 in the pathophysiology of cherubism. Orphanet J. Rare Dis. 2012, 7, S5. [Google Scholar] [CrossRef] [PubMed]
- Chow, W.Y.; Rajan, R.; Muller, K.H.; Reid, D.G.; Skepper, J.N.; Wong, W.C.; Brooks, R.A.; Green, M.; Bihan, D.; Farndale, R.W.; et al. NMR spectroscopy of native and in vitro tissues implicates polyADP ribose in biomineralization. Science 2014, 344, 742–746. [Google Scholar] [CrossRef] [PubMed]
- Fujita, S.; Mukai, T.; Mito, T.; Kodama, S.; Nagasu, A.; Kittaka, M.; Sone, T.; Ueki, Y.; Morita, Y. Pharmacological inhibition of tankyrase induces bone loss in mice by increasing osteoclastogenesis. Bone 2018, 106, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Cummings, S.R.; San Martin, J.; McClung, M.R.; Siris, E.S.; Eastell, R.; Reid, I.R.; Delmas, P.; Zoog, H.B.; Austin, M.; Wang, A.; et al. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. New Engl. J. Med. 2009, 361, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Black, D.M.; Rosen, C.J. Clinical practice. Postmenopausal osteoporosis. New Engl. J. Med. 2016, 374, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Beranger, G.E.; Momier, D.; Guigonis, J.M.; Samson, M.; Carle, G.F.; Scimeca, J.C. Differential binding of poly(ADP-Ribose) polymerase-1 and JunD/Fra2 accounts for RANKL-induced TCIRG1 gene expression during osteoclastogenesis. J. Bone Miner. Res. 2007, 22, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Robaszkiewicz, A.; Qu, C.; Wisnik, E.; Ploszaj, T.; Mirsaidi, A.; Kunze, F.A.; Richards, P.J.; Cinelli, P.; Mbalaviele, G.; Hottiger, M.O. ARTD1 regulates osteoclastogenesis and bone homeostasis by dampening NF-kappaB-dependent transcription of IL-1β. Sci. Rep. 2016, 6, 21131. [Google Scholar] [CrossRef] [PubMed]
- Qu, C.; Bonar, S.L.; Hickman-Brecks, C.L.; Abu-Amer, S.; McGeough, M.D.; Pena, C.A.; Broderick, L.; Yang, C.; Grimston, S.K.; Kading, J.; et al. NLRP3 mediates osteolysis through inflammation-dependent and -independent mechanisms. FASEB J. 2015, 29, 1269–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Qu, C.; Alippe, Y.; Bonar, S.L.; Civitelli, R.; Abu-Amer, Y.; Hottiger, M.O.; Mbalaviele, G. Poly-ADP-ribosylation-mediated degradation of ARTD1 by the NLRP3 inflammasome is a prerequisite for osteoclast maturation. Cell Death Dis. 2016, 7, e2153. [Google Scholar] [CrossRef] [PubMed]
- Mbalaviele, G.; Novack, D.V.; Schett, G.; Teitelbaum, S.L. Inflammatory osteolysis: A conspiracy against bone. J. Clin. Investig. 2017, 127, 2030–2039. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, C.; Robaszkiewicz, A.; Lakatos, P.; Szabo, E.; Virag, L. Poly(ADP-ribose) in the bone: From oxidative stress signal to structural element. Free Radic. Biol. Med. 2015, 82, 179–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robaszkiewicz, A.; Erdelyi, K.; Kovacs, K.; Kovacs, I.; Bai, P.; Rajnavolgyi, E.; Virag, L. Hydrogen peroxide-induced poly(ADP-ribosyl)ation regulates osteogenic differentiation-associated cell death. Free Radic. Biol. Med. 2012, 53, 1552–1564. [Google Scholar] [CrossRef] [PubMed]
- Robaszkiewicz, A.; Valko, Z.; Kovacs, K.; Hegedus, C.; Bakondi, E.; Bai, P.; Virag, L. The role of p38 signaling and poly(ADP-ribosyl)ation-induced metabolic collapse in the osteogenic differentiation-coupled cell death pathway. Free Radic. Biol. Med. 2014, 76, 69–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B. TNF and bone remodeling. Curr. Osteoporos. Rep. 2017, 15, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Adamopoulos, I.E. Inflammation in bone physiology and pathology. Curr. Opin. Rheumatol. 2018, 30, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Majewski, P.M.; Thurston, R.D.; Ramalingam, R.; Kiela, P.R.; Ghishan, F.K. Cooperative role of NF-{kappa}B and poly(ADP-ribose) polymerase 1 (PARP-1) in the TNF-induced inhibition of PHEX expression in osteoblasts. J. Biol. Chem. 2010, 285, 34828–34838. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Gaivin, R.J.; Marth, T.A.; Willard, B.; Levine, M.A.; Lietman, S.A. Cloning and characterization of the human SH3BP2 promoter. Biochem. Biophys. Res. Commun. 2012, 425, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prod’Homme, V.; Boyer, L.; Dubois, N.; Mallavialle, A.; Munro, P.; Mouska, X.; Coste, I.; Rottapel, R.; Tartare-Deckert, S.; Deckert, M. Cherubism allele heterozygosity amplifies microbe-induced inflammatory responses in murine macrophages. J. Clin. Investig. 2015, 125, 1396–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chihara, K.; Kato, Y.; Yoshiki, H.; Takeuchi, K.; Fujieda, S.; Sada, K. Syk-dependent tyrosine phosphorylation of 3BP2 is required for optimal FcRγ-mediated phagocytosis and chemokine expression in U937 cells. Sci. Rep. 2017, 7, 11480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Dimitriou, I.D.; La Rose, J.; Ilangumaran, S.; Yeh, W.C.; Doody, G.; Turner, M.; Gommerman, J.; Rottapel, R. The 3BP2 adapter protein is required for optimal B-cell activation and thymus-independent type 2 humoral response. Mol. Cell Biol 2007, 27, 3109–3122. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente, M.A.; Kumar, L.; Lu, B.; Geha, R.S. 3BP2 deficiency impairs the response of B cells, but not T cells, to antigen receptor ligation. Mol. Cell Biol 2006, 26, 5214–5225. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Katavolos, P.; Nguyen, T.; Lau, T.; Boggs, J.; Sambrone, A.; Kan, D.; Merchant, M.; Harstad, E.; Diaz, D.; et al. Tankyrase inhibition causes reversible intestinal toxicity in mice with a therapeutic index <1. Toxicol. Pathol. 2016, 44, 267–278. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Models | Cells | Drugs | References |
|---|---|---|---|
| Colorectal cancer xenograft model | COLO-320DM SW430 | RK-287107 | [16] |
| Colorectal cancer xenograft model | COLO-320DM HCT-15 | G007-LK | [34] |
| Osteosarcoma xenograft model | MNNG-HOS | IWR-1 | [24] |
| Gastrointestinal stromal tumor model(KitV558Δ/+ mouse model) | Gastrointestinal stromal tumor | G007-LK | [35] |
| Orthotopic lung cancer model | HCC4006 H1650 PC9T790M | AZ1366 | [36] |
| Colorectal cancer xenograft model | Colorectal cancer | AZ1366 | [37] |
| Colorectal cancer xenograft model | Colorectal cancer | NVP-TNKS656 | [38] |
| Colorectal cancer xenograft model | COLO-320DM | G007-LK | [39] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mukai, T.; Fujita, S.; Morita, Y. Tankyrase (PARP5) Inhibition Induces Bone Loss through Accumulation of Its Substrate SH3BP2. Cells 2019, 8, 195. https://doi.org/10.3390/cells8020195
Mukai T, Fujita S, Morita Y. Tankyrase (PARP5) Inhibition Induces Bone Loss through Accumulation of Its Substrate SH3BP2. Cells. 2019; 8(2):195. https://doi.org/10.3390/cells8020195
Chicago/Turabian StyleMukai, Tomoyuki, Shunichi Fujita, and Yoshitaka Morita. 2019. "Tankyrase (PARP5) Inhibition Induces Bone Loss through Accumulation of Its Substrate SH3BP2" Cells 8, no. 2: 195. https://doi.org/10.3390/cells8020195
APA StyleMukai, T., Fujita, S., & Morita, Y. (2019). Tankyrase (PARP5) Inhibition Induces Bone Loss through Accumulation of Its Substrate SH3BP2. Cells, 8(2), 195. https://doi.org/10.3390/cells8020195