Presenilins and γ-Secretase in Membrane Proteostasis



{kind=link}
{kind=link}
Abstract
:1. Introduction
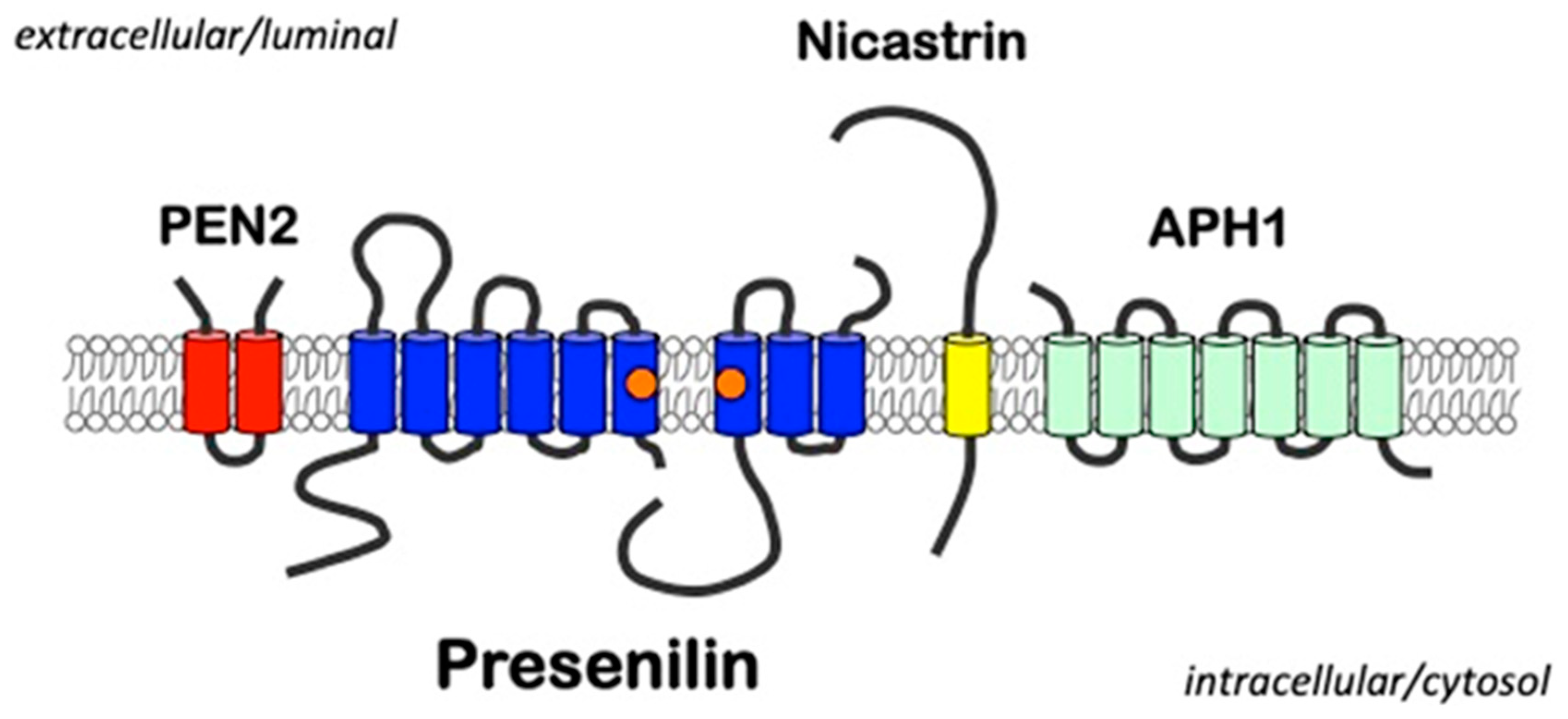
2. Presenilins
3. Functions of Presenilins in Membrane Protein Metabolism
3.1. γ-Secretase as an Intramembrane Cleaving Protease
3.2. Presenilins as Modulators of Vesicle/Protein Trafficking
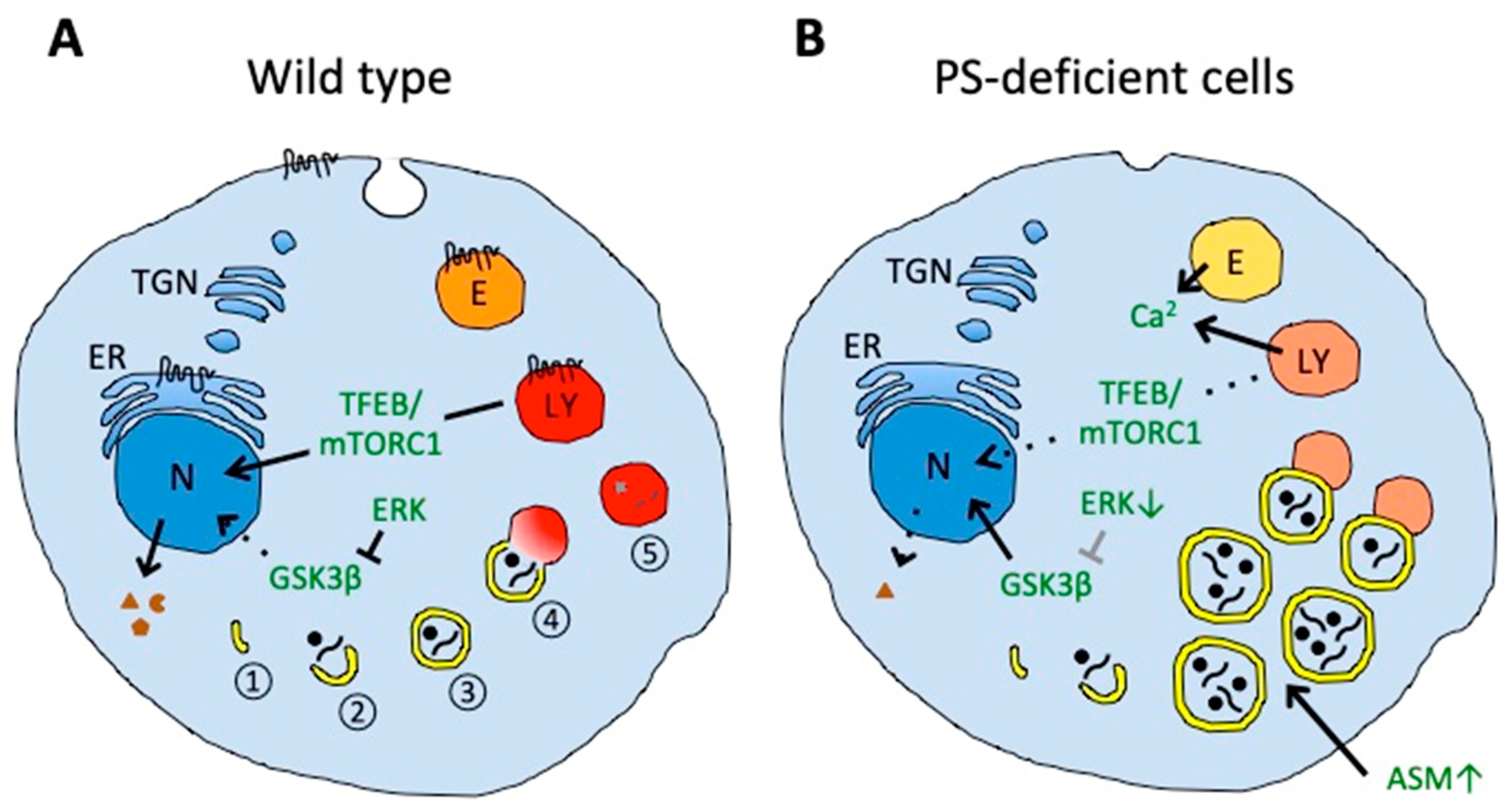
4. Presenilin in Autophagy
5. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Levy-Lahad, E.; Wasco, W.; Poorkaj, P.; Romano, D.M.; Oshima, J.; Pettingell, W.H.; Yu, C.E.; Jondro, P.D.; Schmidt, S.D.; Wang, K. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 1995, 269, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Rogaev, E.I.; Sherrington, R.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Liang, Y.; Chi, H.; Lin, C.; Holman, K.; Tsuda, T. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 1995, 376, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Duggan, S.P.; McCarthy, J.V. Beyond γ-secretase activity: The multifunctional nature of presenilins in cell signalling pathways. Cell Signal. 2016, 28, 1–11. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Iwatsubo, T.; Wolfe, M.S. Presenilins and γ-secretase: Structure, function, and role in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, Y.; Xu, H.; Zhang, Y.W. The γ-secretase complex: From structure to function. Front. Cell. Neurosci. 2014, 8, 427. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, A.J.; Coleman-Vaughan, C.; McCarthy, J.V. Regulated intramembrane proteolysis: Emergent role in cell signalling pathways. Biochem. Soc. Trans. 2017, 45, 1185–1202. [Google Scholar] [CrossRef] [PubMed]
- Herreman, A.; Hartmann, D.; Annaert, W.; Saftig, P.; Craessaerts, K.; Serneels, L.; Umans, L.; Schrijvers, V.; Checler, F.; Vanderstichele, H.; et al. Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proc. Natl. Acad. Sci. USA 1999, 96, 11872–11877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donoviel, D.B.; Hadjantonakis, A.K.; Ikeda, M.; Zheng, H.; Hyslop, P.S.; Bernstein, A. Mice lacking both presenilin genes exhibit early embryonic patterning defects. Genes Dev. 1999, 13, 2801–2810. [Google Scholar] [CrossRef] [PubMed]
- Jurisch-Yaksi, N.; Sannerud, R.; Annaert, W. A fast growing spectrum of biological functions of γ-secretase in development and disease. Biochim. Biophys. Acta 2013, 1828, 2815–2827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenough, M.A. The Role of Presenilin in Protein Trafficking and Degradation-Implications for Metal Homeostasis. J. Mol. Neurosci. 2016, 60, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed]
- Metaxakis, A.; Ploumi, C.; Tavernarakis, N. Autophagy in Age-Associated Neurodegeneration. Cells 2018, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Scrivo, A.; Bourdenx, M.; Pampliega, O.; Cuervo, A.M. Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurol. 2018, 17, 802–815. [Google Scholar] [CrossRef]
- Kovacs, D.M.; Fausett, H.J.; Page, K.J.; Kim, T.W.; Moir, R.D.; Merriam, D.E.; Hollister, R.D.; Hallmark, O.G.; Mancini, R.; Felsenstein, K.M.; et al. Alzheimer-associated presenilins 1 and 2: Neuronal expression in brain and localization to intracellular membranes in mammalian cells. Nat. Med. 1996, 2, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.; Capell, A.; Grünberg, J.; Pesold, B.; Schindzielorz, A.; Prior, R.; Podlisny, M.B.; Fraser, P.; Hyslop, P.S.; Selkoe, D.J.; et al. The Alzheimer’s disease-associated presenilins are differentially phosphorylated proteins located predominantly within the endoplasmic reticulum. Mol. Med. 1996, 2, 673–691. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Beullens, M.; Contreras, B.; Levesque, L.; Craessaerts, K.; Cordell, B.; Moechars, D.; Bollen, M.; Fraser, P.; George-Hyslop, P.S.; et al. Phosphorylation, subcellular localization, and membrane orientation of the Alzheimer’s disease-associated presenilins. J. Biol. Chem. 1997, 272, 3590–3598. [Google Scholar] [CrossRef] [PubMed]
- Thinakaran, G.; Borchelt, D.R.; Lee, M.K.; Slunt, H.H.; Spitzer, L.; Kim, G.; Ratovitsky, T.; Davenport, F.; Nordstedt, C.; Seeger, M.; et al. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron 1996, 17, 181–190. [Google Scholar] [CrossRef]
- Walter, J.; Grünberg, J.; Capell, A.; Pesold, B.; Schindzielorz, A.; Citron, M.; Mendla, K.; George-Hyslop, P.S.; Multhaup, G.; Selkoe, D.J.; et al. Proteolytic processing of the Alzheimer disease-associated presenilin-1 generates an in vivo substrate for protein kinase C. Proc. Natl. Acad. Sci. USA 1997, 94, 5349–5354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfe, M.S.; Xia, W.; Ostaszewski, B.L.; Diehl, T.S.; Kimberly, W.T.; Selkoe, D.J. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature 1999, 398, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Edbauer, D.; Winkler, E.; Regula, J.T.; Pesold, B.; Steiner, H.; Haass, C. Reconstitution of gamma-secretase activity. Nat. Cell Biol. 2003, 5, 486–488. [Google Scholar] [CrossRef] [PubMed]
- Kimberly, W.T.; LaVoie, M.J.; Ostaszewski, B.L.; Ye, W.; Wolfe, M.S.; Selkoe, D.J. Gamma-secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph-1, and Pen-2. Proc. Natl. Acad. Sci. USA 2003, 100, 6382–6387. [Google Scholar] [CrossRef] [PubMed]
- Takasugi, N.; Tomita, T.; Hayashi, I.; Tsuruoka, M.; Niimura, M.; Takahashi, Y.; Thinakaran, G.; Iwatsubo, T. The role of presenilin cofactors in the gamma-secretase complex. Nature 2003, 422, 438–441. [Google Scholar] [CrossRef] [PubMed]
- LaVoie, M.J.; Fraering, P.C.; Ostaszewski, B.L.; Ye, W.; Kimberly, W.T.; Wolfe, M.S.; Selkoe, D.J. Assembly of the gamma-secretase complex involves early formation of an intermediate subcomplex of Aph-1 and nicastrin. J. Biol. Chem. 2003, 278, 37213–37222. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Chen, F.; Sanjo, N.; Kawarai, T.; Hasegawa, H.; Duthie, M.; Li, W.; Ruan, X.; Luthra, A.; Mount, H.T.; et al. APH-1 interacts with mature and immature forms of presenilins and nicastrin and may play a role in maturation of presenilin.nicastrin complexes. J. Biol. Chem. 2003, 278, 7374–7380. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.J.; Wang, H.; Li, H.; Kim, B.S.; Shah, S.; Lee, H.J.; Thinakaran, G.; Kim, T.W.; Yu, G.; Xu, H. PEN-2 and APH-1 coordinately regulate proteolytic processing of presenilin 1. J. Biol. Chem. 2003, 278, 7850–7854. [Google Scholar] [CrossRef] [PubMed]
- Prokop, S.; Shirotani, K.; Edbauer, D.; Haass, C.; Steiner, H. Requirement of PEN-2 for stabilization of the presenilin N-/C-terminal fragment heterodimer within the gamma-secretase complex. J. Biol. Chem. 2004, 279, 23255–23261. [Google Scholar] [CrossRef] [PubMed]
- Spasic, D.; Tolia, A.; Dillen, K.; Baert, V.; De Strooper, B.; Vrijens, S.; Annaert, W. Presenilin-1 maintains a nine-transmembrane topology throughout the secretory pathway. J. Biol. Chem. 2006, 281, 26569–26577. [Google Scholar] [CrossRef] [PubMed]
- Annaert, W.G.; Levesque, L.; Craessaerts, K.; Dierinck, I.; Snellings, G.; Westaway, D.; George-Hyslop, P.S.; Cordell, B.; Fraser, P.; De Strooper, B. Presenilin 1 controls gamma-secretase processing of amyloid precursor protein in pre-golgi compartments of hippocampal neurons. J. Cell Biol. 1999, 147, 277–294. [Google Scholar] [CrossRef] [PubMed]
- Marambaud, P.; Shioi, J.; Serban, G.; Georgakopoulos, A.; Sarner, S.; Nagy, V.; Baki, L.; Wen, P.; Efthimiopoulos, S.; Shao, Z.; et al. A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J. 2002, 21, 1948–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vetrivel, K.S.; Cheng, H.; Lin, W.; Sakurai, T.; Li, T.; Nukina, N.; Wong, P.C.; Xu, H.; Thinakaran, G. Association of gamma-secretase with lipid rafts in post-Golgi and endosome membranes. J. Biol. Chem. 2004, 279, 44945–44954. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, S.H.; Bagshaw, R.D.; Guiral, M.; Zhang, S.; Ackerley, C.A.; Pak, B.J.; Callahan, J.W.; Mahuran, D.J. Presenilin-1, nicastrin, amyloid precursor protein, and gamma-secretase activity are co-localized in the lysosomal membrane. J. Biol. Chem. 2003, 278, 26687–26694. [Google Scholar] [CrossRef] [PubMed]
- Kimura, N.; Nakamura, S.I.; Honda, T.; Takashima, A.; Nakayama, H.; Ono, F.; Sakakibara, I.; Doi, K.; Kawamura, S.; Yoshikawa, Y. Age-related changes in the localization of presenilin-1 in cynomolgus monkey brain. Brain Res. 2001, 922, 30–41. [Google Scholar] [CrossRef]
- Ankarcrona, M.; Hultenby, K. Presenilin-1 is located in rat mitochondria. Biochem. Biophys. Res. Commun. 2002, 295, 766–770. [Google Scholar] [CrossRef]
- Meckler, X.; Checler, F. Presenilin 1 and Presenilin 2 Target γ-Secretase Complexes to Distinct Cellular Compartments. J. Biol. Chem. 2016, 291, 12821–12837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sannerud, R.; Esselens, C.; Ejsmont, P.; Mattera, R.; Rochin, L.; Tharkeshwar, A.K.; De Baets, G.; De Wever, V.; Habets, R.; Baert, V.; et al. Restricted Location of PSEN2/γ-Secretase Determines Substrate Specificity and Generates an Intracellular Aβ Pool. Cell 2016, 166, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.; Grünberg, J.; Schindzielorz, A.; Haass, C. Proteolytic fragments of the Alzheimer’s disease associated presenilins-1 and -2 are phosphorylated in vivo by distinct cellular mechanisms. Biochemistry 1998, 37, 5961–5967. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.; Schindzielorz, A.; Grünberg, J.; Haass, C. Phosphorylation of presenilin-2 regulates its cleavage by caspases and retards progression of apoptosis. Proc. Natl. Acad. Sci. USA 1999, 96, 1391–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeger, M.; Nordstedt, C.; Petanceska, S.; Kovacs, D.M.; Gouras, G.K.; Hahne, S.; Fraser, P.; Levesque, L.; Czernik, A.J.; George-Hyslop, P.S.; et al. Evidence for phosphorylation and oligomeric assembly of presenilin 1. Proc. Natl. Acad. Sci. USA 1997, 94, 5090–5094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirschenbaum, F.; Hsu, S.C.; Cordell, B.; McCarthy, J.V. Substitution of a glycogen synthase kinase-3beta phosphorylation site in presenilin 1 separates presenilin function from beta-catenin signaling. J. Biol. Chem. 2001, 276, 7366–7375. [Google Scholar] [CrossRef] [PubMed]
- Kirschenbaum, F.; Hsu, S.C.; Cordell, B.; McCarthy, J.V. Glycogen synthase kinase-3beta regulates presenilin 1 C-terminal fragment levels. J. Biol. Chem. 2001, 276, 30701–30707. [Google Scholar] [CrossRef] [PubMed]
- Fluhrer, R.; Friedlein, A.; Haass, C.; Walter, J. Phosphorylation of presenilin 1 at the caspase recognition site regulates its proteolytic processing and the progression of apoptosis. J. Biol. Chem. 2004, 279, 1585–1593. [Google Scholar] [CrossRef] [PubMed]
- Prager, K.; Wang-Eckhardt, L.; Fluhrer, R.; Killick, R.; Barth, E.; Hampel, H.; Haass, C.; Walter, J. A structural switch of presenilin 1 by glycogen synthase kinase 3beta-mediated phosphorylation regulates the interaction with beta-catenin and its nuclear signaling. J. Biol. Chem. 2007, 282, 14083–14093. [Google Scholar] [CrossRef] [PubMed]
- Matz, A.; Halamoda-Kenzaoui, B.; Hamelin, R.; Mosser, S.; Alattia, J.R.; Dimitrov, M.; Moniatte, M.; Fraering, P.C. Identification of new Presenilin-1 phosphosites: Implication for γ-secretase activity and Aβ production. J. Neurochem. 2015, 133, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Bustos, V.; Pulina, M.V.; Kelahmetoglu, Y.; Sinha, S.C.; Gorelick, F.S.; Flajolet, M.; Greengard, P. Bidirectional regulation of Aβ levels by Presenilin 1. Proc. Natl. Acad. Sci. USA 2017, 114, 7142–7147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustos, V.; Pulina, M.V.; Bispo, A.; Lam, A.; Flajolet, M.; Gorelick, F.S.; Greengard, P. Phosphorylated Presenilin 1 decreases β-amyloid by facilitating autophagosome-lysosome fusion. Proc. Natl. Acad. Sci. USA 2017, 114, 7148–7153. [Google Scholar] [CrossRef] [PubMed]
- Duggan, S.P.; Yan, R.; McCarthy, J.V. A ubiquitin-binding CUE domain in presenilin-1 enables interaction with K63-linked polyubiquitin chains. FEBS Lett. 2015, 589, 1001–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercken, M.; Takahashi, H.; Honda, T.; Sato, K.; Murayama, M.; Nakazato, Y.; Noguchi, K.; Imahori, K.; Takashima, A. Characterization of human presenilin 1 using N-terminal specific monoclonal antibodies: Evidence that Alzheimer mutations affect proteolytic processing. FEBS Lett. 1996, 389, 297–303. [Google Scholar] [CrossRef] [Green Version]
- Ward, R.V.; Davis, J.B.; Gray, C.W.; Barton, A.J.; Bresciani, L.G.; Caivano, M.; Murphy, V.F.; Duff, K.; Hutton, M.; Hardy, J.; et al. Presenilin-1 is processed into two major cleavage products in neuronal cell lines. Neurodegeneration 1996, 5, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Podlisny, M.B.; Citron, M.; Amarante, P.; Sherrington, R.; Xia, W.; Zhang, J.; Diehl, T.; Levesque, G.; Fraser, P.; Haass, C.; et al. Presenilin proteins undergo heterogeneous endoproteolysis between Thr291 and Ala299 and occur as stable N- and C-terminal fragments in normal and Alzheimer brain tissue. Neurobiol. Dis. 1997, 3, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Marambaud, P.; Ancolio, K.; Lopez-Perez, E.; Checler, F. Proteasome inhibitors prevent the degradation of familial Alzheimer’s disease-linked presenilin 1 and potentiate A beta 42 recovery from human cells. Mol. Med. 1998, 4, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Huynh, D.P.; Vinters, H.V.; Ho, D.H.; Ho, V.V.; Pulst, S.M. Neuronal expression and intracellular localization of presenilins in normal and Alzheimer disease brains. J. Neuropathol. Exp. Neurol. 1997, 56, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Weggen, S.; Diehlmann, A.; Buslei, R.; Beyreuther, K.; Bayer, T.A. Prominent expression of presenilin-1 in senile plaques and reactive astrocytes in Alzheimer’s disease brain. Neuroreport 1998, 9, 3279–3283. [Google Scholar] [PubMed]
- Diehlmann, A.; Ida, N.; Weggen, S.; Grünberg, J.; Haass, C.; Masters, C.L.; Bayer, T.A.; Beyreuther, K. Analysis of presenilin 1 and presenilin 2 expression and processing by newly developed monoclonal antibodies. J. Neurosci. Res. 1999, 56, 405–419. [Google Scholar] [CrossRef]
- Nadler, Y.; Alexandrovich, A.; Grigoriadis, N.; Hartmann, T.; Rao, K.S.; Shohami, E.; Stein, R. Increased expression of the gamma-secretase components presenilin-1 and nicastrin in activated astrocytes and microglia following traumatic brain injury. Glia 2008, 56, 552–567. [Google Scholar] [CrossRef] [PubMed]
- Farfara, D.; Trudler, D.; Segev-Amzaleg, N.; Galron, R.; Stein, R.; Frenkel, D. γ-Secretase component presenilin is important for microglia β-amyloid clearance. Ann. Neurol. 2011, 69, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Jayadev, S.; Case, A.; Eastman, A.J.; Nguyen, H.; Pollak, J.; Wiley, J.C.; Möller, T.; Morrison, R.S.; Garden, G.A. Presenilin 2 is the predominant γ-secretase in microglia and modulates cytokine release. PLoS ONE 2010, 5, e15743. [Google Scholar] [CrossRef] [PubMed]
- Jayadev, S.; Case, A.; Alajajian, B.; Eastman, A.J.; Möller, T.; Garden, G.A. Presenilin 2 influences miR146 level and activity in microglia. J. Neurochem. 2013, 127, 592–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Struhl, G.; Adachi, A. Requirements for presenilin-dependent cleavage of notch and other transmembrane proteins. Mol. Cell 2000, 6, 625–636. [Google Scholar] [CrossRef]
- Funamoto, S.; Sasaki, T.; Ishihara, S.; Nobuhara, M.; Nakano, M.; Watanabe-Takahashi, M.; Saito, T.; Kakuda, N.; Miyasaka, T.; Nishikawa, K.; et al. Substrate ectodomain is critical for substrate preference and inhibition of γ-secretase. Nat. Commun. 2013, 4, 2529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurent, S.A.; Hoffmann, F.S.; Kuhn, P.H.; Cheng, Q.; Chu, Y.; Schmidt-Supprian, M.; Hauck, S.M.; Schuh, E.; Krumbholz, M.; Rübsamen, H.; et al. γ-Secretase directly sheds the survival receptor BCMA from plasma cells. Nat. Commun. 2015, 6, 7333. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Lee, S.F.; Tabuchi, K.; Hao, Y.H.; Yu, C.; LaPlant, Q.; Ball, H.; Dann, C.E.; Südhof, T.; Yu, G. Nicastrin functions as a gamma-secretase-substrate receptor. Cell 2005, 122, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Lichtenthaler, S.F.; Haass, C.; Steiner, H. Regulated intramembrane proteolysis--lessons from amyloid precursor protein processing. J. Neurochem. 2011, 117, 779–796. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Bai, X.C.; Ma, D.; Xie, T.; Yan, C.; Sun, L.; Yang, G.; Zhao, Y.; Zhou, R.; Scheres, S.H.W.; et al. Three-dimensional structure of human γ-secretase. Nature 2014, 512, 166–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolduc, D.M.; Montagna, D.R.; Gu, Y.; Selkoe, D.J.; Wolfe, M.S. Nicastrin functions to sterically hinder γ-secretase-substrate interactions driven by substrate transmembrane domain. Proc. Natl. Acad. Sci. USA 2016, 113, E509–E518. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yang, G.; Guo, X.; Zhou, Q.; Lei, J.; Shi, Y. Recognition of the amyloid precursor protein by human γ-secretase. Science 2019, 363, eaaw0930. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Zhou, R.; Zhou, Q.; Guo, X.; Yan, C.; Ke, M.; Lei, J.; Shi, Y. Structural basis of Notch recognition by human γ-secretase. Nature 2019, 565, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S. Toward the structure of presenilin/γ-secretase and presenilin homologs. Biochim. Biophys. Acta 2013, 1828, 2886–2897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morishima-Kawashima, M. Molecular mechanism of the intramembrane cleavage of the β-carboxyl terminal fragment of amyloid precursor protein by γ-secretase. Front. Physiol. 2014, 5, 463. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, J.V.; Twomey, C.; Wujek, P. Presenilin-dependent regulated intramembrane proteolysis and gamma-secretase activity. Cell. Mol. Life Sci. 2009, 66, 1534–1555. [Google Scholar] [CrossRef] [PubMed]
- Haapasalo, A.; Kovacs, D.M. The many substrates of presenilin/γ-secretase. J. Alzheimers Dis. 2011, 25, 3–28. [Google Scholar] [CrossRef] [PubMed]
- Schauenburg, L.; Liebsch, F.; Eravci, M.; Mayer, M.C.; Weise, C.; Multhaup, G. APLP1 is endoproteolytically cleaved by γ-secretase without previous ectodomain shedding. Sci. Rep. 2018, 8, 1916. [Google Scholar] [CrossRef] [PubMed]
- Lichtenthaler, S.F.; Lemberg, M.K.; Fluhrer, R. Proteolytic ectodomain shedding of membrane proteins in mammals-hardware, concepts, and recent developments. EMBO J. 2018, 37, e99456. [Google Scholar] [CrossRef] [PubMed]
- Borggrefe, T.; Lauth, M.; Zwijsen, A.; Huylebroeck, D.; Oswald, F.; Giaimo, B.D. The Notch intracellular domain integrates signals from Wnt, Hedgehog, TGFβ/BMP and hypoxia pathways. Biochim. Biophys. Acta. 2016, 1863, 303–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, S.J.; Gomez-Lamarca, M. Notch after cleavage. Curr. Opin. Cell Biol. 2018, 51, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Pardossi-Piquard, R.; Checler, F. The physiology of the β-amyloid precursor protein intracellular domain AICD. J. Neurochem. 2012, 120 (Suppl. 1), 109–124. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Mett, J.; Grimm, H.S.; Hartmann, T. APP Function and Lipids: A Bidirectional Link. Front. Mol. Neurosci. 2017, 10, 63. [Google Scholar] [CrossRef] [PubMed]
- Ni, C.Y.; Murphy, M.P.; Golde, T.E.; Carpenter, G. gamma -Secretase cleavage and nuclear localization of ErbB-4 receptor tyrosine kinase. Science 2001, 294, 2179–2181. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Jung, K.M.; Huang, Y.Z.; Bennett, L.B.; Lee, J.S.; Mei, L.; Kim, T.W. Presenilin-dependent gamma-secretase-like intramembrane cleavage of ErbB4. J. Biol. Chem. 2002, 277, 6318–6323. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.; Feng, L. Implication of gamma-secretase in neuregulin-induced maturation of oligodendrocytes. Biochem. Biophys. Res. Commun. 2004, 314, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.C.; Allison, J.G.; Vidal, G.A.; Burow, M.E.; Beckman, B.S.; Marrero, L.; Jones, F.E. The ERBB4/HER4 receptor tyrosine kinase regulates gene expression by functioning as a STAT5A nuclear chaperone. J. Cell Biol. 2004, 167, 469–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal, G.A.; Naresh, A.; Marrero, L.; Jones, F.E. Presenilin-dependent gamma-secretase processing regulates multiple ERBB4/HER4 activities. J. Biol. Chem. 2005, 280, 19777–19783. [Google Scholar] [CrossRef] [PubMed]
- Arasada, R.R.; Carpenter, G. Secretase-dependent tyrosine phosphorylation of Mdm2 by the ErbB-4 intracellular domain fragment. J. Biol. Chem. 2005, 280, 30783–30787. [Google Scholar] [CrossRef] [PubMed]
- Sardi, S.P.; Murtie, J.; Koirala, S.; Patten, B.A.; Corfas, G. Presenilin-dependent ErbB4 nuclear signaling regulates the timing of astrogenesis in the developing brain. Cell 2006, 127, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Lammich, S.; Okochi, M.; Takeda, M.; Kaether, C.; Capell, A.; Zimmer, A.K.; Edbauer, D.; Walter, J.; Steiner, H.; Haass, C. Presenilin-dependent intramembrane proteolysis of CD44 leads to the liberation of its intracellular domain and the secretion of an Abeta-like peptide. J. Biol. Chem. 2002, 277, 44754–44759. [Google Scholar] [CrossRef] [PubMed]
- Murakami, D.; Okamoto, I.; Nagano, O.; Kawano, Y.; Tomita, T.; Iwatsubo, T.; De Strooper, B.; Yumoto, E.; Saya, H. Presenilin-dependent gamma-secretase activity mediates the intramembranous cleavage of CD44. Oncogene 2003, 22, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Ke, J.Z.; Zhang, Q.; Ke, H.Z.; Chalouni, C.; Vignery, A. The intracellular domain of CD44 promotes the fusion of macrophages. Blood 2006, 107, 796–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelletier, L.; Guillaumot, P.; Frêche, B.; Luquain, C.; Christiansen, D.; Brugière, S.; Garin, J.; Manié, S.N. Gamma-secretase-dependent proteolysis of CD44 promotes neoplastic transformation of rat fibroblastic cells. Cancer Res. 2006, 66, 3681–3687. [Google Scholar] [CrossRef] [PubMed]
- De Falco, V.; Tamburrino, A.; Ventre, S.; Castellone, M.D.; Malek, M.; Manié, S.N.; Santoro, M. CD44 proteolysis increases CREB phosphorylation and sustains proliferation of thyroid cancer cells. Cancer Res. 2012, 72, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Lee, H.W.; Kang, H.G.; Kim, H.Y.; Kim, S.J.; Chun, K.H. Cleaved CD44 intracellular domain supports activation of stemness factors and promotes tumorigenesis of breast cancer. Oncotarget 2015, 6, 8709–8721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, K.; Grieger Lindner, C.; Li, Y.; Urbánek, P.; Ruschel, A.; Minnich, K.; Bruder, D.; Gereke, M.; Sechi, A.; Herrlich, P. Gamma secretase dependent release of the CD44 cytoplasmic tail upregulates IFI16 in cd44-/- tumor cells, MEFs and macrophages. PLoS ONE 2018, 13, e0207358. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulos, A.; Litterst, C.; Ghersi, E.; Baki, L.; Xu, C.; Serban, G.; Robakis, N.K. Metalloproteinase/Presenilin1 processing of ephrinB regulates EphB-induced Src phosphorylation and signaling. EMBO J. 2006, 25, 1242–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waschbüsch, D.; Born, S.; Niediek, V.; Kirchgessner, N.; Tamboli, I.Y.; Walter, J.; Merkel, R.; Hoffmann, B. Presenilin 1 affects focal adhesion site formation and cell force generation via c-Src transcriptional and posttranslational regulation. J. Biol. Chem. 2009, 284, 10138–10149. [Google Scholar] [CrossRef] [PubMed]
- Kemmerling, N.; Wunderlich, P.; Theil, S.; Linnartz-Gerlach, B.; Hersch, N.; Hoffmann, B.; Heneka, M.T.; de Strooper, B.; Neumann, H.; Walter, J. Intramembranous processing by γ-secretase regulates reverse signaling of ephrin-B2 in migration of microglia. Glia 2017, 65, 1103–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopan, R.; Ilagan, M.X. Gamma-secretase: Proteasome of the membrane. Nat. Rev. Mol. Cell Biol. 2004, 5, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Tabuchi, K.; Chen, G.; Südhof, T.C.; Shen, J. Conditional forebrain inactivation of nicastrin causes progressive memory impairment and age-related neurodegeneration. J. Neurosci. 2009, 29, 7290–7301. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Iqbal, M.; Zheng, J.; Wines-Samuelson, M.; Shen, J. Partial loss of presenilin impairs age-dependent neuronal survival in the cerebral cortex. J. Neurosci. 2014, 34, 15912–15922. [Google Scholar] [CrossRef] [PubMed]
- Acx, H.; Serneels, L.; Radaelli, E.; Muyldermans, S.; Vincke, C.; Pepermans, E.; Müller, U.; Chávez-Gutiérrez, L.; De Strooper, B. Inactivation of γ-secretases leads to accumulation of substrates and non-Alzheimer neurodegeneration. EMBO Mol. Med. 2017, 9, 1088–1099. [Google Scholar] [CrossRef] [PubMed]
- Oster-Granite, M.L.; McPhie, D.L.; Greenan, J.; Neve, R.L. Age-dependent neuronal and synaptic degeneration in mice transgenic for the C terminus of the amyloid precursor protein. J. Neurosci. 1996, 16, 6732–6741. [Google Scholar] [CrossRef] [PubMed]
- Nalbantoglu, J.; Tirado-Santiago, G.; Lahsaïni, A.; Poirier, J.; Goncalves, O.; Verge, G.; Momoli, F.; Welner, S.A.; Massicotte, G.; Julien, J.P.; et al. Impaired learning and LTP in mice expressing the carboxy terminus of the Alzheimer amyloid precursor protein. Nature 1997, 387, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Pera, M.; Larrea, D.; Guardia-Laguarta, C.; Montesinos, J.; Velasco, K.R.; Agrawal, R.R.; Xu, Y.; Chan, R.B.; Di Paolo, G.; Mehler, M.F.; et al. Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. EMBO J. 2017, 36, 3356–3371. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Mullaney, K.A.; Peterhoff, C.M.; Che, S.; Schmidt, S.D.; Boyer-Boiteau, A.; Ginsberg, S.D.; Cataldo, A.M.; Mathews, P.M.; Nixon, R.A. Alzheimer’s-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proc. Natl. Acad. Sci. USA 2010, 107, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Sato, Y.; Mohan, P.S.; Peterhoff, C.; Pensalfini, A.; Rigoglioso, A.; Jiang, Y.; Nixon, R.A. Evidence that the rab5 effector APPL1 mediates APP-βCTF-induced dysfunction of endosomes in Down syndrome and Alzheimer’s disease. Mol. Psychiatry 2016, 21, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Weissmiller, A.M.; Natera-Naranjo, O.; Reyna, S.M.; Pearn, M.L.; Zhao, X.; Nguyen, P.; Cheng, S.; Goldstein, L.S.; Tanzi, R.E.; Wagner, S.L.; et al. A γ-secretase inhibitor, but not a γ-secretase modulator, induced defects in BDNF axonal trafficking and signaling: Evidence for a role for APP. PLoS ONE 2015, 10, e0118379. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Weissmiller, A.M.; White, J.A.; Fang, F.; Wang, X.; Wu, Y.; Pearn, M.L.; Zhao, X.; Sawa, M.; Chen, S.; et al. Amyloid precursor protein-mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. J Clin. Investig. 2016, 126, 1815–1833. [Google Scholar] [CrossRef] [PubMed]
- Deyts, C.; Clutter, M.; Herrera, S.; Jovanovic, N.; Goddi, A.; Parent, A.T. Loss of presenilin function is associated with a selective gain of APP function. Elife 2016, 5, e15645. [Google Scholar] [CrossRef] [PubMed]
- Barthet, G.; Jordà-Siquier, T.; Rumi-Masante, J.; Bernadou, F.; Müller, U.; Mulle, C. Presenilin-mediated cleavage of APP regulates synaptotagmin-7 and presynaptic plasticity. Nat. Commun. 2018, 9, 4780. [Google Scholar] [CrossRef] [PubMed]
- Tamboli, I.Y.; Prager, K.; Thal, D.R.; Thelen, K.M.; Dewachter, I.; Pietrzik, C.U.; St George-Hyslop, P.; Sisodia, S.S.; De Strooper, B.; Heneka, M.T.; et al. Loss of gamma-secretase function impairs endocytosis of lipoprotein particles and membrane cholesterol homeostasis. J. Neurosci. 2008, 28, 12097–12106. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, P.; Glebov, K.; Kemmerling, N.; Tien, N.T.; Neumann, H.; Walter, J. Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) protein by ectodomain shedding and γ-secretase-dependent intramembranous cleavage. J. Biol. Chem. 2013, 288, 33027–33036. [Google Scholar] [CrossRef] [PubMed]
- Glebov, K.; Wunderlich, P.; Karaca, I.; Walter, J. Functional involvement of γ-secretase in signaling of the triggering receptor expressed on myeloid cells-2 (TREM2). J. Neuroinflamm. 2016, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Kleinberger, G.; Yamanishi, Y.; Suárez-Calvet, M.; Czirr, E.; Lohmann, E.; Cuyvers, E.; Struyfs, H.; Pettkus, N.; Wenninger-Weinzierl, A.; Mazaheri, F.; et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med. 2014, 6, 243ra86. [Google Scholar] [CrossRef] [PubMed]
- Walter, J. The Triggering Receptor Expressed on Myeloid Cells 2: A Molecular Link of Neuroinflammation and Neurodegenerative Diseases. J. Biol. Chem. 2016, 291, 4334–4341. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yang, W.; Wen, W.; Sun, J.; Su, B.; Liu, B.; Ma, D.; Lv, D.; Wen, Y.; Qu, T.; et al. Gamma-secretase gene mutations in familial acne inversa. Science 2010, 330, 1065. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Mattei, P.; You, J.; Sobreira, N.L.; Hinds, G.A. γ-Secretase Mutation in an African American Family with Hidradenitis Suppurativa. JAMA Dermatol. 2015, 151, 668–670. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.A. Concurrent hidradenitis suppurativa and Dowling-Degos disease taken down a ‘Notch’. Br. J. Dermatol. 2018, 178, 328. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sisodia, S.S. Acne inversa caused by missense mutations in NCSTN is not fully compatible with impairments in Notch signaling. J. Investig. Dermatol. 2015, 135, 618–620. [Google Scholar] [CrossRef] [PubMed]
- Dumanchin, C.; Czech, C.; Campion, D.; Cuif, M.H.; Poyot, T.; Martin, C.; Charbonnier, F.; Goud, B.; Pradier, L.; Frebourg, T. Presenilins interact with Rab11, a small GTPase involved in the regulation of vesicular transport. Hum. Mol. Genet. 1999, 8, 1263–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheper, W.; Zwart, R.; van der Sluijs, P.; Annaert, W.; Gool, W.A.; Baas, F. Alzheimer’s presenilin 1 is a putative membrane receptor for rab GDP dissociation inhibitor. Hum. Mol. Genet. 2000, 9, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Netzer, W.J.; Zhong, M.; Lin, Y.; Du, G.; Frohman, M.; Foster, D.A.; Sisodia, S.S.; Xu, H.; Gorelick, F.S.; et al. Presenilin-1 uses phospholipase D1 as a negative regulator of beta-amyloid formation. Proc. Natl. Acad. Sci. USA 2006, 103, 1941–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.K.; Anderson, H.A.; Yu, G.; Robertson, A.G.; Allen, S.J.; Tyler, S.J.; Naylor, R.L.; Mason, G.; Wilcock, G.W.; Roche, P.A.; et al. Identification of syntaxin 1A as a novel binding protein for presenilin-1. Brain Res. Mol. Brain Res. 2000, 78, 100–107. [Google Scholar] [CrossRef]
- Suga, K.; Tomiyama, T.; Mori, H.; Akagawa, K. Syntaxin 5 interacts with presenilin holoproteins, but not with their N- or C-terminal fragments, and affects beta-amyloid peptide production. Biochem. J. 2004, 381, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Lau, K.F.; McLoughlin, D.M.; Standen, C.; Miller, C.C. X11 alpha and x11 beta interact with presenilin-1 via their PDZ domains. Mol. Cell. Neurosci. 2000, 16, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Leem, J.Y.; Greenfield, J.P.; Wang, P.; Kim, B.S.; Wang, R.; Lopes, K.O.; Kim, S.H.; Zheng, H.; Greengard, P.; et al. Presenilin-1 regulates intracellular trafficking and cell surface delivery of beta-amyloid precursor protein. J. Biol. Chem. 2003, 278, 3446–3454. [Google Scholar] [CrossRef] [PubMed]
- Lazarov, O.; Morfini, G.A.; Pigino, G.; Gadadhar, A.; Chen, X.; Robinson, J.; Ho, H.; Brady, S.T.; Sisodia, S.S. Impairments in fast axonal transport and motor neuron deficits in transgenic mice expressing familial Alzheimer’s disease-linked mutant presenilin 1. J. Neurosci. 2007, 27, 7011–7020. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.R.; Nye, J.S.; Lamb, N.J.; Fernandez, A.; Kitzmann, M. Intracellular retention of caveolin 1 in presenilin-deficient cells. J. Biol. Chem. 2005, 280, 6663–6668. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, G.; Reyna, S.M.; Dunlap, M.; Van Der Kant, R.; Callender, J.A.; Young, J.E.; Roberts, E.A.; Goldstein, L.S. Defective Transcytosis of APP and Lipoproteins in Human iPSC-Derived Neurons with Familial Alzheimer’s Disease Mutations. Cell Rep. 2016, 17, 759–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Li, X.; Huang, T.; Jiang, L.L.; Tan, Z.; Zhang, M.; Cheng, I.H.; Wang, X.; Bu, G.; Zhang, Y.W.; et al. Intracellular trafficking of TREM2 is regulated by presenilin 1. Exp. Mol. Med. 2017, 49, e405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naruse, S.; Thinakaran, G.; Luo, J.J.; Kusiak, J.W.; Tomita, T.; Iwatsubo, T.; Qian, X.; Ginty, D.D.; Price, D.L.; Borchelt, D.R.; et al. Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron 1998, 21, 1213–1221. [Google Scholar] [CrossRef]
- Kaether, C.; Lammich, S.; Edbauer, D.; Ertl, M.; Rietdorf, J.; Capell, A.; Steiner, H.; Haass, C. Presenilin-1 affects trafficking and processing of betaAPP and is targeted in a complex with nicastrin to the plasma membrane. J. Cell Biol. 2002, 158, 551–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Yang, D.S.; Petanceska, S.; Yang, A.; Tandon, A.; Yu, G.; Rozmahel, R.; Ghiso, J.; Nishimura, M.; Zhang, D.M.; et al. Carboxyl-terminal fragments of Alzheimer beta-amyloid precursor protein accumulate in restricted and unpredicted intracellular compartments in presenilin 1-deficient cells. J. Biol. Chem. 2000, 275, 36794–36802. [Google Scholar] [CrossRef] [PubMed]
- Gandy, S.; Zhang, Y.W.; Ikin, A.; Schmidt, S.D.; Bogush, A.; Levy, E.; Sheffield, R.; Nixon, R.A.; Liao, F.F.; Mathews, P.M.; et al. Alzheimer’s presenilin 1 modulates sorting of APP and its carboxyl-terminal fragments in cerebral neurons in vivo. J. Neurochem. 2007, 102, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Uemura, K.; Kitagawa, N.; Kohno, R.; Kuzuya, A.; Kageyama, T.; Chonabayashi, K.; Shibasaki, H.; Shimohama, S. Presenilin 1 is involved in maturation and trafficking of N-cadherin to the plasma membrane. J. Neurosci. Res. 2003, 74, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Esselens, C.; Oorschot, V.; Baert, V.; Raemaekers, T.; Spittaels, K.; Serneels, L.; Zheng, H.; Saftig, P.; De Strooper, B.; Klumperman, J.; et al. Presenilin 1 mediates the turnover of telencephalin in hippocampal neurons via an autophagic degradative pathway. J. Cell Biol. 2004, 166, 1041–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saura, C.A.; Choi, S.Y.; Beglopoulos, V.; Malkani, S.; Zhang, D.; Shankaranarayana Rao, B.S.; Chattarji, S.; Kelleher, R.J.; Kandel, E.R.; Duff, K.; et al. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron 2004, 42, 23–36. [Google Scholar] [CrossRef]
- Zhang, M.; Haapasalo, A.; Kim, D.Y.; Ingano, L.A.; Pettingell, W.H.; Kovacs, D.M. Presenilin/gamma-secretase activity regulates protein clearance from the endocytic recycling compartment. FASEB J. 2006, 20, 1176–1178. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Tang, P.; Wang, P.; Boissy, R.E.; Zheng, H. Regulation of tyrosinase trafficking and processing by presenilins: Partial loss of function by familial Alzheimer’s disease mutation. Proc. Natl. Acad. Sci. USA 2006, 103, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Repetto, E.; Yoon, I.S.; Zheng, H.; Kang, D.E. Presenilin 1 regulates epidermal growth factor receptor turnover and signaling in the endosomal-lysosomal pathway. J. Biol. Chem. 2007, 282, 31504–31516. [Google Scholar] [CrossRef] [PubMed]
- Zou, K.; Hosono, T.; Nakamura, T.; Shiraishi, H.; Maeda, T.; Komano, H.; Yanagisawa, K.; Michikawa, M. Novel role of presenilins in maturation and transport of integrin beta 1. Biochemistry 2008, 47, 3370–3378. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Barthet, G.; Dunys, J.; Shao, Z.; Xuan, Z.; Ren, Y.; Xu, J.; Arbez, N.; Mauger, G.; Bruban, J.; Georgakopoulos, A.; et al. Presenilin mediates neuroprotective functions of ephrinB and brain-derived neurotrophic factor and regulates ligand-induced internalization and metabolism of EphB2 and TrkB receptors. Neurobiol. Aging 2013, 34, 499–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Fleming, A.; Ricketts, T.; Pavel, M.; Virgin, H.; Menzies, F.M.; Rubinsztein, D.C. Autophagy regulates Notch degradation and modulates stem cell development and neurogenesis. Nat. Commun. 2016, 7, 10533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, Y.; Dhaliwal, J.S.; Ceizar, M.; Vaculik, M.; Kumar, K.L.; Lagace, D.C. Knockout of Atg5 delays the maturation and reduces the survival of adult-generated neurons in the hippocampus. Cell Death Dis. 2016, 7, e2127. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Cho, M.H.; Shim, W.H.; Kim, J.K.; Jeon, E.Y.; Kim, D.H.; Yoon, S.Y. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol. Psychiatry 2017, 22, 1576–1584. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Codogno, P.; Cuervo, A.M.; Deretic, V.; Elazar, Z.; Fueyo-Margareto, J.; Gewirtz, D.A.; Kroemer, G.; Levine, B.; Mizushima, N.; et al. A comprehensive glossary of autophagy-related molecules and processes. Autophagy 2010, 6, 438–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, C.A.; Murphy, D.D.; Giasson, B.I.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M. Degradative organelles containing mislocalized alpha-and beta-synuclein proliferate in presenilin-1 null neurons. J. Cell Biol. 2004, 165, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; Peterhoff, C.M.; Schmidt, S.D.; Terio, N.B.; Duff, K.; Beard, M.; Mathews, P.M.; Nixon, R.A. Presenilin mutations in familial Alzheimer disease and transgenic mouse models accelerate neuronal lysosomal pathology. J. Neuropathol. Exp. Neurol. 2004, 63, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Coffey, E.E.; Beckel, J.M.; Laties, A.M.; Mitchell, C.H. Lysosomal alkalization and dysfunction in human fibroblasts with the Alzheimer’s disease-linked presenilin 1 A246E mutation can be reversed with cAMP. Neuroscience 2014, 263, 111–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neely, K.M.; Green, K.N.; LaFerla, F.M. Presenilin is necessary for efficient proteolysis through the autophagy-lysosome system in a γ-secretase-independent manner. J. Neurosci. 2011, 31, 2781–2791. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Garbett, K.; Veeraraghavalu, K.; Wilburn, B.; Gilmore, R.; Mirnics, K.; Sisodia, S.S. A role for presenilins in autophagy revisited: Normal acidification of lysosomes in cells lacking PSEN1 and PSEN2. J. Neurosci. 2012, 32, 8633–8648. [Google Scholar] [CrossRef] [PubMed]
- Coen, K.; Flannagan, R.S.; Baron, S.; Carraro-Lacroix, L.R.; Wang, D.; Vermeire, W.; Michiels, C.; Munck, S.; Baert, V.; Sugita, S.; et al. Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J. Cell Biol. 2012, 198, 23–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neely Kayala, K.M.; Dickinson, G.D.; Minassian, A.; Walls, K.C.; Green, K.N.; Laferla, F.M. Presenilin-null cells have altered two-pore calcium channel expression and lysosomal calcium: Implications for lysosomal function. Brain Res. 2012, 1489, 8–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; McBrayer, M.K.; Wolfe, D.M.; Haslett, L.J.; Kumar, A.; Sato, Y.; Lie, P.P.; Mohan, P.; Coffey, E.E.; Kompella, U.; et al. Presenilin 1 Maintains Lysosomal Ca(2+) Homeostasis via TRPML1 by Regulating vATPase-Mediated Lysosome Acidification. Cell Rep. 2015, 12, 1430–1444. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Jin, H.K.; Park, M.H.; Kim, B.R.; Lee, P.H.; Nakauchi, H.; Carter, J.E.; He, X.; Schuchman, E.H.; Bae, J.S. Acid sphingomyelinase modulates the autophagic process by controlling lysosomal biogenesis in Alzheimer’s disease. J. Exp. Med. 2014, 211, 1551–1570. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.; Cusack, C.L.; Nnah, I.C.; Khayati, K.; Saqcena, C.; Huynh, T.B.; Noggle, S.A.; Ballabio, A.; Dobrowolski, R. Dysregulation of Nutrient Sensing and CLEARance in Presenilin Deficiency. Cell Rep. 2016, 14, 2166–2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín-Maestro, P.; Gargini, R.; Sproul, A.; García, E.; Antón, L.C.; Noggle, S.; Arancio, O.; Avila, J.; García-Escudero, V. Mitophagy Failure in Fibroblasts and iPSC-Derived Neurons of Alzheimer’s Disease-Associated Presenilin 1 Mutation. Front. Mol. Neurosci. 2017, 10, 291. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.M.; Ke, M.; Tan, Y.; Huang, Z.; Zhang, K.; Ai, N.; Ge, W.; Qin, D.; Lu, J.H.; Su, H. Presenilin 1 deficiency suppresses autophagy in human neural stem cells through reducing γ-secretase-independent ERK/CREB signaling. Cell Death Dis. 2018, 9, 879. [Google Scholar] [CrossRef] [PubMed]
- Száraz, P.; Bánhegyi, G.; Marcolongo, P.; Benedetti, A. Transient knockdown of presenilin-1 provokes endoplasmic reticulum stress related formation of autophagosomes in HepG2 cells. Arch. Biochem. Biophys. 2013, 538, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Song, B.Q.; Chi, Y.; Li, X.; Du, W.J.; Han, Z.B.; Tian, J.J.; Li, J.J.; Chen, F.; Wu, H.H.; Han, L.X.; et al. Inhibition of Notch Signaling Promotes the Adipogenic Differentiation of Mesenchymal Stem Cells Through Autophagy Activation and PTEN-PI3K/AKT/mTOR Pathway. Cell. Physiol. Biochem. 2015, 36, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Arico, S.; Petiot, A.; Bauvy, C.; Dubbelhuis, P.F.; Meijer, A.J.; Codogno, P.; Ogier-Denis, E. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 2001, 276, 35243–35246. [Google Scholar] [CrossRef] [PubMed]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Presenilin 2 Modulates Endoplasmic Reticulum-Mitochondria Coupling by Tuning the Antagonistic Effect of Mitofusin 2. Cell Rep. 2016, 15, 2226–2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contino, S.; Porporato, P.E.; Bird, M.; Marinangeli, C.; Opsomer, R.; Sonveaux, P.; Bontemps, F.; Dewachter, I.; Octave, J.N.; Bertrand, L.; et al. Presenilin 2-Dependent Maintenance of Mitochondrial Oxidative Capacity and Morphology. Front. Physiol. 2017, 8, 796. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oikawa, N.; Walter, J. Presenilins and γ-Secretase in Membrane Proteostasis. Cells 2019, 8, 209. https://doi.org/10.3390/cells8030209
Oikawa N, Walter J. Presenilins and γ-Secretase in Membrane Proteostasis. Cells. 2019; 8(3):209. https://doi.org/10.3390/cells8030209
Chicago/Turabian StyleOikawa, Naoto, and Jochen Walter. 2019. "Presenilins and γ-Secretase in Membrane Proteostasis" Cells 8, no. 3: 209. https://doi.org/10.3390/cells8030209
APA StyleOikawa, N., & Walter, J. (2019). Presenilins and γ-Secretase in Membrane Proteostasis. Cells, 8(3), 209. https://doi.org/10.3390/cells8030209