Microglial Progranulin: Involvement in Alzheimer’s Disease and Neurodegenerative Diseases



{kind=link}
{kind=link}
{kind=link}
Abstract
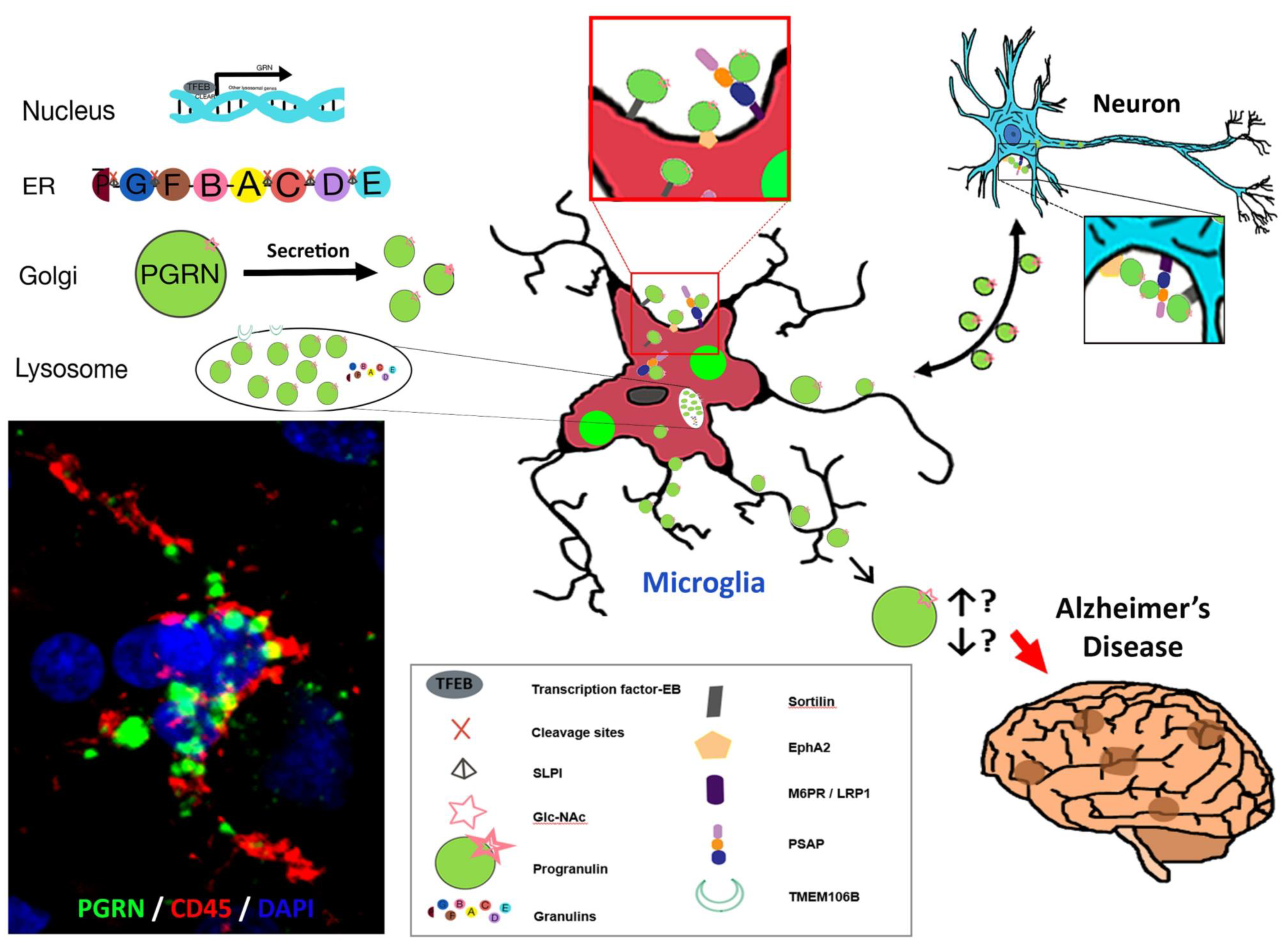
:1. Introduction
2. Significance
3. Features of GRN/Grn Gene and Progranulin Protein
3.1. GRN/Grn Gene Structure and Regulation
3.2. Progranulin Protein
3.3. Progranulin Post-Translational Modification and Secretion
4. Cell Biology of Progranulin
4.1. Types of Cells Expressing Progranulin
4.2. Progranulin Expression in Human Brain-Derived Cells
4.3. Expression of Progranulin by Brain Cells in Vivo
5. Involvement of PGRN in Disease Processes
5.1. PGRN as A Tumor Promoting Factor
5.2. PGRN as A Biomarker for Neurodegenerative Diseases
5.3. Role of PGRN in Inflammation
5.4. Increased Expression of Microglial Progranulin in Acute Injury Animal Models
5.5. Animal Models of FTLD Due to GRN Deletion
5.6. Progranulin in Other Animal Disease Models
5.7. Manipulation of Microglial Progranulin Compared to Neuronal Progranulin
6. Role of PGRN in Alzheimer’s Disease
6.1. PGRN in Animal Models of Alzheimer’s Disease
6.2. Possible Mechanisms of Action of PGRN in Immune Cells
7. Progranulin Effects on Lysosomal Function
8. Is Progranulin a Potential Therapeutic Target for Alzheimer’s Disease, Parkinson’s Disease and FTLD?
9. Summary
10. Conclusions and Future Directions for Studies of PGRN in Alzheimer’s and Parkinson’s Disease and Other Neurodegenerative Diseases
10.1. Biochemical Nature of Pathology Associated Progranulin
10.2. Phenotyping of Microglia Overexpressing Progranulin
10.3. Further Human Brain Studies of PGRN in Other Brain Diseases Involving Microglia
Funding
Conflicts of Interest
References
- Piton, M.; Hirtz, C.; Desmetz, C.; Milhau, J.; Lajoix, A.D.; Bennys, K.; Lehmann, S.; Gabelle, A. Alzheimer’s disease: Advances in drug development. J. Alzheimers. Dis. 2018, 65, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.-J.; Li, J.-Y.; Yang, Z.; Liu, Z.; Feng, J.-S. Efficacy and safety of anti-amyloid-beta immunotherapy for Alzheimer’s disease: A systematic review and network meta-analysis. Ann. Clin. Transl. Neurol. 2017, 4, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Hara, Y.; McKeehan, N.; Fillit, H.M. Translating the biology of aging into novel therapeutics for Alzheimer disease. Neurology 2018, 92, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Qiu, P.; Cui, W.; Zhang, B.; Yan, X.; He, S. Recent advances in multi-target anti-Alzheimer disease compounds (2013 up to present). Curr. Med. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Miguel-Alvarez, M.; Santos-Lozano, A.; Sanchis-Gomar, F.; Fiuza-Luces, C.; Pareja-Galeano, H.; Garatachea, N.; Lucia, A. Non-steroidal anti-inflammatory drugs as a treatment for Alzheimer’s disease: A systematic review and meta-analysis of treatment effect. Drugs Aging 2015, 32, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Kao, A.W.; McKay, A.; Singh, P.P.; Brunet, A.; Huang, E.J. Progranulin, lysosomal regulation and neurodegenerative disease. Nat. Rev. Neurosci. 2017, 18, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Chitramuthu, B.P.; Bennett, H.P.J.; Bateman, A. Progranulin: A new avenue towards the understanding and treatment of neurodegenerative disease. Brain 2017, 140, 3081–3104. [Google Scholar] [CrossRef] [PubMed]
- Paushter, D.H.; Du, H.; Feng, T.; Hu, F. The lysosomal function of progranulin, a guardian against neurodegeneration. Acta Neuropathol. 2018, 136, 1861–1868. [Google Scholar] [CrossRef] [PubMed]
- Lui, H.; Zhang, J.; Makinson, S.R.; Cahill, M.K.; Kelley, K.W.; Huang, H.Y.; Shang, Y.; Oldham, M.C.; Martens, L.H.; Gao, F.; et al. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell 2016, 165, 921–935. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Belcourt, D.; Bennett, H.; Lazure, C.; Solomon, S. Granulins, a novel class of peptide from leukocytes. Biochem. Biophys. Res. Commun. 1990, 173, 1161–1168. [Google Scholar] [CrossRef]
- He, Z.; Ong, C.H.P.; Halper, J.; Bateman, A. Progranulin is a mediator of the wound response. Nat. Med. 2003, 9, 225–229. [Google Scholar] [CrossRef]
- Baker, M.; Mackenzie, I.R.; Pickering-Brown, S.M.; Gass, J.; Rademakers, R.; Lindholm, C.; Snowden, J.; Adamson, J.; Sadovnick, A.D.; Rollinson, S.; et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006, 442, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Gass, J.; Cannon, A.; Mackenzie, I.R.; Boeve, B.; Baker, M.; Adamson, J.; Crook, R.; Melquist, S.; Kuntz, K.; Petersen, R.; et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum. Mol. Genet. 2006, 15, 2988–3001. [Google Scholar] [CrossRef]
- Cruts, M.; Kumar-Singh, S.; Van Broeckhoven, C. Progranulin mutations in ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Curr. Alzheimer Res. 2006, 3, 485–491. [Google Scholar] [CrossRef]
- Ward, M.E.; Chen, R.; Huang, H.-Y.; Ludwig, C.; Telpoukhovskaia, M.; Taubes, A.; Boudin, H.; Minami, S.S.; Reichert, M.; Albrecht, P.; et al. Possible involvement of lysosomal dysfunction in pathological changes of the brain in aged progranulin-deficient mice. Acta Neuropathol. 2012, 287, 32298–32306. [Google Scholar] [CrossRef]
- Smith, K.R.; Damiano, J.; Franceschetti, S.; Carpenter, S.; Canafoglia, L.; Morbin, M.; Rossi, G.; Pareyson, D.; Mole, S.E.; Staropoli, J.F.; et al. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am. J. Hum. Genet. 2012, 90, 1102–1107. [Google Scholar] [CrossRef]
- Jian, J.; Zhao, S.; Tian, Q.Y.; Liu, H.; Zhao, Y.; Chen, W.C.; Grunig, G.; Torres, P.A.; Wang, B.C.; Zeng, B.; et al. Association Between Progranulin and Gaucher Disease. EBioMedicine 2016, 11, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Arvanitakis, Z. Update on frontotemporal dementia. Neurologist 2010, 16, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Kelley, B.J.; Haidar, W.; Boeve, B.F.; Baker, M.; Graff-Radford, N.R.; Krefft, T.; Frank, A.R.; Jack, C.R.J.; Shiung, M.; Knopman, D.S.; et al. Prominent phenotypic variability associated with mutations in Progranulin. Neurobiol. Aging 2009, 30, 739–751. [Google Scholar] [CrossRef]
- Sheng, J.; Su, L.; Xu, Z.; Chen, G. Progranulin polymorphism rs5848 is associated with increased risk of Alzheimer’s disease. Gene 2014, 542, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Kamalainen, A.; Viswanathan, J.; Natunen, T.; Helisalmi, S.; Kauppinen, T.; Pikkarainen, M.; Pursiheimo, J.-P.; Alafuzoff, I.; Kivipelto, M.; Haapasalo, A.; et al. GRN variant rs5848 reduces plasma and brain levels of granulin in Alzheimer’s disease patients. J. Alzheimers. Dis. 2013, 33, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Fenoglio, C.; Galimberti, D.; Cortini, F.; Kauwe, J.S.K.; Cruchaga, C.; Venturelli, E.; Villa, C.; Serpente, M.; Scalabrini, D.; Mayo, K.; et al. Rs5848 variant influences GRN mRNA levels in brain and peripheral mononuclear cells in patients with Alzheimer’s disease. J. Alzheimers. Dis. 2009, 18, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Minami, S.S.; Min, S.-W.; Krabbe, G.; Wang, C.; Zhou, Y.; Asgarov, R.; Li, Y.; Martens, L.H.; Elia, L.P.; Ward, M.E.; et al. Progranulin protects against amyloid beta deposition and toxicity in Alzheimer’s disease mouse models. Nat. Med. 2014, 20, 1157–1164. [Google Scholar] [CrossRef]
- Palfree, R.G.E.; Bennett, H.P.J.; Bateman, A. The Evolution of the Secreted Regulatory Protein Progranulin. PLoS ONE 2015, 10, e0133749. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, V.; Daniel, R.; Siew LIM, P.; Bateman, A. Structural and functional analysis of a promoter of the human granulin/epithelin gene. Biochem. J. 1996, 319, 441–447. [Google Scholar] [CrossRef]
- Ong, C.H.P.; He, Z.; Kriazhev, L.; Shan, X.; Palfree, R.G.E.; Bateman, A. Regulation of progranulin expression in myeloid cells. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R1602–R1612. [Google Scholar] [CrossRef]
- Lu, R.; Serrero, G. Stimulation of PC cell-derived growth factor (epithelin/granulin precursor) expression by estradiol in human breast cancer cells. Biochem. Biophys. Res. Commun. 1999, 256, 204–207. [Google Scholar] [CrossRef]
- Suzuki, M.; Yonezawa, T.; Fujioka, H.; Matuamuro, M.; Nishihara, M. Induction of granulin precursor gene expression by estrogen treatment in neonatal rat hypothalamus. Neurosci. Lett. 2001, 297, 199–202. [Google Scholar] [CrossRef]
- Diaz-Cueto, L.; Arechavaleta-Velasco, F.; Diaz-Arizaga, A.; Dominguez-Lopez, P.; Robles-Flores, M. PKC signaling is involved in the regulation of progranulin (acrogranin/PC-cell-derived growth factor/granulin-epithelin precursor) protein expression in human ovarian cancer cell lines. Int. J. Gynecol. Cancer 2012, 22, 945–950. [Google Scholar] [CrossRef]
- Piscopo, P.; Rivabene, R.; Adduci, A.; Mallozzi, C.; Malvezzi-Campeggi, L.; Crestini, A.; Confaloni, A. Hypoxia induces up-regulation of progranulin in neuroblastoma cell lines. Neurochem. Int. 2010, 57, 893–898. [Google Scholar] [CrossRef]
- Luo, L.; Lu, L.; Lu, Y.; Zhang, L.; Li, B.; Guo, K.; Chen, L.; Wang, Y.; Shao, Y.; Xu, J. Effects of hypoxia on progranulin expression in HT22 mouse hippocampal cells. Mol. Med. Rep. 2014, 9, 1675–1680. [Google Scholar] [CrossRef]
- Jiao, J.; Herl, L.D.; Farese, R.V.; Gao, F.-B. MicroRNA-29b regulates the expression level of human progranulin, a secreted glycoprotein implicated in frontotemporal dementia. PLoS ONE 2010, 5, e10551. [Google Scholar] [CrossRef]
- Wang, W.-X.; Kyprianou, N.; Wang, X.; Nelson, P.T. Dysregulation of the mitogen granulin in human cancer through the miR-15/107 microRNA gene group. Cancer Res. 2010, 70, 9137–9142. [Google Scholar] [CrossRef]
- Kocerha, J.; Kouri, N.; Baker, M.; Finch, N.; DeJesus-Hernandez, M.; Gonzalez, J.; Chidamparam, K.; Josephs, K.A.; Boeve, B.F.; Graff-Radford, N.R.; et al. Altered microRNA expression in frontotemporal lobar degeneration with TDP-43 pathology caused by progranulin mutations. Bmc Genom. 2011, 12, 527. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, W.; Yang, F.; Feng, T.; Zhou, M.; Yu, Y.; Yu, X.; Zhao, W.; Yi, F.; Tang, W.; et al. Interleukin-6-stimulated progranulin expression contributes to the malignancy of hepatocellular carcinoma cells by activating mTOR signaling. Sci. Rep. 2016, 6, 21260. [Google Scholar] [CrossRef]
- Tanaka, Y.; Matsuwaki, T.; Yamanouchi, K.; Nishihara, M. Increased lysosomal biogenesis in activated microglia and exacerbated neuronal damage after traumatic brain injury in progranulin-deficient mice. Neuroscience 2013, 250, 8–19. [Google Scholar] [CrossRef]
- Holler, C.J.; Taylor, G.; McEachin, Z.T.; Deng, Q.; Watkins, W.J.; Hudson, K.; Easley, C.A.; Hu, W.T.; Hales, C.M.; Rossoll, W.; et al. Trehalose upregulates progranulin expression in human and mouse models of GRN haploinsufficiency: A novel therapeutic lead to treat frontotemporal dementia. Mol. Neurodegener. 2016, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, V.; Bateman, A. Structure and chromosomal location of the human granulin gene. Biochem. Biophys. Res. Commun. 1992, 188, 57–63. [Google Scholar] [CrossRef]
- Baba, T.; Hoff, H.B.; Nemoto, H.; Lee, H.; Orth, J.; Arai, Y.; Gerton, G.L. Acrogranin, an acrosomal cysteine-rich glycoprotein, is the precursor of the growth-modulating peptides, granulins, and epithelins, and is expressed in somatic as well as male germ cells. Mol. Reprod. Dev. 1993, 34, 233–243. [Google Scholar] [CrossRef]
- Cenik, B.; Sephton, C.F.; Cenik, B.K.; Herz, J.; Yu, G. Progranulin: A proteolytically processed protein at the crossroads of inflammation and neurodegeneration. J. Biol. Chem. 2012, 287, 32298–32306. [Google Scholar] [CrossRef] [PubMed]
- Salazar, D.A.; Butler, V.J.; Argouarch, A.R.; Hsu, T.-Y.; Mason, A.; Nakamura, A.; McCurdy, H.; Cox, D.; Ng, R.; Pan, G.; et al. The Progranulin Cleavage Products, Granulins, Exacerbate TDP-43 Toxicity and Increase TDP-43 Levels. J. Neurosci. 2015, 35, 9315–9328. [Google Scholar] [CrossRef] [PubMed]
- Holler, C.J.; Taylor, G.; Deng, Q.; Kukar, T. Intracellular Proteolysis of Progranulin Generates Stable, Lysosomal Granulins That Are Haploinsufficient in Patients with Frontotemporal Dementia Caused by GRN Mutations. eNeuro 2017, 4, 1–49. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Padukkavidana, T.; Vaegter, C.B.; Brady, O.A.; Zheng, Y.; Mackenzie, I.R.; Feldman, H.H.; Nykjaer, A.; Strittmatter, S.M. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron 2010, 68, 654–667. [Google Scholar] [CrossRef] [PubMed]
- Almeida, S.; Zhou, L.; Gao, F.B. Progranulin, a glycoprotein deficient in frontotemporal dementia, is a novel substrate of several protein disulfide isomerase family proteins. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Ungurs, M.J.; Sinden, N.J.; Stockley, R.A. Progranulin is a substrate for neutrophil-elastase and proteinase-3 in the airway and its concentration correlates with mediators of airway inflammation in COPD. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L80–L87. [Google Scholar] [CrossRef] [PubMed]
- Suh, H.-S.; Choi, N.; Tarassishin, L.; Lee, S.C. Regulation of progranulin expression in human microglia and proteolysis of progranulin by matrix metalloproteinase-12 (MMP-12). PLoS ONE 2012, 7, e35115. [Google Scholar] [CrossRef] [PubMed]
- Hanby, H.A.; Zheng, X.L. Biochemistry and physiological functions of ADAMTS7 metalloprotease. Adv. Biochem. 2013, 1, 43–50. [Google Scholar] [CrossRef]
- Zhu, J.; Nathan, C.; Jin, W.; Sim, D.; Ashcroft, G.S.; Wahl, S.M.; Lacomis, L.; Erdjument-Bromage, H.; Tempst, P.; Wright, C.D.; et al. Conversion of proepithelin to epithelins: Roles of SLPI and elastase in host defense and wound repair. Cell 2002, 111, 867–878. [Google Scholar] [CrossRef]
- Petoukhov, E.; Fernando, S.; Mills, F.; Shivji, F.; Hunter, D.; Krieger, C.; Silverman, M.A.; Bamji, S.X. Activity-dependent secretion of progranulin from synapses. J. Cell Sci. 2013, 126, 5412–5421. [Google Scholar] [CrossRef]
- Nguyen, A.D.; Nguyen, T.A.; Cenik, B.; Yu, G.; Herz, J.; Walther, T.C.; Davidson, W.S.; Farese, R.V. Secreted progranulin is a homodimer and is not a component of high density lipoproteins (HDL). J. Biol. Chem. 2013, 288, 8627–8635. [Google Scholar] [CrossRef]
- Benussi, L.; Ciani, M.; Tonoli, E.; Morbin, M.; Palamara, L.; Albani, D.; Fusco, F.; Forloni, G.; Glionna, M.; Baco, M.; et al. Loss of exosomes in progranulin-associated frontotemporal dementia. Neurobiol. Aging 2016, 40, 41–49. [Google Scholar] [CrossRef]
- Coppola, G.; Karydas, A.; Rademakers, R.; Wang, Q.; Baker, M.; Hutton, M.; Miller, B.L.; Geschwind, D.H. Gene expression study on peripheral blood identifies progranulin mutations. Ann. Neurol. 2008, 64, 92–96. [Google Scholar] [CrossRef]
- Daniel, R.; He, Z.; Carmichael, K.P.; Halper, J.; Bateman, A. Cellular localization of gene expression for progranulin. J. Histochem. Cytochem. 2000, 48, 999–1009. [Google Scholar] [CrossRef]
- Toh, H.; Cao, M.; Daniels, E.; Bateman, A. Expression of the growth factor progranulin in endothelial cells influences growth and development of blood vessels: A novel mouse model. PLoS ONE 2013, 8, e64989. [Google Scholar] [CrossRef]
- Bhandari, V.; Giaid, A.; Bateman, A. The complementary deoxyribonucleic acid sequence, tissue distribution, and cellular localization of the rat granulin precursor. Endocrinology 1993, 133, 2682–2689. [Google Scholar] [CrossRef]
- Wei, J.; Hettinghouse, A.; Liu, C. The role of progranulin in arthritis. Ann. NY Acad. Sci. 2016, 1383, 5–20. [Google Scholar] [CrossRef]
- Mundra, J.J.; Jian, J.; Bhagat, P.; Liu, C.-J. Progranulin inhibits expression and release of chemokines CXCL9 and CXCL10 in a TNFR1 dependent manner. Sci. Rep. 2016, 6, 21115. [Google Scholar] [CrossRef]
- Jian, J.; Li, G.; Hettinghouse, A.; Liu, C. Progranulin: A key player in autoimmune diseases. Cytokine 2018, 101, 48–55. [Google Scholar] [CrossRef]
- Zhu, S.; Tai, C.; Petkau, T.L.; Zhang, S.; Liao, C.; Dong, Z.; Wen, W.; Chang, Q.; Wang, Y.T.; Macvicar, B.A.; et al. Progranulin promotes activation of microglia/macrophage after pilocarpine-induced status epilepticus. Brain Res. 2013, 1530, 54–65. [Google Scholar] [CrossRef]
- Kawase, R.; Ohama, T.; Matsuyama, A.; Matsuwaki, T.; Okada, T.; Yamashita, T.; Yuasa-Kawase, M.; Nakaoka, H.; Nakatani, K.; Inagaki, M.; et al. Deletion of progranulin exacerbates atherosclerosis in ApoE knockout mice. Cardiovasc. Res. 2013, 100, 125–133. [Google Scholar] [CrossRef]
- Petkau, T.L.; Neal, S.J.; Orban, P.C.; MacDonald, J.L.; Hill, A.M.; Lu, G.; Feldman, H.H.; Mackenzie, I.R.A.; Leavitt, B.R. Progranulin expression in the developing and adult murine brain. J. Comp. Neurol. 2010, 518, 3931–3947. [Google Scholar] [CrossRef]
- Matsuwaki, T.; Asakura, R.; Suzuki, M.; Yamanouchi, K.; Nishihara, M. Age-dependent changes in progranulin expression in the mouse brain. J. Reprod. Dev. 2011, 57, 113–119. [Google Scholar] [CrossRef]
- Naphade, S.B.; Kigerl, K.A.; Jakeman, L.B.; Kostyk, S.K.; Popovich, P.G.; Kuret, J. Progranulin expression is upregulated after spinal contusion in mice. Acta Neuropathol. 2010, 119, 123–133. [Google Scholar] [CrossRef]
- Martens, L.H.; Zhang, J.; Barmada, S.J.; Zhou, P.; Kamiya, S.; Sun, B.; Min, S.-W.; Gan, L.; Finkbeiner, S.; Huang, E.J.; et al. Progranulin deficiency promotes neuroinflammation and neuron loss following toxin-induced injury. J. Clin. Invest. 2012, 122, 3955–3959. [Google Scholar] [CrossRef]
- Beel, S.; Herdewyn, S.; Fazal, R.; De Decker, M.; Moisse, M.; Robberecht, W.; Van Den Bosch, L.; Van Damme, P. Progranulin reduces insoluble TDP-43 levels, slows down axonal degeneration and prolongs survival in mutant TDP-43 mice. Mol. Neurodegener. 2018, 13, 55. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Colonna, M.; Schwartz, M.; Amit, I.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290. [Google Scholar] [CrossRef]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef]
- He, Z.; Bateman, A. Progranulin gene expression regulates epithelial cell growth and promotes tumor growth in vivo. Cancer Res. 1999, 59, 3222–3229. [Google Scholar]
- Rhost, S.; Hughes, E.; Harrison, H.; Rafnsdottir, S.; Jacobsson, H.; Gregersson, P.; Magnusson, Y.; Fitzpatrick, P.; Andersson, D.; Berger, K.; et al. Sortilin inhibition limits secretion-induced progranulin-dependent breast cancer progression and cancer stem cell expansion. Breast Cancer Res. 2018, 20, 137. [Google Scholar] [CrossRef]
- Koo, D.-H.; Do, I.-G.; Oh, S.; Lee, Y.-G.; Kim, K.; Sohn, J.H.; Park, S.-K.; Yang, H.-J.; Jung, Y.S.; Park, D.I.; et al. Prognostic Value of Progranulin in Patients with Colorectal Cancer Treated with Curative Resection. Pathol. Oncol. Res. 2018. [Google Scholar] [CrossRef]
- Kimura, A.; Takemura, M.; Serrero, G.; Yoshikura, N.; Hayashi, Y.; Saito, K.; Inuzuka, T. Higher levels of progranulin in cerebrospinal fluid of patients with lymphoma and carcinoma with CNS metastasis. J. Neurooncol. 2018, 137, 455–462. [Google Scholar] [CrossRef]
- Buraschi, S.; Xu, S.-Q.; Stefanello, M.; Moskalev, I.; Morcavallo, A.; Genua, M.; Tanimoto, R.; Birbe, R.; Peiper, S.C.; Gomella, L.G.; et al. Suppression of progranulin expression inhibits bladder cancer growth and sensitizes cancer cells to cisplatin. Oncotarget 2016, 7, 39980–39995. [Google Scholar] [CrossRef]
- Nielsen, S.R.; Quaranta, V.; Linford, A.; Emeagi, P.; Rainer, C.; Santos, A.; Ireland, L.; Sakai, T.; Sakai, K.; Kim, Y.-S.; et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat. Cell Biol. 2016, 18, 549–560. [Google Scholar] [CrossRef]
- Suarez-Calvet, M.; Capell, A.; Araque Caballero, M.A.; Morenas-Rodriguez, E.; Fellerer, K.; Franzmeier, N.; Kleinberger, G.; Eren, E.; Deming, Y.; Piccio, L.; et al. CSF progranulin increases in the course of Alzheimer’s disease and is associated with sTREM2, neurodegeneration and cognitive decline. Embo Mol. Med. 2018, 10. [Google Scholar] [CrossRef]
- Morenas-Rodriguez, E.; Cervera-Carles, L.; Vilaplana, E.; Alcolea, D.; Carmona-Iragui, M.; Dols-Icardo, O.; Ribosa-Nogue, R.; Munoz-Llahuna, L.; Sala, I.; Belen Sanchez-Saudinos, M.; et al. Progranulin Protein Levels in Cerebrospinal Fluid in Primary Neurodegenerative Dementias. J. Alzheimers. Dis. 2016, 50, 539–546. [Google Scholar] [CrossRef]
- Abella, V.; Pino, J.; Scotece, M.; Conde, J.; Lago, F.; Gonzalez-Gay, M.A.; Mera, A.; Gomez, R.; Mobasheri, A.; Gualillo, O. Progranulin as a biomarker and potential therapeutic agent. Drug Discov. Today 2017, 22, 1557–1564. [Google Scholar] [CrossRef]
- Chen, X.; Liu, J.; Zhu, M.; Hu, B.; Bai, F.; Jin, W.; Zhou, J.; Chen, C.; Song, Y.; Pan, J. Progranulin is a novel biomarker for predicting an acute exacerbation of chronic obstructive pulmonary disease. Clin. Respir. J. 2018, 12, 2525–2533. [Google Scholar] [CrossRef]
- Nicoletto, B.B.; Sarmento, R.A.; Pedrollo, E.F.; Krolikowski, T.C.; Canani, L.H. Association between progranulin serum levels and dietary intake. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Alissa, E.M.; Sutaih, R.H.; Kamfar, H.Z.; Alagha, A.E.; Marzouki, Z.M. Serum progranulin levels in relation to insulin resistance in childhood obesity. J. Pediatr. Endocrinol. Metab. 2017, 30, 1251–1256. [Google Scholar] [CrossRef]
- Daxer, J.; Herttrich, T.; Zhao, Y.Y.; Vogel, M.; Hiemisch, A.; Scheuermann, K.; Korner, A.; Kratzsch, J.; Kiess, W.; Quante, M. Nocturnal levels of chemerin and progranulin in adolescents: Influence of sex, body mass index, glucose metabolism and sleep. J. Pediatr. Endocrinol. Metab. 2017, 30, 57–61. [Google Scholar] [CrossRef]
- Vercellino, M.; Grifoni, S.; Romagnolo, A.; Masera, S.; Mattioda, A.; Trebini, C.; Chiavazza, C.; Caligiana, L.; Capello, E.; Mancardi, G.L.; et al. Progranulin expression in brain tissue and cerebrospinal fluid levels in multiple sclerosis. Mult. Scler. 2011, 17, 1194–1201. [Google Scholar] [CrossRef]
- Carecchio, M.; Fenoglio, C.; De Riz, M.; Guidi, I.; Comi, C.; Cortini, F.; Venturelli, E.; Restelli, I.; Cantoni, C.; Bresolin, N.; et al. Progranulin plasma levels as potential biomarker for the identification of GRN deletion carriers. A case with atypical onset as clinical amnestic Mild Cognitive Impairment converted to Alzheimer’s disease. J. Neurol. Sci. 2009, 287, 291–293. [Google Scholar] [CrossRef]
- Ghidoni, R.; Paterlini, A.; Benussi, L. Circulating progranulin as a biomarker for neurodegenerative diseases. Am. J. Neurodegener. Dis. 2012, 1, 180–190. [Google Scholar]
- Kortvelyessy, P.; Gukasjan, A.; Sweeney-Reed, C.M.; Heinze, H.-J.; Thurner, L.; Bittner, D.M. Progranulin and Amyloid-beta Levels: Relationship to Neuropsychology in Frontotemporal and Alzheimer’s Disease. J. Alzheimers. Dis. 2015, 46, 375–380. [Google Scholar] [CrossRef]
- Antonell, A.; Gil, S.; Sanchez-Valle, R.; Balasa, M.; Bosch, B.; Prat, M.C.; Chiollaz, A.-C.; Fernandez, M.; Yague, J.; Molinuevo, J.L.; et al. Serum progranulin levels in patients with frontotemporal lobar degeneration and Alzheimer’s disease: Detection of GRN mutations in a Spanish cohort. J. Alzheimers. Dis. 2012, 31, 581–591. [Google Scholar] [CrossRef]
- Wilke, C.; Gillardon, F.; Deuschle, C.; Dubois, E.; Hobert, M.A.; Muller vom Hagen, J.; Kruger, S.; Biskup, S.; Blauwendraat, C.; Hruscha, M.; et al. Serum Levels of Progranulin Do Not Reflect Cerebrospinal Fluid Levels in Neurodegenerative Disease. Curr. Alzheimer Res. 2016, 13, 654–662. [Google Scholar] [CrossRef]
- Galimberti, D.; Fumagalli, G.G.; Fenoglio, C.; Cioffi, S.M.G.; Arighi, A.; Serpente, M.; Borroni, B.; Padovani, A.; Tagliavini, F.; Masellis, M.; et al. Progranulin plasma levels predict the presence of GRN mutations in asymptomatic subjects and do not correlate with brain atrophy: Results from the GENFI study. Neurobiol. Aging 2018, 62, 245. [Google Scholar] [CrossRef]
- Wilke, C.; Gillardon, F.; Deuschle, C.; Hobert, M.A.; Jansen, I.E.; Metzger, F.G.; Heutink, P.; Gasser, T.; Maetzler, W.; Blauwendraat, C.; et al. Cerebrospinal Fluid Progranulin, but Not Serum Progranulin, Is Reduced in GRN-Negative Frontotemporal Dementia. Neurodegener. Dis. 2017, 17, 83–88. [Google Scholar] [CrossRef]
- Kojima, Y.; Ono, K.; Inoue, K.; Takagi, Y.; Kikuta, K.; Nishimura, M.; Yoshida, Y.; Nakashima, Y.; Matsumae, H.; Furukawa, Y.; et al. Progranulin expression in advanced human atherosclerotic plaque. Atherosclerosis 2009, 206, 102–108. [Google Scholar] [CrossRef]
- Suh, H.-S.; Gelman, B.B.; Lee, S.C. Potential roles of microglial cell progranulin in HIV-associated CNS pathologies and neurocognitive impairment. J. Neuroimmune Pharm. 2014, 9, 117–132. [Google Scholar] [CrossRef]
- Luo, Q.; Yan, X.; Tu, H.; Yin, Y.; Cao, J. Progranulin aggravates pulmonary immunopathology during influenza virus infection. Thorax 2018. [Google Scholar] [CrossRef]
- Wagner, H. New vistas on TLR9 activation. Eur. J. Immunol. 2011, 41, 2814–2816. [Google Scholar] [CrossRef]
- Tang, W.; Lu, Y.; Tian, Q.-Y.; Zhang, Y.; Guo, F.-J.; Liu, G.-Y.; Syed, N.M.; Lai, Y.; Lin, E.A.; Kong, L.; et al. The growth factor progranulin binds to TNF receptors and is therapeutic against inflammatory arthritis in mice. Science 2011, 332, 478–484. [Google Scholar] [CrossRef]
- Tian, Q.; Zhao, Y.; Mundra, J.J.; Gonzalez-Gugel, E.; Jian, J.; Uddin, S.M.Z.; Liu, C. Three TNFR-binding domains of PGRN act independently in inhibition of TNF-alpha binding and activity. Front. Biosci. 2014, 19, 1176–1185. [Google Scholar] [CrossRef]
- Chen, X.; Chang, J.; Deng, Q.; Xu, J.; Nguyen, T.A.; Martens, L.H.; Cenik, B.; Taylor, G.; Hudson, K.F.; Chung, J.; et al. Progranulin does not bind tumor necrosis factor (TNF) receptors and is not a direct regulator of TNF-dependent signaling or bioactivity in immune or neuronal cells. J. Neurosci. 2013, 33, 9202–9213. [Google Scholar] [CrossRef]
- Lang, I.; Füllsack, S.; Wajant, H. Lack of evidence for a direct interaction of progranulin and tumor necrosis factor receptor-1 and tumor necrosis factor receptor-2 from cellular binding studies. Front. Immunol. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Wang, B.C.; Liu, H.; Talwar, A.; Jian, J. New discovery rarely runs smooth: An update on progranulin/TNFR interactions. Protein Cell 2015, 6, 792–803. [Google Scholar] [CrossRef]
- Wei, F.; Zhang, Y.; Jian, J.; Mundra, J.J.; Tian, Q.; Lin, J.; Lafaille, J.J.; Tang, W.; Zhao, W.; Yu, X.; et al. PGRN protects against colitis progression in mice in an IL-10 and TNFR2 dependent manner. Sci. Rep. 2014, 4, 7023. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Takemura, M.; Serrero, G.; Hayashi, J.; Yue, B.; Tsuboi, A.; Kubo, H.; Mitsuhashi, T.; Mannami, K.; Sato, M.; et al. Increased serum GP88 (Progranulin) concentrations in rheumatoid arthritis. Inflammation 2014, 37, 1806–1813. [Google Scholar] [CrossRef]
- Johnson, J.; Yeter, K.; Rajbhandary, R.; Neal, R.; Tian, Q.; Jian, J.; Fadle, N.; Thurner, L.; Liu, C.; Stohl, W. Serum progranulin levels in Hispanic rheumatoid arthritis patients treated with TNF antagonists: A prospective, observational study. Clin. Rheumatol. 2017, 36, 507–516. [Google Scholar] [CrossRef]
- Park, B.; Buti, L.; Lee, S.; Matsuwaki, T.; Spooner, E.; Brinkmann, M.M.; Nishihara, M.; Ploegh, H.L. Granulin is a soluble cofactor for toll-like receptor 9 signaling. Immunity 2011, 34, 505–513. [Google Scholar] [CrossRef]
- Zheng, Y.; Brady, O.A.; Meng, P.S.; Mao, Y.; Hu, F. C-terminus of progranulin interacts with the beta-propeller region of sortilin to regulate progranulin trafficking. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Neill, T.; Buraschi, S.; Goyal, A.; Sharpe, C.; Natkanski, E.; Schaefer, L.; Morrione, A.; Iozzo, R. V EphA2 is a functional receptor for the growth factor progranulin. J. Cell Biol. 2016, 215, 687–703. [Google Scholar] [CrossRef]
- Chen, J.; Saito, S.; Kobayashi, N.; Sato, K.; Terashita, T.; Shimokawa, T.; Mominoki, K.; Miyawaki, K.; Sano, A.; Matsuda, S. Expression patterns in alternative splicing forms of prosaposin mRNA in the rat facial nerve nucleus after facial nerve transection. Neurosci. Res. 2008, 60, 82–94. [Google Scholar] [CrossRef]
- Moisse, K.; Volkening, K.; Leystra-Lantz, C.; Welch, I.; Hill, T.; Strong, M.J. Divergent patterns of cytosolic TDP-43 and neuronal progranulin expression following axotomy: Implications for TDP-43 in the physiological response to neuronal injury. Brain Res. 2009, 1249, 202–211. [Google Scholar] [CrossRef]
- Egashira, Y.; Suzuki, Y.; Azuma, Y.; Takagi, T.; Mishiro, K.; Sugitani, S.; Tsuruma, K.; Shimazawa, M.; Yoshimura, S.; Kashimata, M.; et al. The growth factor progranulin attenuates neuronal injury induced by cerebral ischemia-reperfusion through the suppression of neutrophil recruitment. J. Neuroinflammation 2013, 10, 105. [Google Scholar] [CrossRef]
- Kayasuga, Y.; Chiba, S.; Suzuki, M.; Kikusui, T.; Matsuwaki, T.; Yamanouchi, K.; Kotaki, H.; Horai, R.; Iwakura, Y.; Nishihara, M. Alteration of behavioural phenotype in mice by targeted disruption of the progranulin gene. Behav. Brain Res. 2007, 185, 110–118. [Google Scholar] [CrossRef]
- Ahmed, Z.; Sheng, H.; Xu, Y.F.; Lin, W.L.; Innes, A.E.; Gass, J.; Yu, X.; Hou, H.; Chiba, S.; Yamanouchi, K.; et al. Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am. J. Pathol. 2010, 177, 311–324. [Google Scholar] [CrossRef]
- Yin, F.; Dumont, M.; Banerjee, R.; Ma, Y.; Li, H.; Lin, M.T.; Beal, M.F.; Nathan, C.; Thomas, B.; Ding, A. Behavioral deficits and progressive neuropathology in progranulin-deficient mice: A mouse model of frontotemporal dementia. FASEB J. 2010, 24, 4639–4647. [Google Scholar] [CrossRef]
- Petkau, T.L.; Neal, S.J.; Milnerwood, A.; Mew, A.; Hill, A.M.; Orban, P.; Gregg, J.; Lu, G.; Feldman, H.H.; Mackenzie, I.R.A.; et al. Synaptic dysfunction in progranulin-deficient mice. Neurobiol. Dis. 2012, 45, 711–722. [Google Scholar] [CrossRef]
- Philips, T.; De Muynck, L.; Thu, H.N.T.; Weynants, B.; Vanacker, P.; Dhondt, J.; Sleegers, K.; Schelhaas, H.J.; Verbeek, M.; Vandenberghe, R.; et al. Microglial upregulation of progranulin as a marker of motor neuron degeneration. J. Neuropathol. Exp. Neurol. 2010, 69, 1191–1200. [Google Scholar] [CrossRef]
- Petkau, T.L.; Kosior, N.; de Asis, K.; Connolly, C.C.; Leavitt, B.R. Selective depletion of microglial progranulin in mice is not sufficient to cause neuronal ceroid lipofuscinosis or neuroinflammation. J. Neuroinflammation 2017, 14, 225. [Google Scholar] [CrossRef]
- Arrant, A.E.; Filiano, A.J.; Patel, A.R.; Hoffmann, M.Q.; Boyle, N.R.; Kashyap, S.N.; Onyilo, V.C.; Young, A.H.; Roberson, E.D. Reduction of microglial progranulin does not exacerbate pathology or behavioral deficits in neuronal progranulin-insufficient mice. Neurobiol. Dis. 2018, 124, 152–162. [Google Scholar] [CrossRef]
- Arrant, A.E.; Filiano, A.J.; Unger, D.E.; Young, A.H.; Roberson, E.D. Restoring neuronal progranulin reverses deficits in a mouse model of frontotemporal dementia. Brain 2017, 140, 1447–1465. [Google Scholar] [CrossRef]
- Wieghofer, P.; Knobeloch, K.P.; Prinz, M. Genetic targeting of microglia. Glia 2015, 63, 1–22. [Google Scholar] [CrossRef]
- Blank, T.; Prinz, M. CatacLysMic specificity when targeting myeloid cells? Eur. J. Immunol. 2016, 46, 1340–1342. [Google Scholar] [CrossRef]
- Krabbe, G.; Minami, S.S.; Etchegaray, J.I.; Taneja, P.; Djukic, B.; Davalos, D.; Le, D.; Lo, I.; Zhan, L.; Reichert, M.C.; et al. Microglial NFkappaB-TNFalpha hyperactivation induces obsessive-compulsive behavior in mouse models of progranulin-deficient frontotemporal dementia. Proc. Natl. Acad. Sci. USA 2017, 114, 5029–5034. [Google Scholar] [CrossRef]
- Elmore, M.R.P.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef]
- Spangenberg, E.E.; Lee, R.J.; Najafi, A.R.; Rice, R.A.; Elmore, M.R.P.; Blurton-Jones, M.; West, B.L.; Green, K.N. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-beta pathology. Brain 2016, 139, 1265–1281. [Google Scholar] [CrossRef]
- Luo, J.; Elwood, F.; Britschgi, M.; Villeda, S.; Zhang, H.; Ding, Z.; Zhu, L.; Alabsi, H.; Getachew, R.; Narasimhan, R.; et al. Colony-stimulating factor 1 receptor (CSF1R) signaling in injured neurons facilitates protection and survival. J. Exp. Med. 2013, 210, 157–172. [Google Scholar] [CrossRef]
- Satoh, J.-I.; Kino, Y.; Kawana, N.; Yamamoto, Y.; Ishida, T.; Saito, Y.; Arima, K. TMEM106B expression is reduced in Alzheimer’s disease brains. Alzheimers. Res. 2014, 6, 17. [Google Scholar] [CrossRef]
- Pereson, S.; Wils, H.; Kleinberger, G.; McGowan, E.; Vandewoestyne, M.; Van Broeck, B.; Joris, G.; Cuijt, I.; Deforce, D.; Hutton, M.; et al. Progranulin expression correlates with dense-core amyloid plaque burden in Alzheimer disease mouse models. J. Pathol. 2009, 219, 173–181. [Google Scholar] [CrossRef]
- Gowrishankar, S.; Yuan, P.; Wu, Y.; Schrag, M.; Paradise, S.; Grutzendler, J.; De Camilli, P.; Ferguson, S.M. Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques. Proc. Natl. Acad. Sci. USA 2015, 112, E3699–E3708. [Google Scholar] [CrossRef]
- Gliebus, G.; Rosso, A.; Lippa, C.F. Progranulin and beta-amyloid distribution: A case report of the brain from preclinical PS-1 mutation carrier. Am. J. Alzheimers. Dis. Other Demen. 2009, 24, 456–460. [Google Scholar] [CrossRef]
- Ahmed, Z.; Mackenzie, I.R.A.; Hutton, M.L.; Dickson, D.W. Progranulin in frontotemporal lobar degeneration and neuroinflammation. J. Neuroinflammation 2007, 4, 7. [Google Scholar] [CrossRef]
- Takahashi, H.; Klein, Z.A.; Bhagat, S.M.; Kaufman, A.C.; Kostylev, M.A.; Ikezu, T.; Strittmatter, S.M. Opposing effects of progranulin deficiency on amyloid and tau pathologies via microglial TYROBP network. Acta Neuropathol. 2017, 133, 785–807. [Google Scholar] [CrossRef]
- Hosokawa, M.; Tanaka, Y.; Arai, T.; Kondo, H.; Akiyama, H.; Hasegawa, M. Progranulin haploinsufficiency reduces amyloid beta deposition in Alzheimer’s disease model mice. Exp. Anim. 2018, 67, 63–70. [Google Scholar] [CrossRef]
- Pickford, F.; Marcus, J.; Camargo, L.M.; Xiao, Q.; Graham, D.; Mo, J.-R.R.; Burkhardt, M.; Kulkarni, V.; Crispino, J.; Hering, H.; et al. Progranulin is a chemoattractant for microglia and stimulates their endocytic activity. Am. J. Pathol. 2011, 178, 284–295. [Google Scholar] [CrossRef]
- Hosokawa, M.; Arai, T.; Masuda-Suzukake, M.; Kondo, H.; Matsuwaki, T.; Nishihara, M.; Hasegawa, M.; Akiyama, H. Progranulin reduction is associated with increased tau phosphorylation in P301L tau transgenic mice. J. Neuropathol. Exp. Neurol. 2015, 74, 158–165. [Google Scholar] [CrossRef]
- Nicholson, A.M.; Finch, N.A.C.A.; Almeida, M.; Perkerson, R.B.; van Blitterswijk, M.; Wojtas, A.; Cenik, B.; Rotondo, S.; Inskeep, V.; Almasy, L.; et al. Prosaposin is a regulator of progranulin levels and oligomerization. Nat. Commun. 2016, 7, 11992. [Google Scholar] [CrossRef]
- Zhou, F.-Q.; Jiang, J.; Griffith, C.M.; Patrylo, P.R.; Cai, H.; Chu, Y.; Yan, X.-X. Lack of human-like extracellular sortilin neuropathology in transgenic Alzheimer’s disease model mice and macaques. Alzheimers. Res. 2018, 10, 40. [Google Scholar] [CrossRef]
- Scholtzova, H.; Chianchiano, P.; Pan, J.; Sun, Y.; Goni, F.; Mehta, P.D.; Wisniewski, T. Amyloid beta and Tau Alzheimer’s disease related pathology is reduced by Toll-like receptor 9 stimulation. Acta Neuropathol. Commun. 2014, 2, 101. [Google Scholar] [CrossRef]
- Walker, D.G.; Tang, T.M.; Lue, L.-F. Increased expression of toll-like receptor 3, an anti-viral signaling molecule, and related genes in Alzheimer’s disease brains. Exp. Neurol. 2018, 309, 91–106. [Google Scholar] [CrossRef]
- Zhou, X.; Sun, L.; Bastos de Oliveira, F.; Qi, X.; Brown, W.J.; Smolka, M.B.; Sun, Y.; Hu, F. Prosaposin facilitates sortilin-independent lysosomal trafficking of progranulin. J. Cell Biol. 2015, 210, 991–1002. [Google Scholar] [CrossRef]
- Zhou, X.; Sullivan, P.M.; Sun, L.; Hu, F. The interaction between progranulin and prosaposin is mediated by granulins and the linker region between saposin B and C. J. Neurochem. 2017, 143, 236–243. [Google Scholar] [CrossRef]
- Gotzl, J.K.; Colombo, A.-V.; Fellerer, K.; Reifschneider, A.; Werner, G.; Tahirovic, S.; Haass, C.; Capell, A. Early lysosomal maturation deficits in microglia triggers enhanced lysosomal activity in other brain cells of progranulin knockout mice. Mol. Neurodegener. 2018, 13, 48. [Google Scholar] [CrossRef]
- Götzl, J.K.; Mori, K.; Damme, M.; Fellerer, K.; Tahirovic, S.; Kleinberger, G.; Janssens, J.; Van Der Zee, J.; Lang, C.M.; Kremmer, E.; et al. Common pathobiochemical hallmarks of progranulin-associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis. Acta Neuropathol. 2014, 127, 845–860. [Google Scholar] [CrossRef]
- Mackenzie, I.R.A.; Baker, M.; Pickering-Brown, S.; Hsiung, G.-Y.R.; Lindholm, C.; Dwosh, E.; Gass, J.; Cannon, A.; Rademakers, R.; Hutton, M.; et al. The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain 2006, 129, 3081–3090. [Google Scholar] [CrossRef]
- Mackenzie, I.R.A. The neuropathology and clinical phenotype of FTD with progranulin mutations. Acta Neuropathol. 2007, 114, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Sole-Domenech, S.; Cruz, D.L.; Capetillo-Zarate, E.; Maxfield, F.R. The endocytic pathway in microglia during health, aging and Alzheimer’s disease. Ageing Res. Rev. 2016, 32, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Mamun, A.A.; Labu, Z.K.; Hidalgo-Lanussa, O.; Barreto, G.E.; Ashraf, G.M. Autophagic dysfunction in Alzheimer’s disease: Cellular and molecular mechanistic approaches to halt Alzheimer’s pathogenesis. J. Cell. Physiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ward, M.E.; Chen, R.; Huang, H.-Y.; Ludwig, C.; Telpoukhovskaia, M.; Taubes, A.; Boudin, H.; Minami, S.S.; Reichert, M.; Albrecht, P.; et al. Individuals with progranulin haploinsufficiency exhibit features of neuronal ceroid lipofuscinosis. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Lee, C.W.; Stankowski, J.N.; Chew, J.; Cook, C.N.; Lam, Y.-W.W.; Almeida, S.; Carlomagno, Y.; Lau, K.-F.F.; Prudencio, M.; Gao, F.-B.B.; et al. The lysosomal protein cathepsin L is a progranulin protease. Mol. Neurodegener. 2017, 12, 55. [Google Scholar] [CrossRef] [PubMed]
- Klein, Z.A.; Takahashi, H.; Ma, M.; Stagi, M.; Zhou, M.; Lam, T.T.; Strittmatter, S.M. Loss of TMEM106B Ameliorates Lysosomal and Frontotemporal Dementia-Related Phenotypes in Progranulin-Deficient Mice. Neuron 2017, 95, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Arrant, A.E.; Onyilo, V.C.; Unger, D.E.; Roberson, E.D. Progranulin Gene Therapy Improves Lysosomal Dysfunction and Microglial Pathology Associated with Frontotemporal Dementia and Neuronal Ceroid Lipofuscinosis. J. Neurosci. 2018, 38, 2341–2358. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Suzuki, G.; Matsuwaki, T.; Hosokawa, M.; Serrano, G.; Beach, T.G.; Yamanouchi, K.; Hasegawa, M.; Nishihara, M. Progranulin regulates lysosomal function and biogenesis through acidification of lysosomes. Hum. Mol. Genet. 2017, 26, 969–988. [Google Scholar] [CrossRef]
- Finch, N.; Carrasquillo, M.M.; Baker, M.; Rutherford, N.J.; Coppola, G.; Dejesus-Hernandez, M.; Crook, R.; Hunter, T.; Ghidoni, R.; Benussi, L.; et al. TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology 2011, 76, 467–474. [Google Scholar] [CrossRef]
- Zhou, X.; Sun, L.; Brady, O.A.; Murphy, K.A.; Hu, F. Elevated TMEM106B levels exaggerate lipofuscin accumulation and lysosomal dysfunction in aged mice with progranulin deficiency. Acta Neuropathol. Commun. 2017, 5, 9. [Google Scholar] [CrossRef]
- Van Kampen, J.M.; Kay, D.G. Progranulin gene delivery reduces plaque burden and synaptic atrophy in a mouse model of Alzheimer’s disease. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Van Kampen, J.M.; Baranowski, D.; Kay, D.G. Progranulin gene delivery protects dopaminergic neurons in a mouse model of Parkinson’s disease. PLoS ONE 2014, 9, e97032. [Google Scholar] [CrossRef]
- Amado, D.A.; Rieders, J.M.; Diatta, F.; Hernandez-con, P.; Singer, A.; Mak, J.T.; Zhang, J.; Lancaster, E.; Davidson, B.L.; Chen-plotkin, A.S. AAV-Mediated Progranulin Delivery to a Mouse Model of Progranulin Deficiency Causes T Cell-Mediated Toxicity. Mol. 2019, 27, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sikora, J.; Harzer, K.; Elleder, M. Neurolysosomal pathology in human prosaposin deficiency suggests essential neurotrophic function of prosaposin. Acta Neuropathol. 2007, 113, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Chen-Plotkin, A.S.; Xiao, J.; Geser, F.; Martinez-Lage, M.; Grossman, M.; Unger, T.; Wood, E.M.; Van Deerlin, V.M.; Trojanowski, J.Q.; Lee, V.M.-Y. Brain progranulin expression in GRN-associated frontotemporal lobar degeneration. Acta Neuropathol. 2010, 119, 111–122. [Google Scholar] [CrossRef]
- Chen, Y.; Li, S.; Su, L.; Sheng, J.; Lv, W.; Chen, G.; Xu, Z. Association of progranulin polymorphism rs5848 with neurodegenerative diseases: A meta-analysis. J. Neurol. 2015, 262, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Mateo, I.; Gonzalez-Aramburu, I.; Pozueta, A.; Vazquez-Higuera, J.L.; Rodriguez-Rodriguez, E.; Sanchez-Juan, P.; Calero, M.; Dobato, J.L.; Infante, J.; Berciano, J.; et al. Reduced serum progranulin level might be associated with Parkinson’s disease risk. Eur. J. Neurol. 2013, 20, 1571–1573. [Google Scholar] [CrossRef]
- Tansey, M.G.; Romero-Ramos, M. Immune system responses in Parkinson’s disease: Early and dynamic. Eur. J. Neurosci. 2018. [Google Scholar] [CrossRef]
- Joe, E.-H.; Choi, D.-J.; An, J.; Eun, J.-H.; Jou, I.; Park, S. Astrocytes, Microglia, and Parkinson’s Disease. Exp. Neurobiol. 2018, 27, 77–87. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendsaikhan, A.; Tooyama, I.; Walker, D.G. Microglial Progranulin: Involvement in Alzheimer’s Disease and Neurodegenerative Diseases. Cells 2019, 8, 230. https://doi.org/10.3390/cells8030230
Mendsaikhan A, Tooyama I, Walker DG. Microglial Progranulin: Involvement in Alzheimer’s Disease and Neurodegenerative Diseases. Cells. 2019; 8(3):230. https://doi.org/10.3390/cells8030230
Chicago/Turabian StyleMendsaikhan, Anarmaa, Ikuo Tooyama, and Douglas G. Walker. 2019. "Microglial Progranulin: Involvement in Alzheimer’s Disease and Neurodegenerative Diseases" Cells 8, no. 3: 230. https://doi.org/10.3390/cells8030230
APA StyleMendsaikhan, A., Tooyama, I., & Walker, D. G. (2019). Microglial Progranulin: Involvement in Alzheimer’s Disease and Neurodegenerative Diseases. Cells, 8(3), 230. https://doi.org/10.3390/cells8030230