Adaptations in Mitochondrial Enzymatic Activity Occurs Independent of Genomic Dosage in Response to Aerobic Exercise Training and Deconditioning in Human Skeletal Muscle

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics
2.2. Subjects and Initial Tests
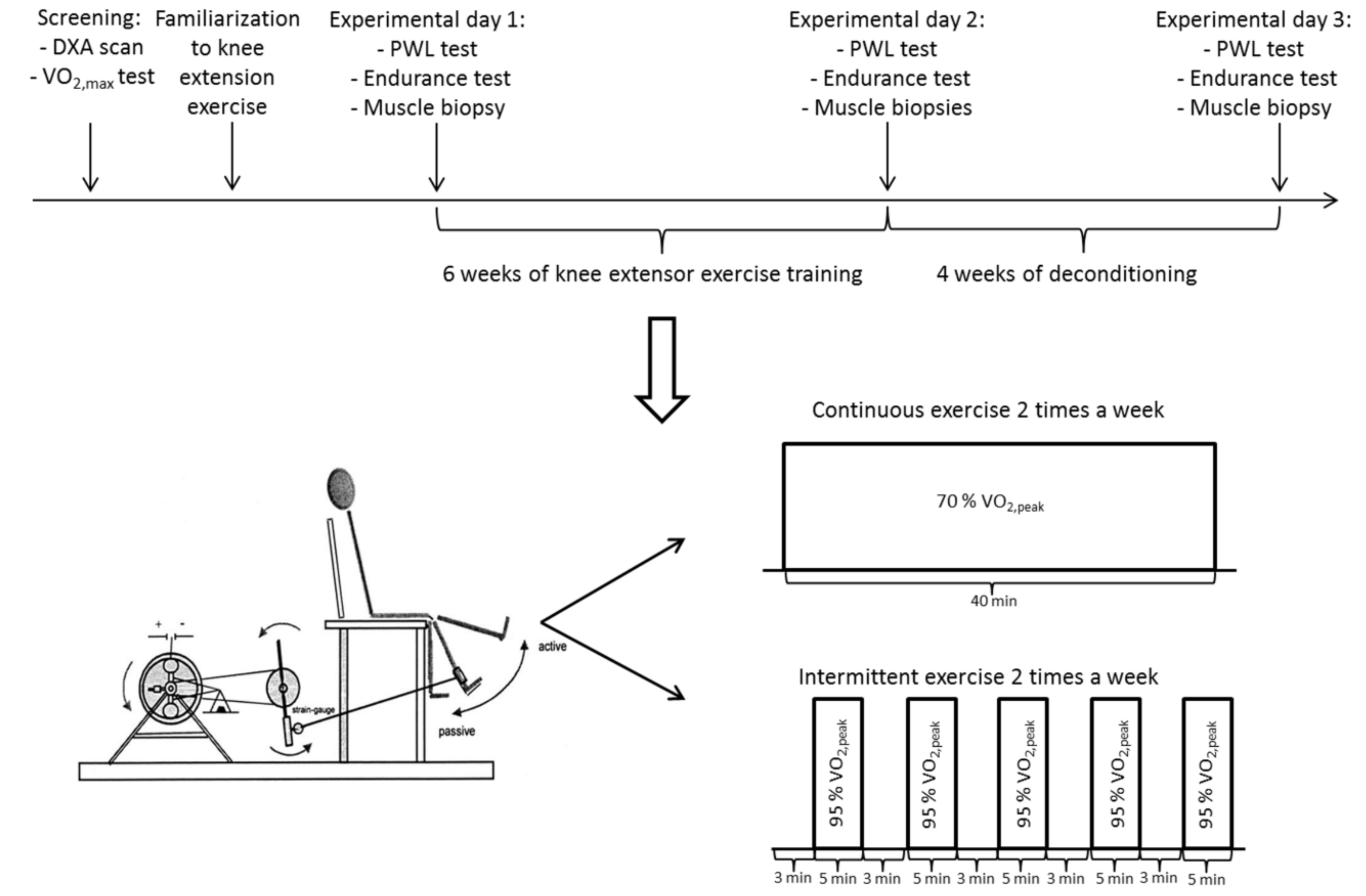
2.3. Experimental Days
2.4. Training and Deconditioning
2.5. Muscle Biopsy
2.6. Quantitative PCR to Determine mtDNA Copy Number per Diploid Nuclear Genome
2.7. Enzyme Activities
2.8. Cardiolipin
2.9. Western Blotting
2.10. Morphological Measurements
2.11. Statistics
3. Results
3.1. Subject Characteristics and Performance Data
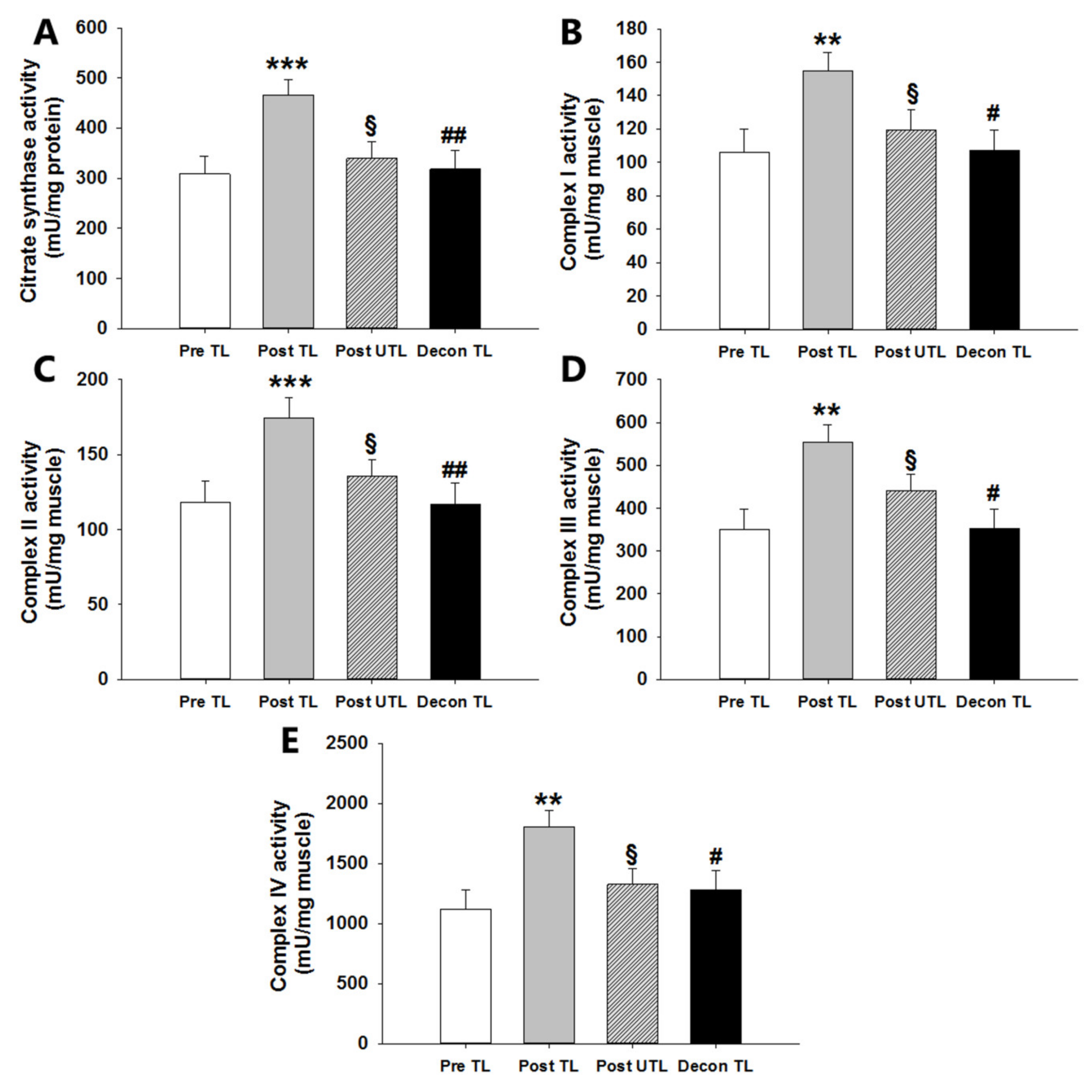
3.2. Mitochondrial Enzyme Complex Activities
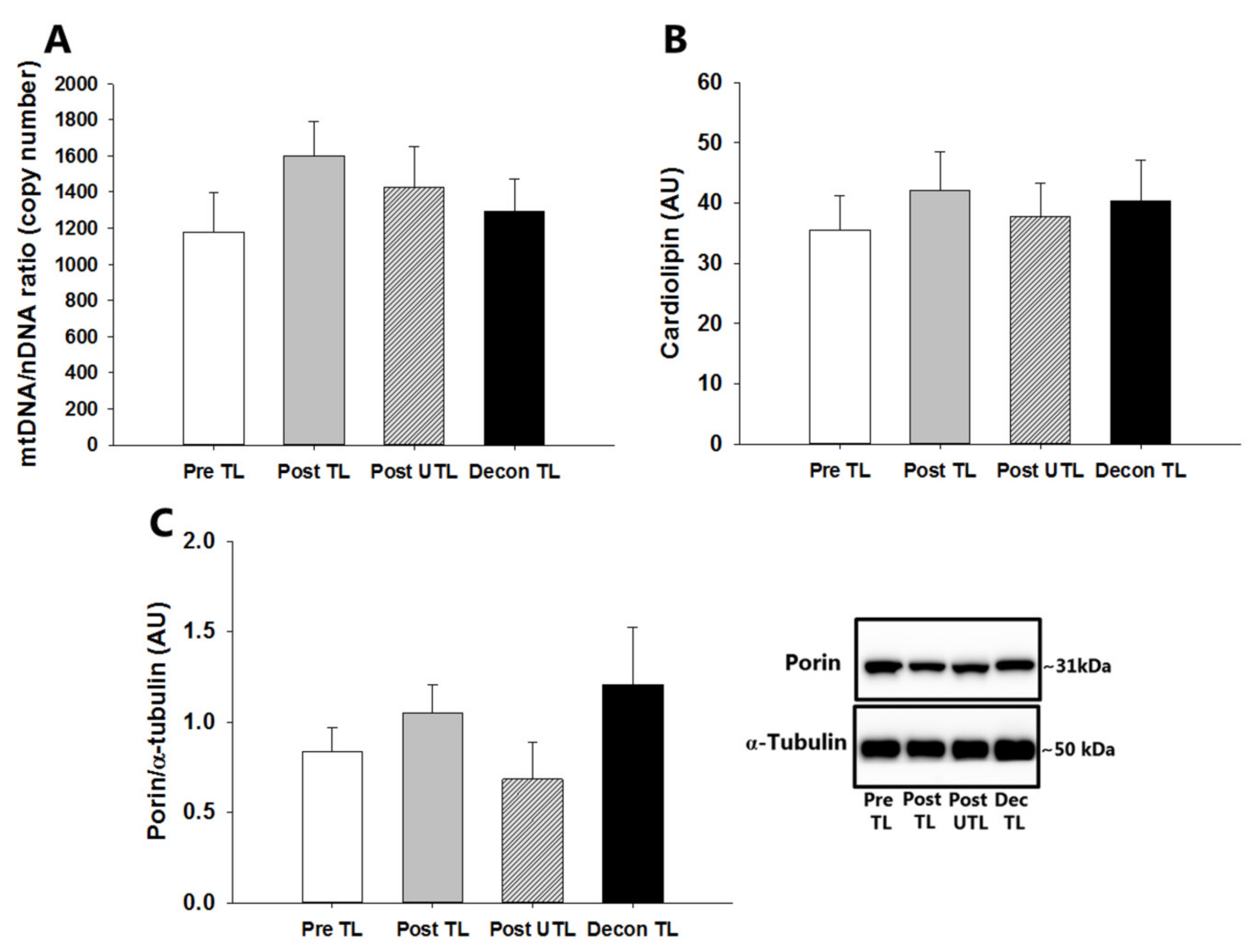
3.3. mtDNA Content
3.4. Cardiolipin and Porin Contents
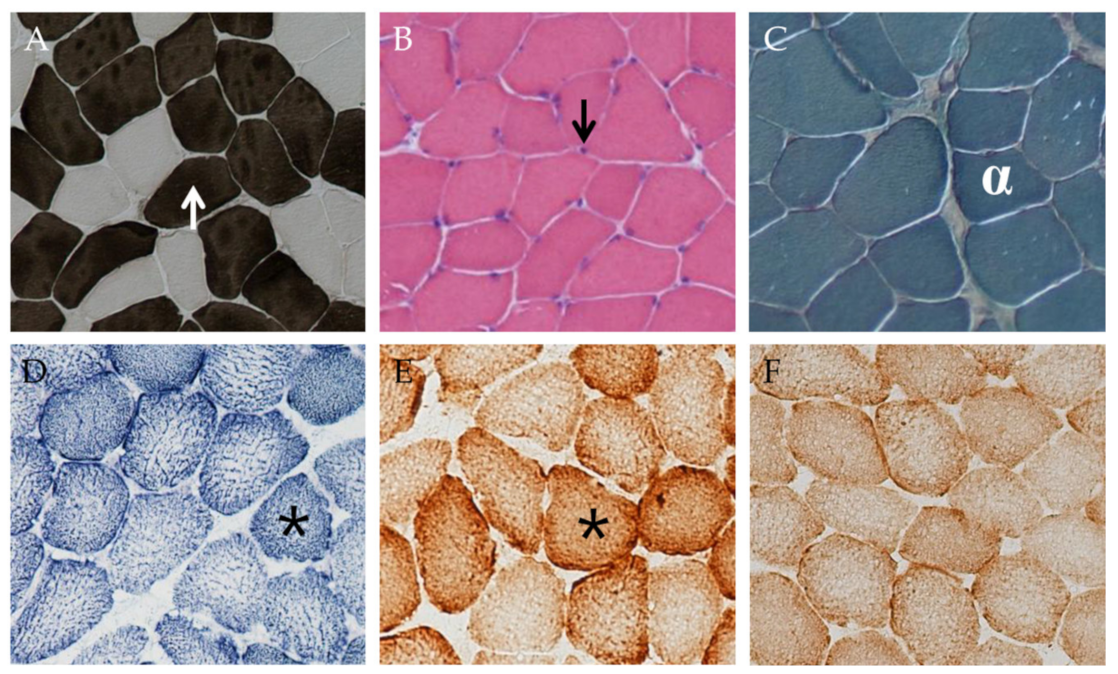
3.5. Muscle Morphology
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Williams, R.S. Mitochondrial gene expression in mammalian striated muscle. Evidence that variation in gene dosage is the major regulatory event. J. Biol. Chem. 1986, 261, 12390–12394. [Google Scholar] [PubMed]
- Menshikova, E.V.; Ritov, V.B.; Toledo, F.G.; Ferrell, R.E.; Goodpaster, B.H.; Kelley, D.E. Effects of weight loss and physical activity on skeletal muscle mitochondrial function in obesity. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E818–E825. [Google Scholar] [CrossRef] [PubMed]
- Menshikova, E.V.; Ritov, V.B.; Ferrell, R.E.; Azuma, K.; Goodpaster, B.H.; Kelley, D.E. Characteristics of skeletal muscle mitochondrial biogenesis induced by moderate-intensity exercise and weight loss in obesity. J. Appl. Physiol. 2007, 103, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Toledo, F.G.; Menshikova, E.V.; Azuma, K.; Radikova, Z.; Kelley, C.A.; Ritov, V.B.; Kelley, D.E. Mitochondrial capacity in skeletal muscle is not stimulated by weight loss despite increases in insulin action and decreases in intramyocellular lipid content. Diabetes 2008, 57, 987–994. [Google Scholar] [CrossRef]
- Boushel, R.; Gnaiger, E.; Schjerling, P.; Skovbro, M.; Kraunsoe, R.; Dela, F. Patients with type 2 diabetes have normal mitochondrial function in skeletal muscle. Diabetologia 2007, 50, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Phielix, E.; Schrauwen-Hinderling, V.B.; Mensink, M.; Lenaers, E.; Meex, R.; Hoeks, J.; Kooi, M.E.; Moonen-Kornips, E.; Sels, J.P.; Hesselink, M.K.; et al. Lower intrinsic ADP-stimulated mitochondrial respiration underlies in vivo mitochondrial dysfunction in muscle of male type 2 diabetic patients. Diabetes 2008, 57, 2943–2949. [Google Scholar] [CrossRef]
- Ritov, V.B.; Menshikova, E.V.; He, J.; Ferrell, R.E.; Goodpaster, B.H.; Kelley, D.E. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes 2005, 54, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Ritov, V.B.; Menshikova, E.V.; Azuma, K.; Wood, R.; Toledo, F.G.; Goodpaster, B.H.; Ruderman, N.B.; Kelley, D.E. Deficiency of electron transport chain in human skeletal muscle mitochondria in type 2 diabetes mellitus and obesity. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E49–E58. [Google Scholar] [CrossRef]
- Balakrishnan, V.S.; Rao, M.; Menon, V.; Gordon, P.L.; Pilichowska, M.; Castaneda, F.; Castaneda-Sceppa, C. Resistance training increases muscle mitochondrial biogenesis in patients with chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 996–1002. [Google Scholar] [CrossRef]
- Puente-Maestu, L.; Lazaro, A.; Tejedor, A.; Camano, S.; Fuentes, M.; Cuervo, M.; Navarro, B.O.; Agusti, A. Effects of exercise on mitochondrial DNA content in skeletal muscle of patients with COPD. Thorax 2011, 66, 121–127. [Google Scholar] [CrossRef]
- Puntschart, A.; Claassen, H.; Jostarndt, K.; Hoppeler, H.; Billeter, R. mRNAs of enzymes involved in energy metabolism and mtDNA are increased in endurance-trained athletes. Am. J. Physiol. 1995, 269, C619–C625. [Google Scholar] [CrossRef]
- Costford, S.R.; Bajpeyi, S.; Pasarica, M.; Albarado, D.C.; Thomas, S.C.; Xie, H.; Church, T.S.; Jubrias, S.A.; Conley, K.E.; Smith, S.R. Skeletal muscle NAMPT is induced by exercise in humans. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E117–E126. [Google Scholar] [CrossRef]
- Murakami, T.; Shimomura, Y.; Fujitsuka, N.; Nakai, N.; Sugiyama, S.; Ozawa, T.; Sokabe, M.; Horai, S.; Tokuyama, K.; Suzuki, M. Enzymatic and genetic adaptation of soleus muscle mitochondria to physical training in rats. Am. J. Physiol. 1994, 267, E388–E395. [Google Scholar] [CrossRef]
- Chow, L.S.; Greenlund, L.J.; Asmann, Y.W.; Short, K.R.; McCrady, S.K.; Levine, J.A.; Nair, K.S. Impact of endurance training on murine spontaneous activity, muscle mitochondrial DNA abundance, gene transcripts, and function. J. Appl. Physiol. 2007, 102, 1078–1089. [Google Scholar] [CrossRef]
- Crane, J.D.; Abadi, A.; Hettinga, B.P.; Ogborn, D.I.; MacNeil, L.G.; Steinberg, G.R.; Tarnopolsky, M.A. Elevated mitochondrial oxidative stress impairs metabolic adaptations to exercise in skeletal muscle. PLoS ONE 2013, 8, e81879. [Google Scholar] [CrossRef]
- Civitarese, A.E.; Carling, S.; Heilbronn, L.K.; Hulver, M.H.; Ukropcova, B.; Deutsch, W.A.; Smith, S.R.; Ravussin, E. Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. 2007, 4, e76. [Google Scholar] [CrossRef]
- Zoladz, J.A.; Grassi, B.; Majerczak, J.; Szkutnik, Z.; Korostynski, M.; Grandys, M.; Jarmuszkiewicz, W.; Korzeniewski, B. Mechanisms responsible for the acceleration of pulmonary VO2 on-kinetics in humans after prolonged endurance training. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R1101–R1114. [Google Scholar] [CrossRef]
- Menshikova, E.V.; Ritov, V.B.; Fairfull, L.; Ferrell, R.E.; Kelley, D.E.; Goodpaster, B.H. Effects of exercise on mitochondrial content and function in aging human skeletal muscle. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 534–540. [Google Scholar] [CrossRef]
- Perry, C.G.; Lally, J.; Holloway, G.P.; Heigenhauser, G.J.; Bonen, A.; Spriet, L.L. Repeated transient mRNA bursts precede increases in transcriptional and mitochondrial proteins during training in human skeletal muscle. J. Physiol. 2010, 588, 4795–4810. [Google Scholar] [CrossRef]
- Freyssenet, D.; Berthon, P.; Denis, C. Mitochondrial biogenesis in skeletal muscle in response to endurance exercises. Arch. Physiol. Biochem. 1996, 104, 129–141. [Google Scholar] [CrossRef]
- Jeppesen, T.D.; Schwartz, M.; Olsen, D.B.; Wibrand, F.; Krag, T.; Duno, M.; Hauerslev, S.; Vissing, J. Aerobic training is safe and improves exercise capacity in patients with mitochondrial myopathy. Brain 2006, 129, 3402–3412. [Google Scholar] [CrossRef]
- Yeo, W.K.; Paton, C.D.; Garnham, A.P.; Burke, L.M.; Carey, A.L.; Hawley, J.A. Skeletal muscle adaptation and performance responses to once a day versus twice every second day endurance training regimens. J. Appl. Physiol. 2008, 105, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Egan, B.; O’Connor, P.L.; Zierath, J.R.; O’Gorman, D.J. Time course analysis reveals gene-specific transcript and protein kinetics of adaptation to short-term aerobic exercise training in human skeletal muscle. PLoS ONE 2013, 8, e74098. [Google Scholar] [CrossRef] [PubMed]
- Pesta, D.; Hoppel, F.; Macek, C.; Messner, H.; Faulhaber, M.; Kobel, C.; Parson, W.; Burtscher, M.; Schocke, M.; Gnaiger, E. Similar qualitative and quantitative changes of mitochondrial respiration following strength and endurance training in normoxia and hypoxia in sedentary humans. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1078–R1087. [Google Scholar] [CrossRef]
- Andersen, P.; Adams, R.P.; Sjogaard, G.; Thorboe, A.; Saltin, B. Dynamic knee extension as model for study of isolated exercising muscle in humans. J. Appl. Physiol. 1985, 59, 1647–1653. [Google Scholar] [CrossRef]
- Wibrand, F.; Jeppesen, T.D.; Frederiksen, A.L.; Olsen, D.B.; Duno, M.; Schwartz, M.; Vissing, J. Limited diagnostic value of enzyme analysis in patients with mitochondrial tRNA mutations. Muscle Nerve 2010, 41, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Krag, T.O.; Pinos, T.; Nielsen, T.L.; Brull, A.; Andreu, A.L.; Vissing, J. Differential Muscle Involvement in Mice and Humans Affected by McArdle Disease. J. Neuropathol. Exp. Neurol. 2016, 75, 441–454. [Google Scholar] [CrossRef]
- Kiens, B.; Essen-Gustavsson, B.; Christensen, N.J.; Saltin, B. Skeletal muscle substrate utilization during submaximal exercise in man: Effect of endurance training. J. Physiol. 1993, 469, 459–478. [Google Scholar] [CrossRef]
- Klausen, K.; Andersen, L.B.; Pelle, I. Adaptive changes in work capacity, skeletal muscle capillarization and enzyme levels during training and detraining. Acta Physiol. Scand. 1981, 113, 9–16. [Google Scholar] [CrossRef]
- Miyachi, M.; Tanaka, H.; Yamamoto, K.; Yoshioka, A.; Takahashi, K.; Onodera, S. Effects of one-legged endurance training on femoral arterial and venous size in healthy humans. J. Appl. Physiol. 2001, 90, 2439–2444. [Google Scholar] [CrossRef]
- Ray, C.A. Sympathetic adaptations to one-legged training. J. Appl. Physiol. 1999, 86, 1583–1587. [Google Scholar] [CrossRef] [PubMed]
- Saltin, B.; Nazar, K.; Costill, D.L.; Stein, E.; Jansson, E.; Essen, B.; Gollnick, D. The nature of the training response; peripheral and central adaptations of one-legged exercise. Acta Physiol. Scand. 1976, 96, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Blomstrand, E.; Radegran, G.; Saltin, B. Maximum rate of oxygen uptake by human skeletal muscle in relation to maximal activities of enzymes in the Krebs cycle. J. Physiol. 1997, 501, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, U.F.; Rasmussen, H.N.; Krustrup, P.; Quistorff, B.; Saltin, B.; Bangsbo, J. Aerobic metabolism of human quadriceps muscle: In vivo data parallel measurements on isolated mitochondria. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E301–E307. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.L.; Hoppeler, H.; Claassen, H.; Vock, P.; Kayser, B.; Schena, F.; Ferretti, G. Effects of endurance training on oxidative capacity and structural composition of human arm and leg muscles. Acta Physiol. Scand. 1997, 161, 459–464. [Google Scholar] [CrossRef]
- Hoppeler, H.; Howald, H.; Conley, K.; Lindstedt, S.L.; Claassen, H.; Vock, P.; Weibel, E.R. Endurance training in humans: Aerobic capacity and structure of skeletal muscle. J. Appl. Physiol. 1985, 59, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Holloszy, J.O. Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J. Biol. Chem. 1967, 242, 2278–2282. [Google Scholar]
- Tarnopolsky, M.A.; Rennie, C.D.; Robertshaw, H.A.; Fedak-Tarnopolsky, S.N.; Devries, M.C.; Hamadeh, M.J. Influence of endurance exercise training and sex on intramyocellular lipid and mitochondrial ultrastructure, substrate use, and mitochondrial enzyme activity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1271–R1278. [Google Scholar] [CrossRef]
- Marcuello, A.; Gonzalez-Alonso, J.; Calbet, J.A.; Damsgaard, R.; Lopez-Perez, M.J.; Diez-Sanchez, C. Skeletal muscle mitochondrial DNA content in exercising humans. J. Appl. Physiol. 2005, 99, 1372–1377. [Google Scholar] [CrossRef]
- Larsen, S.; Nielsen, J.; Hansen, C.N.; Nielsen, L.B.; Wibrand, F.; Stride, N.; Schroder, H.D.; Boushel, R.; Helge, J.W.; Dela, F.; et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 2012, 590, 3349–3360. [Google Scholar] [CrossRef]
- Neufer, P.D.; Dohm, G.L. Exercise induces a transient increase in transcription of the GLUT-4 gene in skeletal muscle. Am. J. Physiol. 1993, 265, C1597–C1603. [Google Scholar] [CrossRef]
- Miller, F.J.; Rosenfeldt, F.L.; Zhang, C.; Linnane, A.W.; Nagley, P. Precise determination of mitochondrial DNA copy number in human skeletal and cardiac muscle by a PCR-based assay: Lack of change of copy number with age. Nucleic Acids Res. 2003, 31, e61. [Google Scholar] [CrossRef]
- Grady, J.P.; Murphy, J.L.; Blakely, E.L.; Haller, R.G.; Taylor, R.W.; Turnbull, D.M.; Tuppen, H.A. Accurate measurement of mitochondrial DNA deletion level and copy number differences in human skeletal muscle. PLoS ONE 2014, 9, e114462. [Google Scholar] [CrossRef]
- Coyle, E.F.; Martin, W.H.; Sinacore, D.R., III; Joyner, M.J.; Hagberg, J.M.; Holloszy, J.O. Time course of loss of adaptations after stopping prolonged intense endurance training. J. Appl. Physiol. 1984, 57, 1857–1864. [Google Scholar] [CrossRef]
- Coyle, E.F.; Martin, W.H.; Bloomfield, S.A., III; Lowry, O.H.; Holloszy, J.O. Effects of detraining on responses to submaximal exercise. J. Appl. Physiol. 1985, 59, 853–859. [Google Scholar] [CrossRef]
- van, M.D.; Hansen, J.; Havekes, B.; Scheer, F.A.; Jorgensen, J.A.; Hoeks, J.; Schrauwen-Hinderling, V.B.; Duez, H.; Lefebvre, P.; Schaper, N.C.; et al. Demonstration of a day-night rhythm in human skeletal muscle oxidative capacity. Mol. Metab. 2016, 5, 635–645. [Google Scholar]
- Pfeiffer, K.; Gohil, V.; Stuart, R.A.; Hunte, C.; Brandt, U.; Greenberg, M.L.; Schagger, H. Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem. 2003, 278, 52873–52880. [Google Scholar] [CrossRef]
- Menshikova, E.V.; Ritov, V.B.; Dube, J.J.; Amati, F.; Stefanovic-Racic, M.; Toledo, F.G.S.; Coen, P.M.; Goodpaster, B.H. Calorie Restriction-induced Weight Loss and Exercise Have Differential Effects on Skeletal Muscle Mitochondria Despite Similar Effects on Insulin Sensitivity. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 73, 81–87. [Google Scholar] [CrossRef]
- Nielsen, J.; Gejl, K.D.; Hey-Mogensen, M.; Holmberg, H.C.; Suetta, C.; Krustrup, P.; Elemans, C.P.H.; Ortenblad, N. Plasticity in mitochondrial cristae density allows metabolic capacity modulation in human skeletal muscle. J. Physiol. 2017, 595, 2839–2847. [Google Scholar] [CrossRef]
- Meinild Lundby, A.K.; Jacobs, R.A.; Gehrig, S.; de, L.J.; Hauser, M.; Bonne, T.C.; Fluck, D.; Dandanell, S.; Kirk, N.; Kaech, A.; et al. Exercise training increases skeletal muscle mitochondrial volume density by enlargement of existing mitochondria and not de novo biogenesis. Acta Physiol. 2017, 222, e12905. [Google Scholar] [CrossRef]
- Spina, R.J.; Chi, M.M.; Hopkins, M.G.; Nemeth, P.M.; Lowry, O.H.; Holloszy, J.O. Mitochondrial enzymes increase in muscle in response to 7–10 days of cycle exercise. J. Appl. Physiol. 1996, 80, 2250–2254. [Google Scholar] [CrossRef]
- Olesen, J.; Kiilerich, K.; Pilegaard, H. PGC−1alpha-mediated adaptations in skeletal muscle. Pflugers Arch. 2010, 460, 153–162. [Google Scholar] [CrossRef]
- Fischer, B.; Schottl, T.; Schempp, C.; Fromme, T.; Hauner, H.; Klingenspor, M.; Skurk, T. Inverse relationship between body mass index and mitochondrial oxidative phosphorylation capacity in human subcutaneous adipocytes. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E380–E387. [Google Scholar] [CrossRef]
- Fisher-Wellman, K.H.; Weber, T.M.; Cathey, B.L.; Brophy, P.M.; Gilliam, L.A.; Kane, C.L.; Maples, J.M.; Gavin, T.P.; Houmard, J.A.; Neufer, P.D. Mitochondrial respiratory capacity and content are normal in young insulin-resistant obese humans. Diabetes 2014, 63, 132–141. [Google Scholar] [CrossRef]
- Toledo, F.G.; Watkins, S.; Kelley, D.E. Changes induced by physical activity and weight loss in the morphology of intermyofibrillar mitochondria in obese men and women. J. Clin. Endocrinol. Metab. 2006, 91, 3224–3227. [Google Scholar] [CrossRef]
- Hey-Mogensen, M.; Hojlund, K.; Vind, B.F.; Wang, L.; Dela, F.; Beck-Nielsen, H.; Fernstrom, M.; Sahlin, K. Effect of physical training on mitochondrial respiration and reactive oxygen species release in skeletal muscle in patients with obesity and type 2 diabetes. Diabetologia 2010, 53, 1976–1985. [Google Scholar] [CrossRef]
- Meex, R.C.; Schrauwen-Hinderling, V.B.; Moonen-Kornips, E.; Schaart, G.; Mensink, M.; Phielix, E.; van de Weijer, T.; Sels, J.P.; Schrauwen, P.; Hesselink, M.K. Restoration of muscle mitochondrial function and metabolic flexibility in type 2 diabetes by exercise training is paralleled by increased myocellular fat storage and improved insulin sensitivity. Diabetes 2010, 59, 572–579. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subject Characteristics | Mean ± SD | Range |
|---|---|---|
| Age, yr | 22.7 ± 2.8 | (18–27) |
| Body mass, kg | 74.2 ± 15.7 | (60–107) |
| Body fat, % | 20.1 ± 7.2 | (10.8–37.2) |
| BMI, kg∙m−2 | 22 ± 4.2 | (18.3–32.7) |
| VO2,max, ml·kg BM−1·min−1 | 46.8 ± 6.8 | (34.4–53.4) |
| Morphological Data | PRE TL | POST TL | Post UTL | Decon TL |
|---|---|---|---|---|
| Fiber type I, % | 38.79 ± 5.32 | 43 ± 5.32 | 47.65 ± 5.59 | 35.72 ± 3.27 |
| Fiber type II % | 59.21 ± 5.32 | 57 ± 5.32 | 52.35 ± 5.59 | 64.28 ± 3.27 |
| Fiber size type I, μm2 | 3432 ± 739 | 4652 ± 556 | 4093 ± 262 | 4572 ± 741 |
| Fiber size type II, μm2 | 3562 ± 669 | 4875 ± 669 | 4998 ± 523 | 4946 ± 414 |
| Central nuclei, % | 0.65 ± 1.11 | 0.25 ± 1.11 | 0.68 ± 2.02 | 1.44 ± 1.84 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fritzen, A.M.; Thøgersen, F.B.; Thybo, K.; Vissing, C.R.; Krag, T.O.; Ruiz-Ruiz, C.; Risom, L.; Wibrand, F.; Høeg, L.D.; Kiens, B.; et al. Adaptations in Mitochondrial Enzymatic Activity Occurs Independent of Genomic Dosage in Response to Aerobic Exercise Training and Deconditioning in Human Skeletal Muscle. Cells 2019, 8, 237. https://doi.org/10.3390/cells8030237
Fritzen AM, Thøgersen FB, Thybo K, Vissing CR, Krag TO, Ruiz-Ruiz C, Risom L, Wibrand F, Høeg LD, Kiens B, et al. Adaptations in Mitochondrial Enzymatic Activity Occurs Independent of Genomic Dosage in Response to Aerobic Exercise Training and Deconditioning in Human Skeletal Muscle. Cells. 2019; 8(3):237. https://doi.org/10.3390/cells8030237
Chicago/Turabian StyleFritzen, Andreas M., Frank B. Thøgersen, Kasper Thybo, Christoffer R. Vissing, Thomas O. Krag, Cristina Ruiz-Ruiz, Lotte Risom, Flemming Wibrand, Louise D. Høeg, Bente Kiens, and et al. 2019. "Adaptations in Mitochondrial Enzymatic Activity Occurs Independent of Genomic Dosage in Response to Aerobic Exercise Training and Deconditioning in Human Skeletal Muscle" Cells 8, no. 3: 237. https://doi.org/10.3390/cells8030237
APA StyleFritzen, A. M., Thøgersen, F. B., Thybo, K., Vissing, C. R., Krag, T. O., Ruiz-Ruiz, C., Risom, L., Wibrand, F., Høeg, L. D., Kiens, B., Duno, M., Vissing, J., & Jeppesen, T. D. (2019). Adaptations in Mitochondrial Enzymatic Activity Occurs Independent of Genomic Dosage in Response to Aerobic Exercise Training and Deconditioning in Human Skeletal Muscle. Cells, 8(3), 237. https://doi.org/10.3390/cells8030237