Niclosamide Triggers Non-Canonical LC3 Lipidation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Antibodies
2.2. Cell Culture and Transfection
2.3. Immunoblot Assay
2.4. Immunostaining Assay
2.5. Molecule Capture
2.6. Statistical Analysis
3. Results
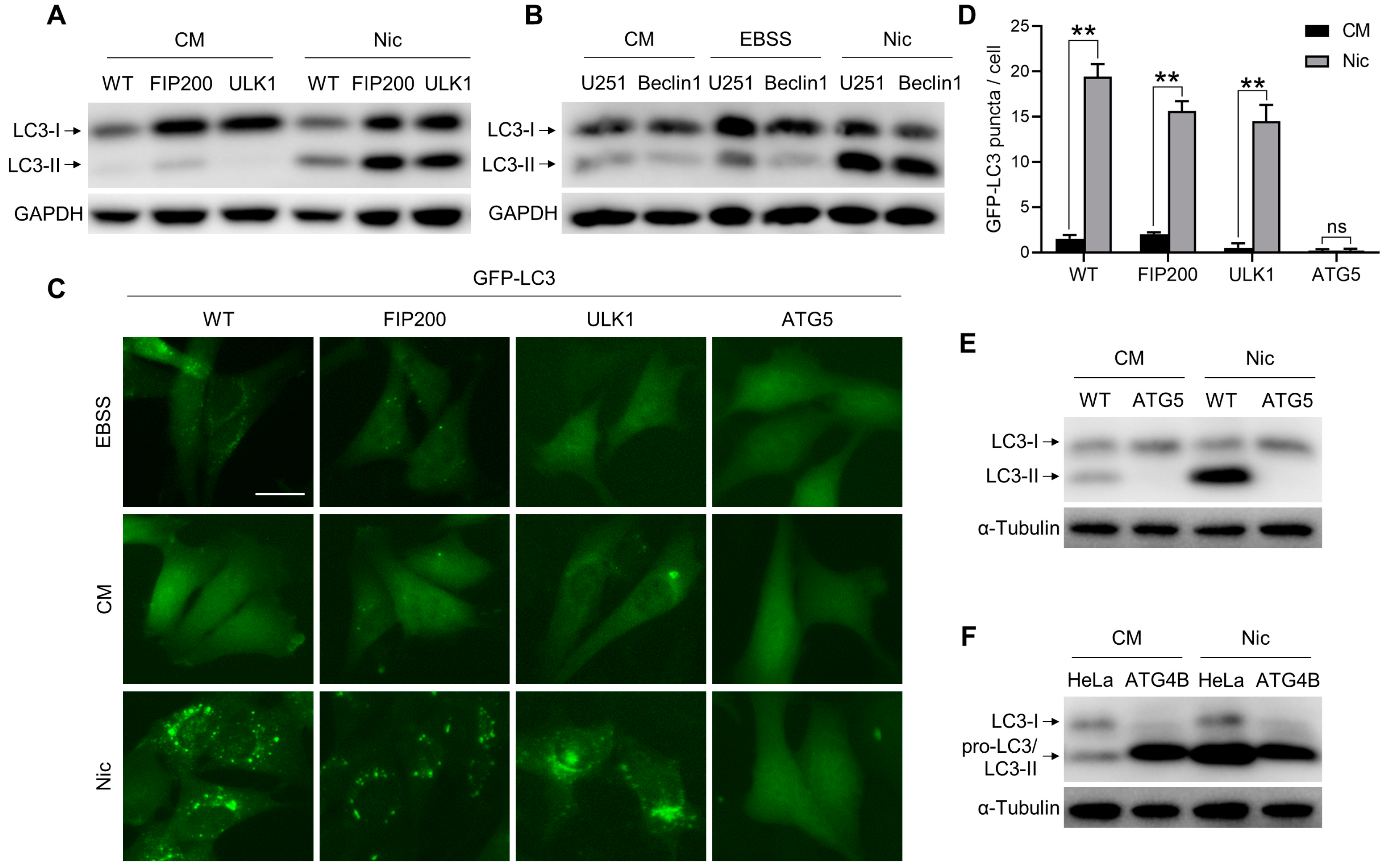
3.1. Nic-Induced NCLL Is Independent of ULK1 Complex and Beclin 1 Complex, But Dependent on Ubiquitin-Like Conjugation Systems
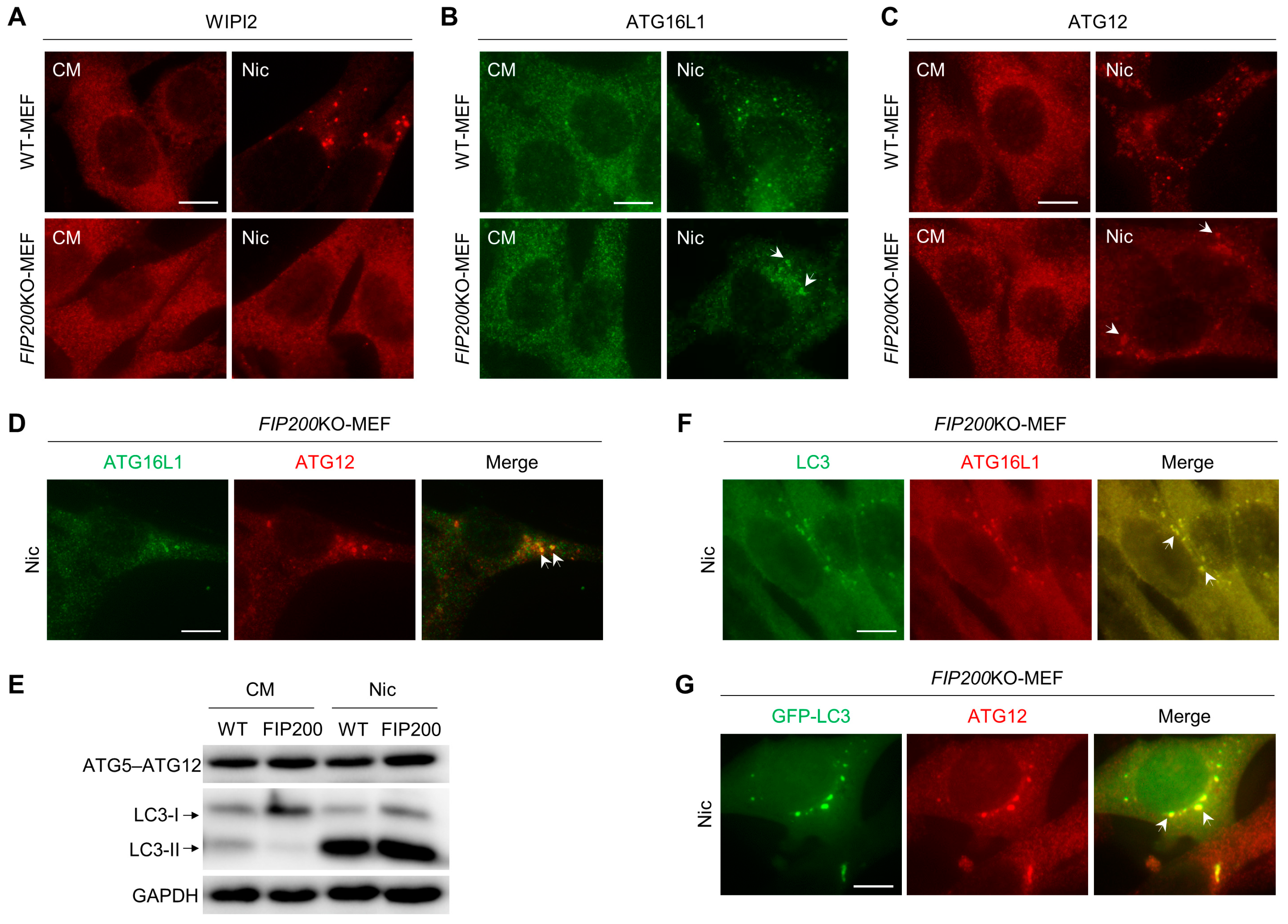
3.2. Nic Recruits the ATG12-ATG5-ATG16L1 Complex via a WIPI2-Independent Pathway
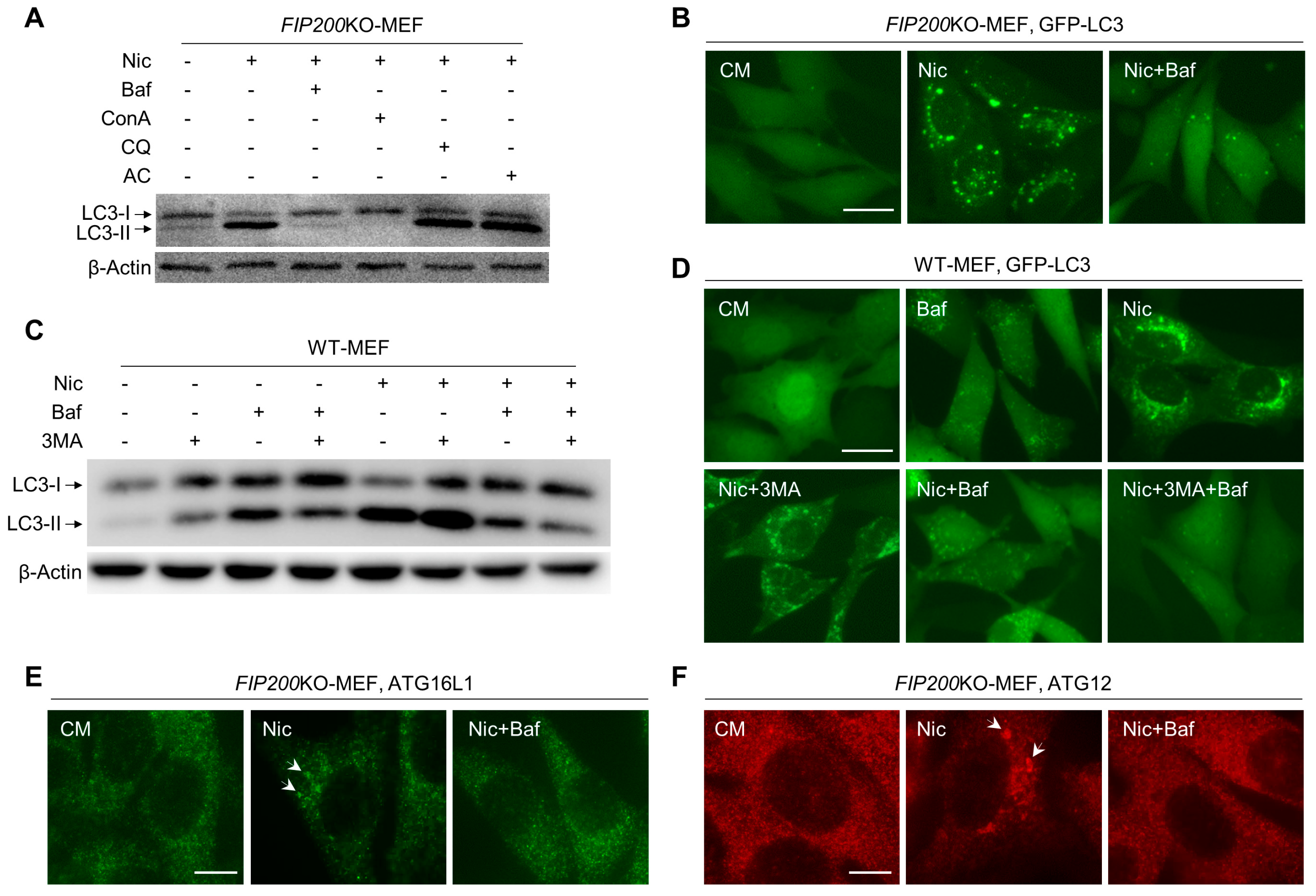
3.3. Baf Is Able to Inhibit Nic-Induced NCLL
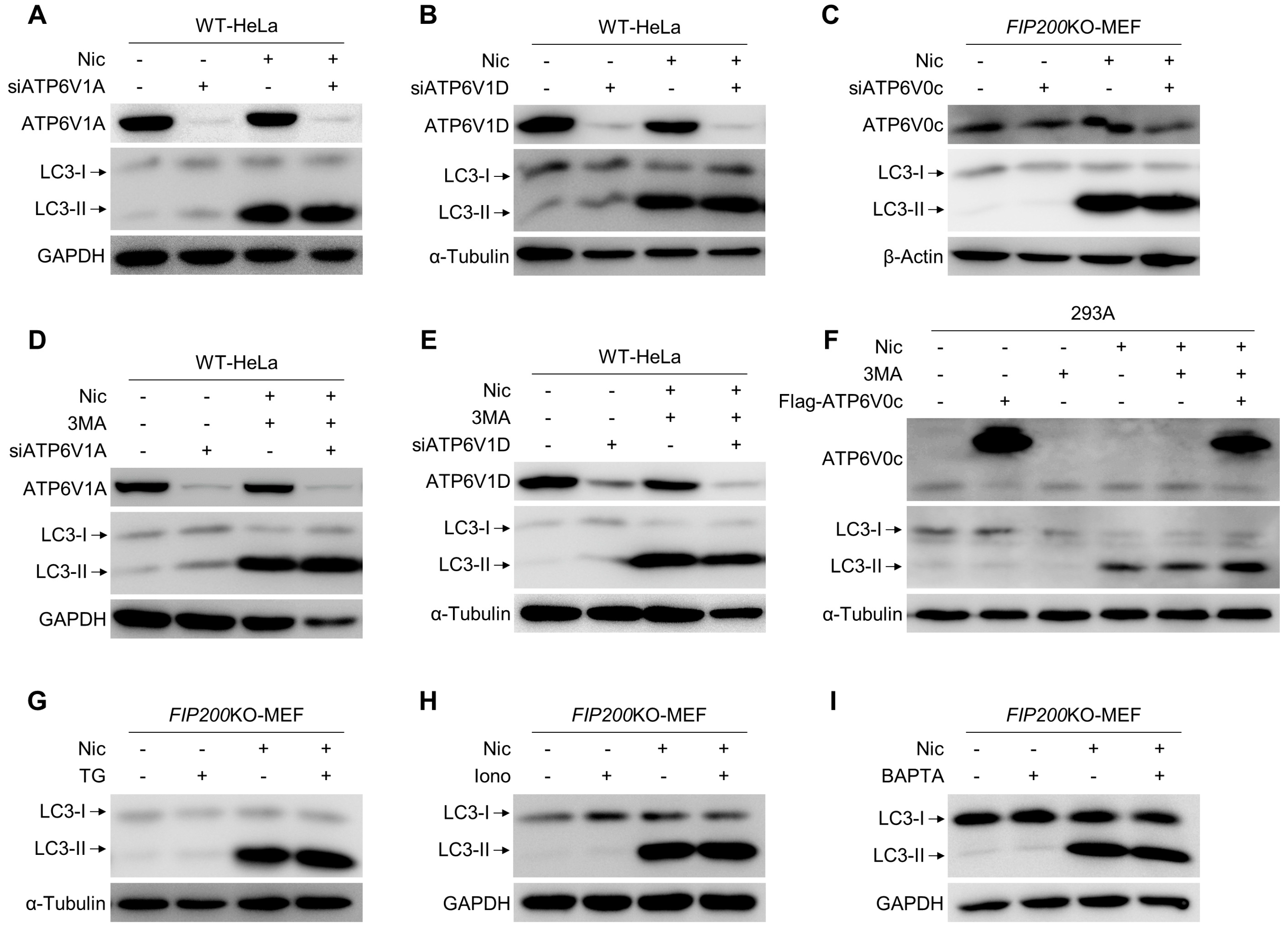
3.4. Nic-Induced NCLL Is Independent of V-ATPase and Ca2+
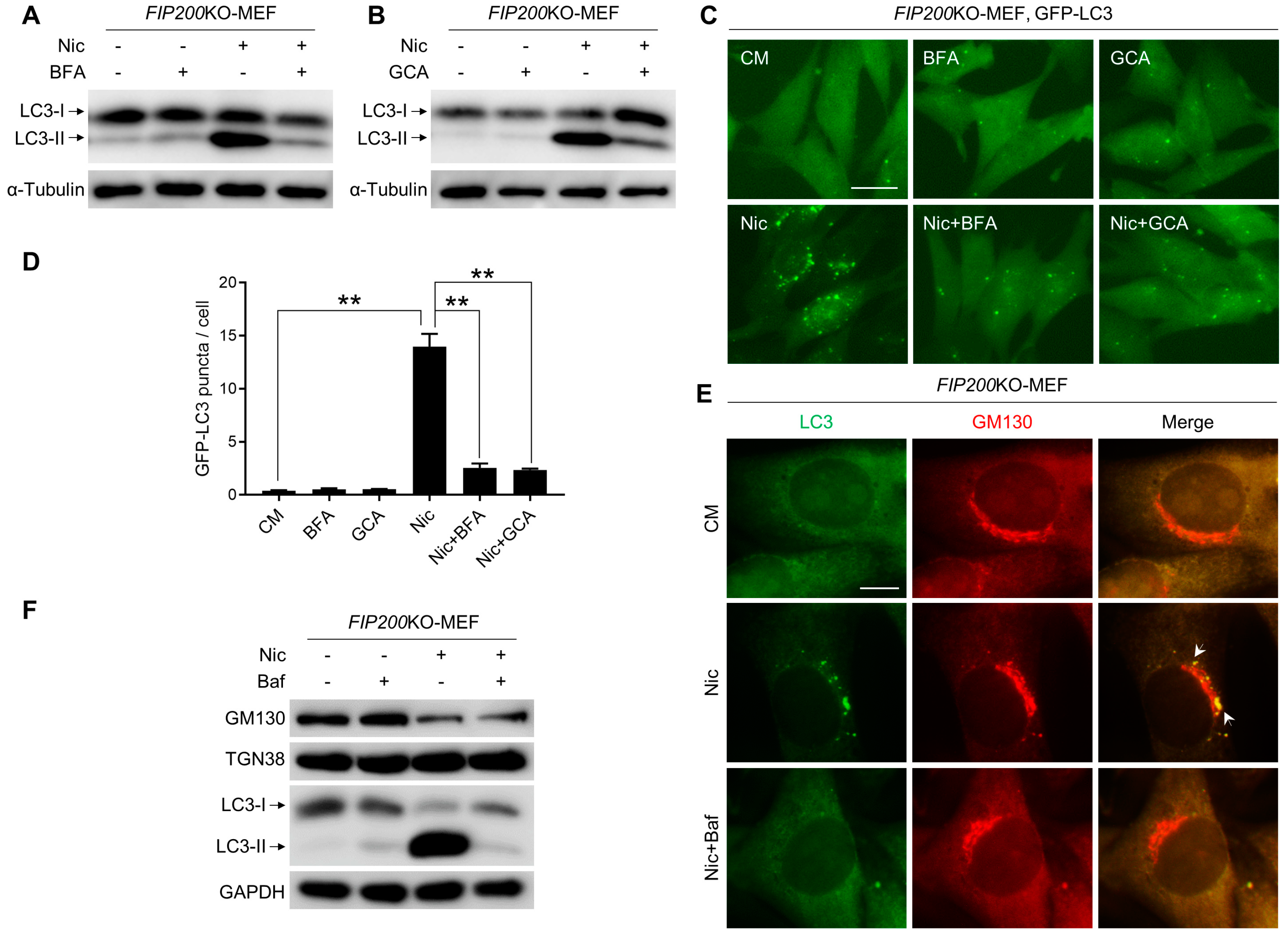
3.5. Nic-Induced NCLL Requires Intact Golgi Complex
3.6. Vimentin Is Involved in Nic-Induced NCLL
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug. Discov. 2018. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, P.K.; Roberts, M.J.; Arend, R.C.; Samant, R.S.; Buchsbaum, D.J. Multi-targeted therapy of cancer by niclosamide: A new application for an old drug. Cancer Lett. 2014, 349, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Mook, R.A., Jr.; Premont, R.T.; Wang, J. Niclosamide: Beyond an antihelminthic drug. Cell Signal. 2018, 41, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Khambu, B.; Zhang, H.; Kang, J.H.; Chen, X.; Chen, D.; Vollmer, L.; Liu, P.Q.; Vogt, A.; Yin, X.M. Suppression of lysosome function induces autophagy via a feedback down-regulation of MTOR complex 1 (MTORC1) activity. J. Biol. Chem. 2013, 288, 35769–35780. [Google Scholar] [CrossRef] [PubMed]
- Balgi, A.D.; Fonseca, B.D.; Donohue, E.; Tsang, T.C.; Lajoie, P.; Proud, C.G.; Nabi, I.R.; Roberge, M. Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. PLoS ONE 2009, 4, e7124. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ren, X.R.; Piao, H.; Zhao, S.; Osada, T.; Premont, R.T.; Mook, R.A., Jr.; Morse, M.A.; Lyerly, H.K.; Chen, W. Niclosamide-induced wnt signaling inhibition in colorectal cancer is mediated by autophagy. Biochem. J. 2019, 476, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and diversity in autophagy mechanisms: Lessons from yeast. Nat. Rev. Mol. Cell Biol. 2009, 10, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 2018, 20, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Packer, M.; Codogno, P. Development of autophagy inducers in clinical medicine. J. Clin. Investig. 2015, 125, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Codogno, P.; Mehrpour, M.; Proikas-Cezanne, T. Canonical and non-canonical autophagy: Variations on a common theme of self-eating? Nat. Rev. Mol. Cell Bio. 2012, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.H.; Horbinski, C.; Guo, F.; Watkins, S.; Uchiyama, Y.; Chu, C.T. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyI-4-phenylpyridinium-induced cell death. Am. J. Pathol. 2007, 170, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Lin, J.; Zhou, J.; Wang, X.; Li, Y.; Ren, X.; Yu, W.; Zhong, W.; Xiao, J.; Sheng, F.; et al. Beclin 1-independent autophagy induced by a Bcl-XL/Bcl-2 targeting compound, Z18. Autophagy 2010, 6, 1032–1041. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Bauvy, C.; Souquere, S.; Tonelli, G.; Liu, L.; Zhu, Y.; Qiao, Z.; Bakula, D.; Proikas-Cezanne, T.; Pierron, G.; et al. The Bcl-2 homology domain 3 mimetic gossypol induces both Beclin 1-dependent and Beclin 1-independent cytoprotective autophagy in cancer cells. J. Biol. Chem. 2010, 285, 25570–25581. [Google Scholar] [CrossRef] [PubMed]
- Grishchuk, Y.; Ginet, V.; Truttmann, A.C.; Clarke, P.G.; Puyal, J. Beclin 1-independent autophagy contributes to apoptosis in cortical neurons. Autophagy 2011, 7, 1115–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheong, H.; Lindsten, T.; Wu, J.; Lu, C.; Thompson, C.B. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc. Natl. Acad. Sci. USA 2011, 108, 11121–11126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, J. LAP it up, fuzz ball: A short history of LC3-associated phagocytosis. Curr. Opin. Immunol. 2018, 55, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Hong, L.; Xu, J.; Zhong, G.; Gu, Q.; Gu, Q.; Guan, Y.; Zheng, X.; Dai, Q.; Luo, X.; et al. Discovery of a small molecule targeting autophagy via ATG4B inhibition and cell death of colorectal cancer cells in vitro and in vivo. Autophagy 2018, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Liu, Y.; Hong, L.; Yang, Z.; Cai, X.; Chen, X.; Fu, Y.; Lin, Y.; Wen, W.; Li, S.; et al. Golgi-associated LC3 lipidation requires V-ATPase in noncanonical autophagy. Cell Death Dis. 2016, 7, e2330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Zhang, B.; Yang, M.; Zhu, J.; Li, H. Systematic Profiling of Histone Readers in Arabidopsis thaliana. Cell Rep. 2018, 22, 1090–1102. [Google Scholar] [CrossRef]
- Dall’Armi, C.; Devereaux, K.A.; Di Paolo, G. The role of lipids in the control of autophagy. Curr. Biol. 2013, 23, R33–R45. [Google Scholar] [CrossRef] [PubMed]
- Dooley, H.C.; Razi, M.; Polson, H.E.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol. Cell 2014, 55, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed]
- Drose, S.; Altendorf, K. Bafilomycins and concanamycins as inhibitors of V-ATPases and P-ATPases. J. Exp. Biol. 1997, 200, 1–8. [Google Scholar] [PubMed]
- Bowman, E.J.; Siebers, A.; Altendorf, K. Bafilomycins: A class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc. Natl. Acad. Sci. USA 1988, 85, 7972–7976. [Google Scholar] [CrossRef] [PubMed]
- Lytton, J.; Westlin, M.; Hanley, M.R. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J. Biol. Chem. 1991, 266, 17067–17071. [Google Scholar] [PubMed]
- Ganley, I.G.; Wong, P.M.; Gammoh, N.; Jiang, X. Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol. Cell 2011, 42, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Niso-Santano, M.; Malik, S.A.; Pietrocola, F.; Bravo-San Pedro, J.M.; Marino, G.; Cianfanelli, V.; Ben-Younes, A.; Troncoso, R.; Markaki, M.; Sica, V.; et al. Unsaturated fatty acids induce non-canonical autophagy. EMBO J. 2015, 34, 1025–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florey, O.; Gammoh, N.; Kim, S.E.; Jiang, X.; Overholtzer, M. V-ATPase and osmotic imbalances activate endolysosomal LC3 lipidation. Autophagy 2015, 11, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Chen, X.; Li, M.; Zhang, H.; Ding, W.X.; Yin, X.M. CCCP-Induced LC3 lipidation depends on Atg9 whereas FIP200/Atg13 and Beclin 1/Atg14 are dispensable. Biochem. Biophys. Res. Commun. 2013, 432, 226–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Wang, L.; Hasegawa, H.; Amin, P.; Han, B.X.; Kaneko, S.; He, Y.; Wang, F. Deletion of PIK3C3/Vps34 in sensory neurons causes rapid neurodegeneration by disrupting the endosomal but not the autophagic pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 9424–9429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLeod, I.X.; Zhou, X.; Li, Q.J.; Wang, F.; He, Y.W. The class III kinase Vps34 promotes T lymphocyte survival through regulating IL-7Ralpha surface expression. J. Immunol. 2011, 187, 5051–5061. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Saitoh, T.; Kageyama, S.; Akira, S.; Noda, T.; Yoshimori, T. Differential involvement of Atg16L1 in Crohn disease and canonical autophagy: Analysis of the organization of the Atg16L1 complex in fibroblasts. J. Biol. Chem. 2009, 284, 32602–32609. [Google Scholar] [CrossRef] [PubMed]
- Gammoh, N.; Florey, O.; Overholtzer, M.; Jiang, X. Interaction between FIP200 and ATG16L1 distinguishes ULK1 complex-dependent and -independent autophagy. Nat. Struct. Mol. Biol. 2013, 20, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, K.; Ulferts, R.; Jacquin, E.; Veith, T.; Gammoh, N.; Arasteh, J.M.; Mayer, U.; Carding, S.R.; Wileman, T.; Beale, R.; et al. The WD40 domain of ATG16L1 is required for its non-canonical role in lipidation of LC3 at single membranes. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.M. The Golgi complex: A common platform for canonical and non-canonical autophagy? Cell Cycle 2013, 12, 12. [Google Scholar] [CrossRef] [PubMed]
- Naydenov, N.G.; Harris, G.; Morales, V.; Ivanov, A.I. Loss of a membrane trafficking protein alphaSNAP induces non-canonical autophagy in human epithelia. Cell Cycle 2012, 11, 4613–4625. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Mei, M.; Li, Q.; Roboti, P.; Pang, Q.; Ying, Z.; Gao, F.; Lowe, M.; Bao, S. Loss of the golgin GM130 causes Golgi disruption, Purkinje neuron loss, and ataxia in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 346–351. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Luo, X.; Shan, H.; Fu, Y.; Gu, Q.; Zheng, X.; Dai, Q.; Xia, F.; Zheng, Z.; Liu, P.; et al. Niclosamide Triggers Non-Canonical LC3 Lipidation. Cells 2019, 8, 248. https://doi.org/10.3390/cells8030248
Liu Y, Luo X, Shan H, Fu Y, Gu Q, Zheng X, Dai Q, Xia F, Zheng Z, Liu P, et al. Niclosamide Triggers Non-Canonical LC3 Lipidation. Cells. 2019; 8(3):248. https://doi.org/10.3390/cells8030248
Chicago/Turabian StyleLiu, Yajun, Xia Luo, Hao Shan, Yuanyuan Fu, Qianqian Gu, Xueping Zheng, Qi Dai, Fan Xia, Zhihua Zheng, Peiqing Liu, and et al. 2019. "Niclosamide Triggers Non-Canonical LC3 Lipidation" Cells 8, no. 3: 248. https://doi.org/10.3390/cells8030248
APA StyleLiu, Y., Luo, X., Shan, H., Fu, Y., Gu, Q., Zheng, X., Dai, Q., Xia, F., Zheng, Z., Liu, P., Yin, X. -M., Hong, L., & Li, M. (2019). Niclosamide Triggers Non-Canonical LC3 Lipidation. Cells, 8(3), 248. https://doi.org/10.3390/cells8030248