3D Telomere Structure Analysis to Detect Genomic Instability and Cytogenetic Evolution in Myelodysplastic Syndromes


Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Samples
2.2. Co-Immuno FISH and Imaging
2.3. Statistical Analyses
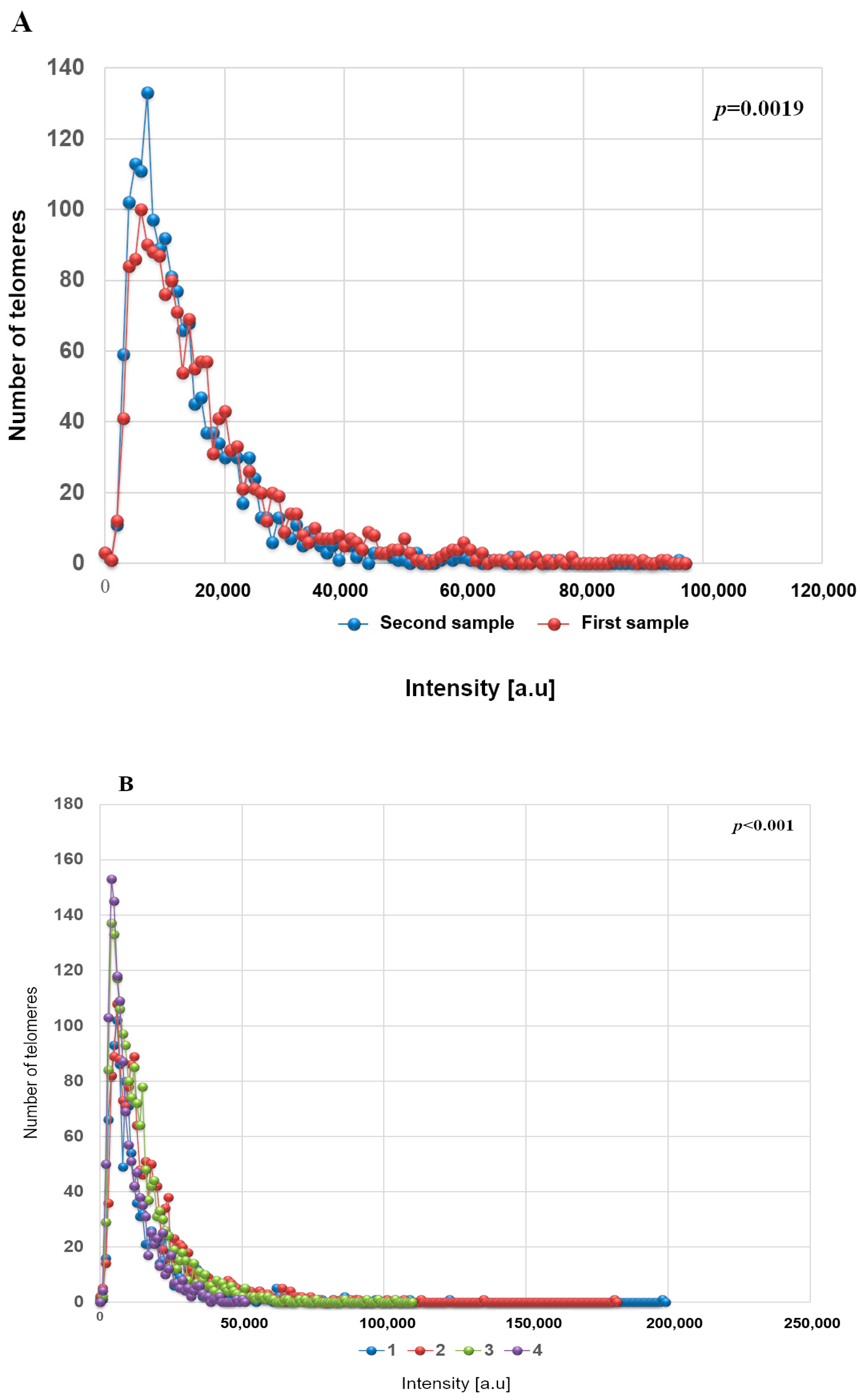
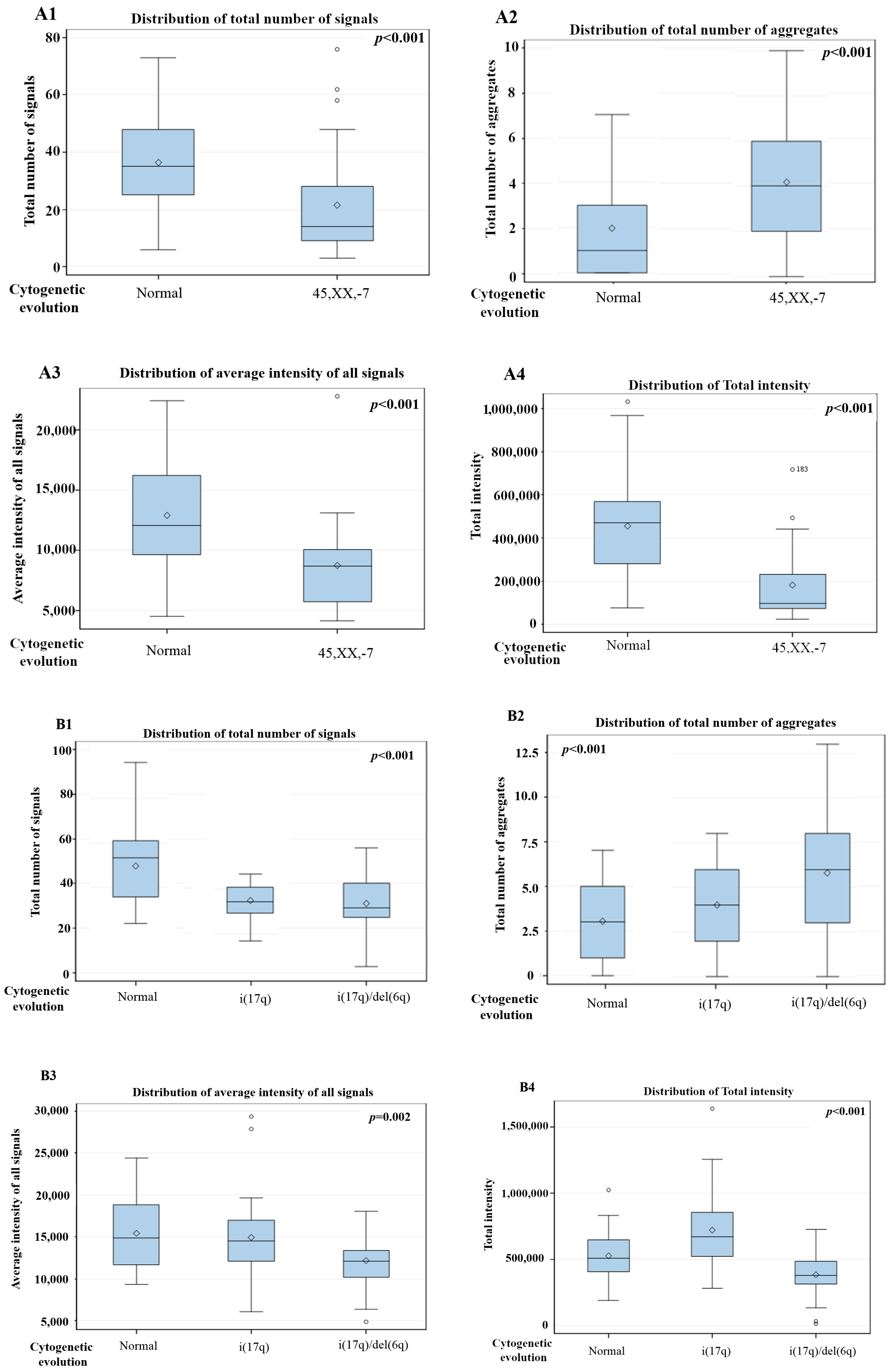
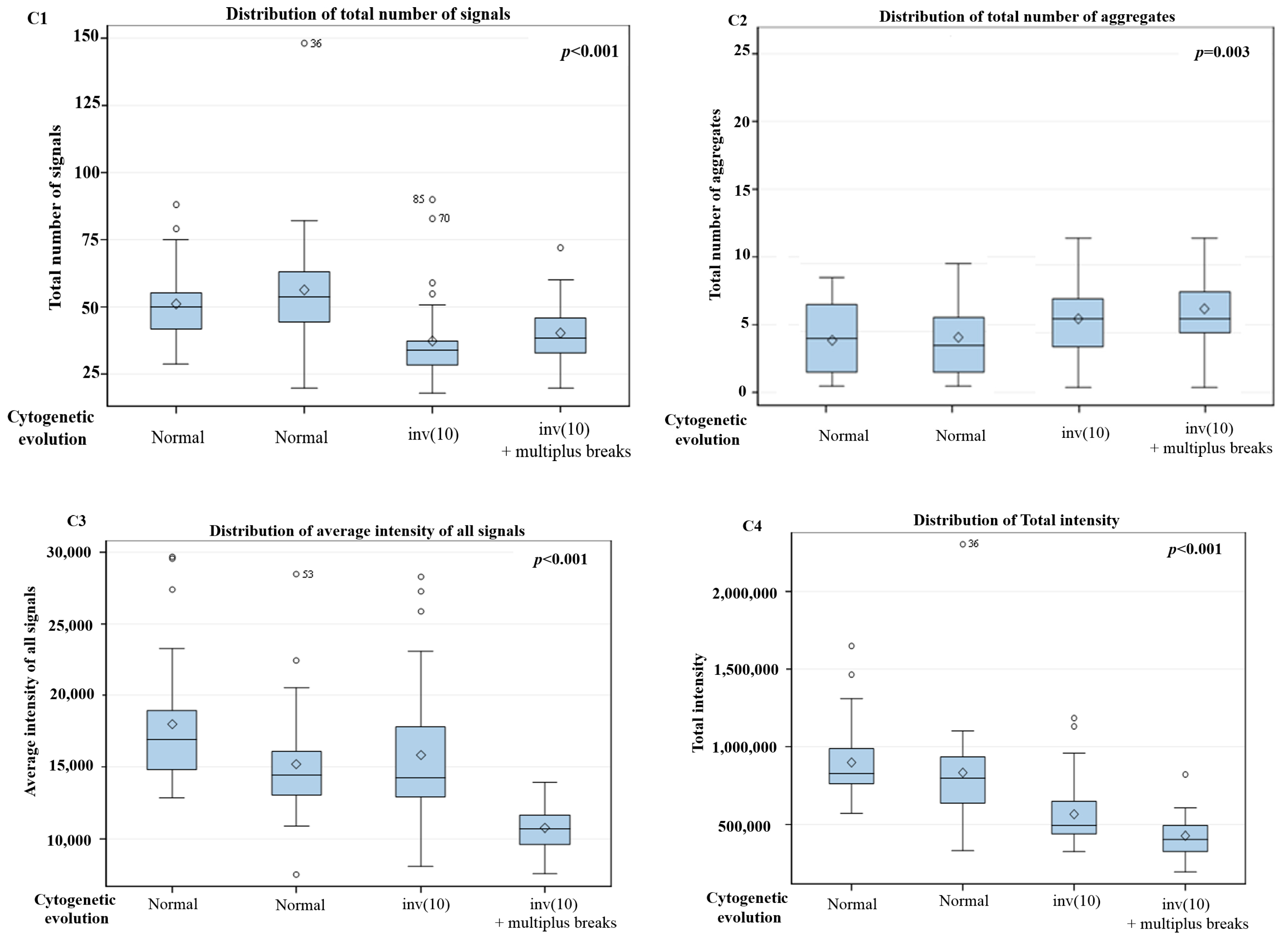
3. Results
3D Nuclear Architecture of Telomeres Has Changed During Cytogenetic Evolution
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellström-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 2009, 114. [Google Scholar] [CrossRef]
- Ertz-Archambault, N.; Kelemen, K. Relapse and cytogenetic evolution in myeloid neoplasms. Panminerva Med. 2017, 59, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Bernasconi, P.; Klersy, C.; Boni, M.; Cavigliano, P.M.; Giardini, I.; Rocca, B.; Zappatore, R.; Dambruoso, I.; Calvello, C.; Caresana, M.; et al. Does cytogenetic evolution have any prognostic relevance in myelodysplastic syndromes? A study on 153 patients from a single institution. Ann. Hematol. 2010, 89, 545–551. [Google Scholar] [CrossRef]
- Tefferi, A.; Vardiman, J.W. Myelodysplastic syndromes. N. Engl. J. Med. 2009, 361, 1872–1885. [Google Scholar] [CrossRef] [PubMed]
- Louis, S.F.; Vermolen, B.J.; Garini, Y.; Young, I.T.; Guffei, A.; Lichtensztejn, Z.; Kuttler, F.; Chuang, T.C.; Moshir, S.; Mougey, V.; et al. c-Myc induces chromosomal rearrangements through telomere and chromosome remodeling in the interphase nucleus. Proc. Natl. Acad. Sci. USA 2005, 102, 9613–9618. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.; Garini, Y. Oncogenic remodeling of the three-dimensional organization of the interphase nucleus: c-Myc induces telomeric aggregates whose formation precedes chromosomal rearrangements. Cell Cycle 2005, 4, 1327–1331. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.; Garini, Y. The significance of telomeric aggregates in the interphase nuclei of tumor cells. J. Cell Biochem. 2006, 97, 904–915. [Google Scholar] [CrossRef]
- Gadji, M.; Adebayo Awe, J.; Rodrigues, P.; Kumar, R.; Houston, D.S.; Klewes, L.; Dièye, T.N.; Rego, E.M.; Passetto, R.F.; de Oliveira, F.M.; et al. Profiling three-dimensional nuclear telomeric architecture of myelodysplastic syndromes and acute myeloid leukemia defines patient subgroups. Clin. Cancer Res. 2012, 18, 3293–3304. [Google Scholar] [CrossRef]
- Awe, J.A.; Xu, M.C.; Wechsler, J.; Benali-Furet, N.; Cayre, Y.E.; Saranchuk, J.; Drachenberg, D.; Mai, S. Three-Dimensional Telomeric Analysis of Isolated Circulating Tumor Cells (CTCs) Defines CTC Subpopulations. Transl. Oncol. 2013, 6, 51–65. [Google Scholar]
- Simons, A.; Shaffer, L.G.; Hastings, R.J. Cytogenetic Nomenclature: Changes in the ISCN 2013 Compared to the 2009 Edition. Cytogenet. Genome Res. 2013, 141, 1–6, Review. [Google Scholar] [CrossRef]
- Vermolen, B.J.; Garini, Y.; Mai, S.; Mougey, V.; Fest, T.; Chuang, T.C.; Chuang, A.Y.; Wark, L.; Young, I.T. Characterizing the three–dimensional organization of telomeres. Cytometry A 2005, 67, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Mathur, S.; Glogowska, A.; McAvoy, E.; Righolt, C.; Rutherford, J.; Willing, C.; Banik, U.; Ruthirakuhan, M.; Mai, S.; Garcia, A. Three-dimensional quantitative imaging of telomeres in buccal cells identifies mild, moderate, and severe Alzheimer’s disease patients. J. Alzheimers Dis. 2014, 39, 35–48. [Google Scholar] [CrossRef]
- Mai, S. Initiation of telomere–mediated chromosomal rearrangements in cancer. J. Cell Biochem. 2010, 109, 1095–1102. [Google Scholar] [CrossRef]
- Gadji, M.; Fortin, D.; Tsanaclis, A.M.; Garini, Y.; Katzir, N.; Wienburg, Y.; Yan, J.; Klewes, L.; Klonisch, T.; Drouin, R.; et al. Three-dimensional nuclear telomere architecture is associated with differential time to progression and overall survival in glioblastoma patients. Neoplasia 2010, 12, 183–191. [Google Scholar] [CrossRef]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Gu, Z.H.; Li, Y.; Zhang, J.L.; Chang, C.K.; Pan, C.M.; Shi, J.Y.; Shen, Y.; Chen, B.; Wang, Y.Y.; et al. Genomic landscape of CD34+ hematopoietic cells in myelodysplastic syndrome and gene mutation profiles as prognostic markers. Proc. Natl. Acad. Sci. USA 2014, 111, 8589–8594. [Google Scholar] [CrossRef] [PubMed]
- Meeker, A.K.; Hicks, J.L.; Iacobuzio-Donahue, C.A.; Montgomery, E.A.; Westra, W.H.; Chan, T.Y.; Ronnett, B.M.; De Marzo, A.M. Telomere length abnormalities occur early in the initiation of epithelial carcinogenesis. Clin. Cancer Res. 2004, 10, 3317–3326. [Google Scholar] [CrossRef]
- Chuang, T.C.; Moshir, S.; Garini, Y.; Chuang, A.Y.; Young, I.T.; Vermolen, B.; van den Doel, R.; Mougey, V.; Perrin, M.; Braun, M.; et al. The three–dimensional organization of telomeres in the nucleus of mammalian cells. BMC Biol. 2004, 2. [Google Scholar] [CrossRef]
- Jabbour, E.; Takahashi, K.; Wang, X.; Cornelison, A.M.; Abruzzo, L.; Kadia, T.; Borthakur, G.; Estrov, Z.; O’Brien, S.; Mallo, M.; et al. Acquisition of cytogenetic abnormalities in patients with IPSS defined lower-risk myelodysplastic syndrome is associated with poor prognosis and transformation to acute myelogenous leukemia. Am. J. Hematol. 2013, 88, 831–837. [Google Scholar] [CrossRef]
- Makishima, H.; Yoshizato, T.; Yoshida, K.; Sekeres, M.A.; Radivoyevitch, T.; Suzuki, H.; Przychodzen, B.; Nagata, Y.; Meggendorfer, M.; Sanada, M.; et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat. Genet. 2017, 49, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Bochtler, T.; Stölzel, F.; Heilig, C.E.; Kunz, C.; Mohr, B.; Jauch, A.; Janssen, J.W.; Kramer, M.; Benner, A.; Bornhäuser, M.; et al. Clonal heterogeneity as detected by metaphase karyotyping is an indicator of poor prognosis in acute myeloid leukemia. J. Clin. Oncol. 2013, 31, 3898–3905. [Google Scholar] [CrossRef]
- Neukirchen, J.; Lauseker, M.; Hildebrandt, B.; Nolting, A.C.; Kaivers, J.; Kobbe, G.; Gattermann, N.; Haas, R.; Germing, U. Cytogenetic clonal evolution in myelodysplastic syndromes is associated with inferior prognosis. Cancer 2017, 123, 4608–4616. [Google Scholar] [CrossRef]
- Tricot, G.; Vlietinck, R.; Boogaerts, M.A.; Hendrickx, B.; De Wolf-Peeters, C.; Van den Berghe, H.; Verwilghen, R.L. Prognostic factors in the myelodysplastic syndromes: importance of initial data on peripheral blood counts, bone marrow cytology, trephine biopsy and chromosomal analysis. Br. J. Haematol. 1985, 60, 19–32. [Google Scholar] [CrossRef]
- Schanz, J.; Cevik, N.; Fonatsch, C.; Braulke, F.; Shirneshan, K.; Bacher, U.; Haase, D. Detailed analysis of clonal evolution and cytogenetic evolution patterns in patients with myelodysplastic syndromes (MDS) and related myeloid disorders. Blood Cancer J. 2018, 8. [Google Scholar] [CrossRef]
- Bandara, M.S.; Goonasekera, H.W.W.; Dissanayake, V.H.W. The utility of hematopoietic stem cell karyotyping in the diagnosis of de novo myelodysplastic syndromes. J. Hematopathol. 2016, 9, 121. [Google Scholar] [CrossRef]
- Horiike, S.; Taniwaki, M.; Misawa, S.; Abe, T. Chromosome abnormalities and karyotypic evolution in 83 patients with myelodysplastic syndrome and predictive value for prognosis. Cancer 1988, 62, 1129–1138. [Google Scholar] [CrossRef]
- Tien, H.F.; Wang, C.H.; Chuang, S.M.; Lee, F.Y.; Liu, M.C.; Chen, Y.C.; Shen, M.C.; Lin, K.H.; Lin, D.T. Acute leukemic transformation of myelodysplastic syndrome—immunophenotypic, genotypic, and cytogenetic studies. Leuk. Res. 1995, 19, 595–603. [Google Scholar] [CrossRef]
- Wang, H.; Wang, X.Q.; Xu, X.P.; Lin, G.W. Cytogenetic evolution correlates with poor prognosis in myelodysplastic syndrome. Cancer Genet. Cytogenet. 2010, 196, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Benitez, J.; Carbonell, F.; Sanchez Fayos, J.; Heimpel, H. Karyotypic evolution in patients with myelodysplastic syndromes. Cancer Genet. Cytogenet. 1985, 16, 157–167. [Google Scholar] [CrossRef]
- Lai, Y.Y.; Qiu, J.Y.; Zhang, Y.; Zhang, Y.; Shi, Y.; He, Q.; Dang, H.; Lu, D.P.; Lu, Z.Y.; Huang, J.X.; et al. Clinical significance of continuous karyotyping in myelodysplastic syndromes. Zhonghua Xue Ye Xue Za Zhi 2004, 25, 645–648. [Google Scholar]
- Raskind, W.H.; Tirumali, N.; Jacobson, R.; Singer, J.; Fialkow, P.J. Evidence for a multistep pathogenesis of a myelodysplastic syndrome. Blood 1984, 63, 1318–1323. [Google Scholar]
- Cermák, J.; Belicková, M.; Krejcová, H.; Michalová, K.; Zilovcová, S.; Zemanová, Z.; Brezinová, J.; Sieglová, Z. The presence of clonal cell subpopulations in peripheral blood and bone marrow of patients with refractory cytopenia with multilineage dysplasia but not in patients with refractory anemia may reflect a multistep pathogenesis of myelodysplasia. Leuk. Res. 2005, 29, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Galili, N.; Cerny, J.; Raza, A. Current treatment options: impact of cytogenetics on the course of myelodysplasia. Curr. Treat. Opt. Oncol. 2007, 8, 8–117. [Google Scholar] [CrossRef]
- Amiel, T.; Sharony, R.; Goldberg-Bittman, L.; Biron-Shental, T.; Fejgin, M.D.; Amiel, A. Telomere aggregates in amniocytes with karyotype of balanced chromosomal rearrangements. Hereditas 2010, 147, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Knecht, H.; Sawan, B.; Lichtensztejn, D.; Lemieux, B.; Wellinger, R.J.; Mai, S. The 3D nuclear organization of telomeres marks the transition from Hodgkin to Reed-Sternberg cells. Leukemia 2009, 23, 565–573. [Google Scholar] [CrossRef]
- Samassekou, O.; Hébert, J.; Mai, S.; Yan, J. Nuclear remodeling of telomeres in chronic myeloid leukemia. Genes Chromosomes Cancer 2013, 52, 495–502. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Number | % |
|---|---|---|
| Age at diagnosis | ||
| Adults range (19–71) | 11/15 | 73.33% |
| Children (1–18) | 4/15 | 26.67% |
| Gender | ||
| Male | 10/15 | 66.67% |
| Female | 5/15 | 33.33% |
| WHO subtype | ||
| RCUD | 2/15 | 13.33% |
| RARS | 1/15 | 6.67% |
| RCMD | 6/15 | 40% |
| RAEB-1 | 3/15 | 20% |
| RAEB-2 | 2/15 | 13.33% |
| MDS-U | 0/15 | 0 |
| MDS with isolated del(5q) | 1/15 | 6.67% |
| IPSS-R | ||
| Very low | 0/15 | 0 |
| Low | 0/15 | 0 |
| Intermediate | 13/15 | 86.68 |
| High | 1/15 | 6.66% |
| Very high | 1/15 | 6.66% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rangel-Pozzo, A.; de Souza, D.C.; Schmid-Braz, A.T.; Azambuja, A.P.d.; Ferraz-Aguiar, T.; Borgonovo, T.; Mai, S. 3D Telomere Structure Analysis to Detect Genomic Instability and Cytogenetic Evolution in Myelodysplastic Syndromes. Cells 2019, 8, 304. https://doi.org/10.3390/cells8040304
Rangel-Pozzo A, de Souza DC, Schmid-Braz AT, Azambuja APd, Ferraz-Aguiar T, Borgonovo T, Mai S. 3D Telomere Structure Analysis to Detect Genomic Instability and Cytogenetic Evolution in Myelodysplastic Syndromes. Cells. 2019; 8(4):304. https://doi.org/10.3390/cells8040304
Chicago/Turabian StyleRangel-Pozzo, Aline, Daiane Corrêa de Souza, Ana Teresa Schmid-Braz, Ana Paula de Azambuja, Thais Ferraz-Aguiar, Tamara Borgonovo, and Sabine Mai. 2019. "3D Telomere Structure Analysis to Detect Genomic Instability and Cytogenetic Evolution in Myelodysplastic Syndromes" Cells 8, no. 4: 304. https://doi.org/10.3390/cells8040304
APA StyleRangel-Pozzo, A., de Souza, D. C., Schmid-Braz, A. T., Azambuja, A. P. d., Ferraz-Aguiar, T., Borgonovo, T., & Mai, S. (2019). 3D Telomere Structure Analysis to Detect Genomic Instability and Cytogenetic Evolution in Myelodysplastic Syndromes. Cells, 8(4), 304. https://doi.org/10.3390/cells8040304