Small-Molecule Host-Defense Peptide Mimetic Antibacterial and Antifungal Agents Activate Human and Mouse Mast Cells via Mas-Related GPCRs


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Materials
2.3. Fungus MIC Assay
2.4. Bacterial MIC Assay
2.5. Cytotoxicity Assays
2.6. Mast Cell Culture
2.7. Lentivirus-Mediated Knockdown of MRGPRX2 in LAD2 Cells
2.8. Western Blotting to Determine MRGPRX2 Expression
2.9. Transfection of RBL-2H3 Cells and Flow Cytometry
2.10. Degranulation Assay
2.11. Statistical Analysis
3. Results
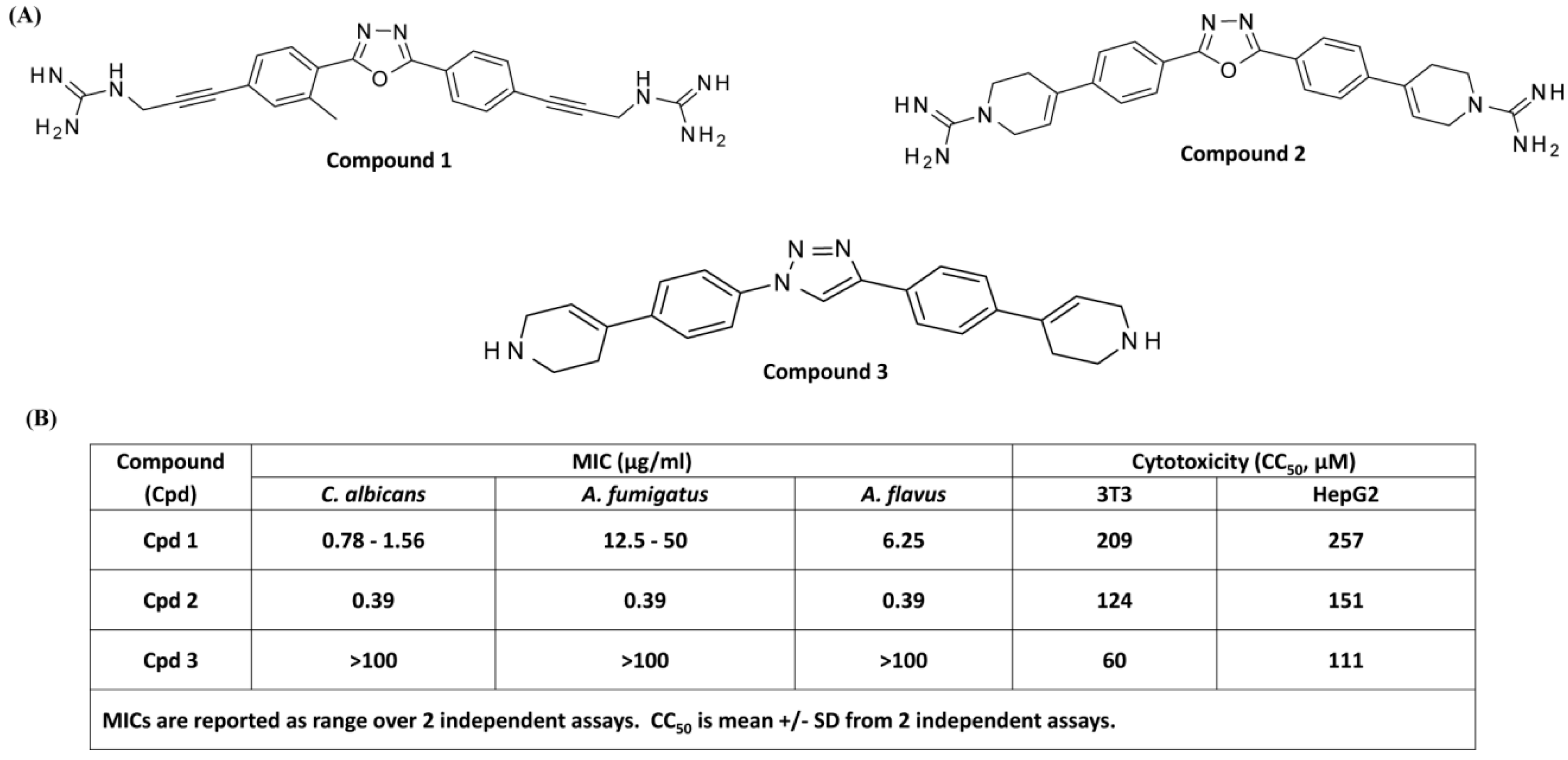
3.1. Antifungal and Cytotoxic Activities of smHDPMs (Compounds 1, 2, and 3) against Candida albicans, Aspergillus fumigatus, and Aspergillus flavus
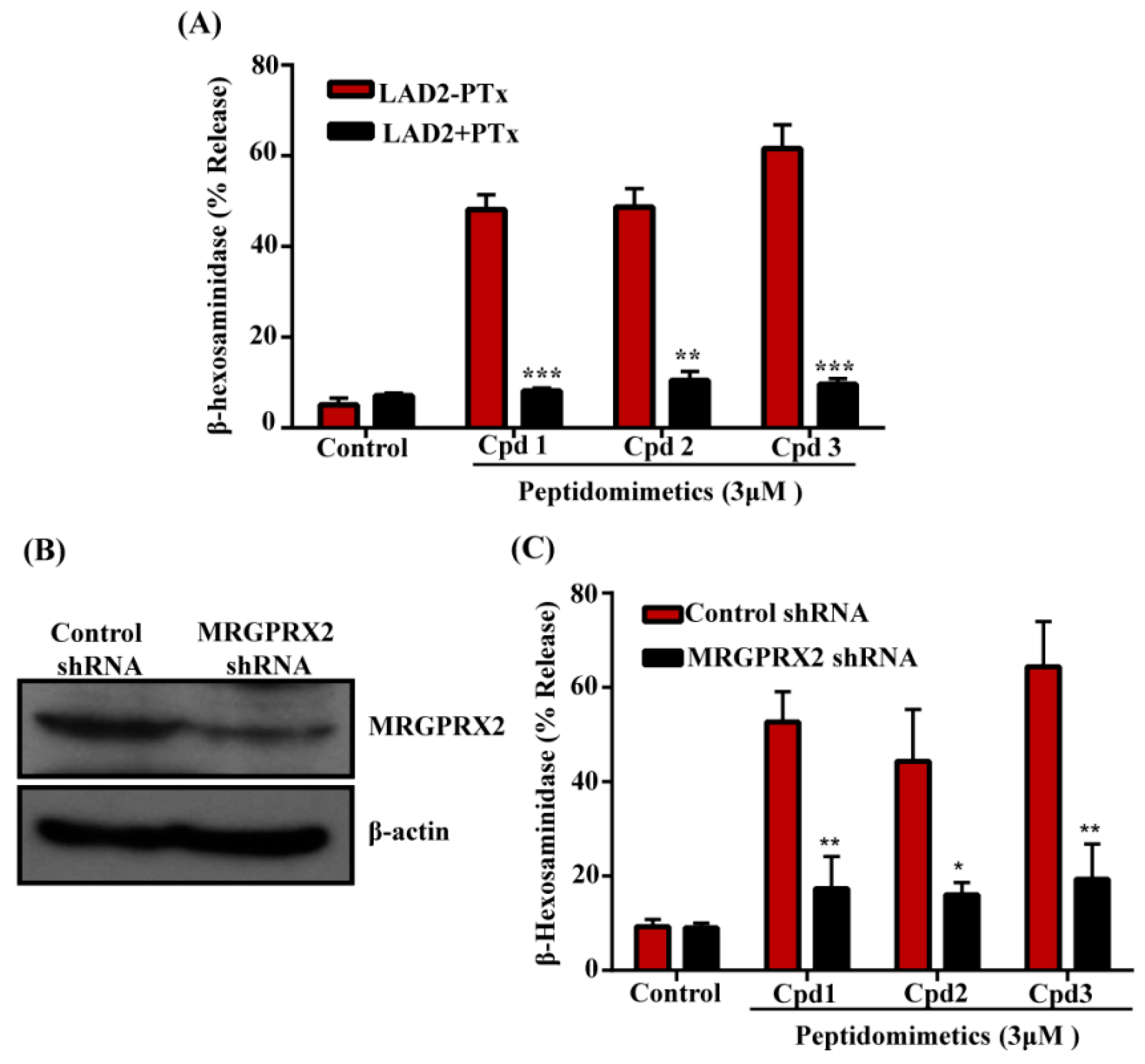
3.2. smHDPMs (Compounds 1, 2, and 3) Activate Human MCs via MRGPRX2
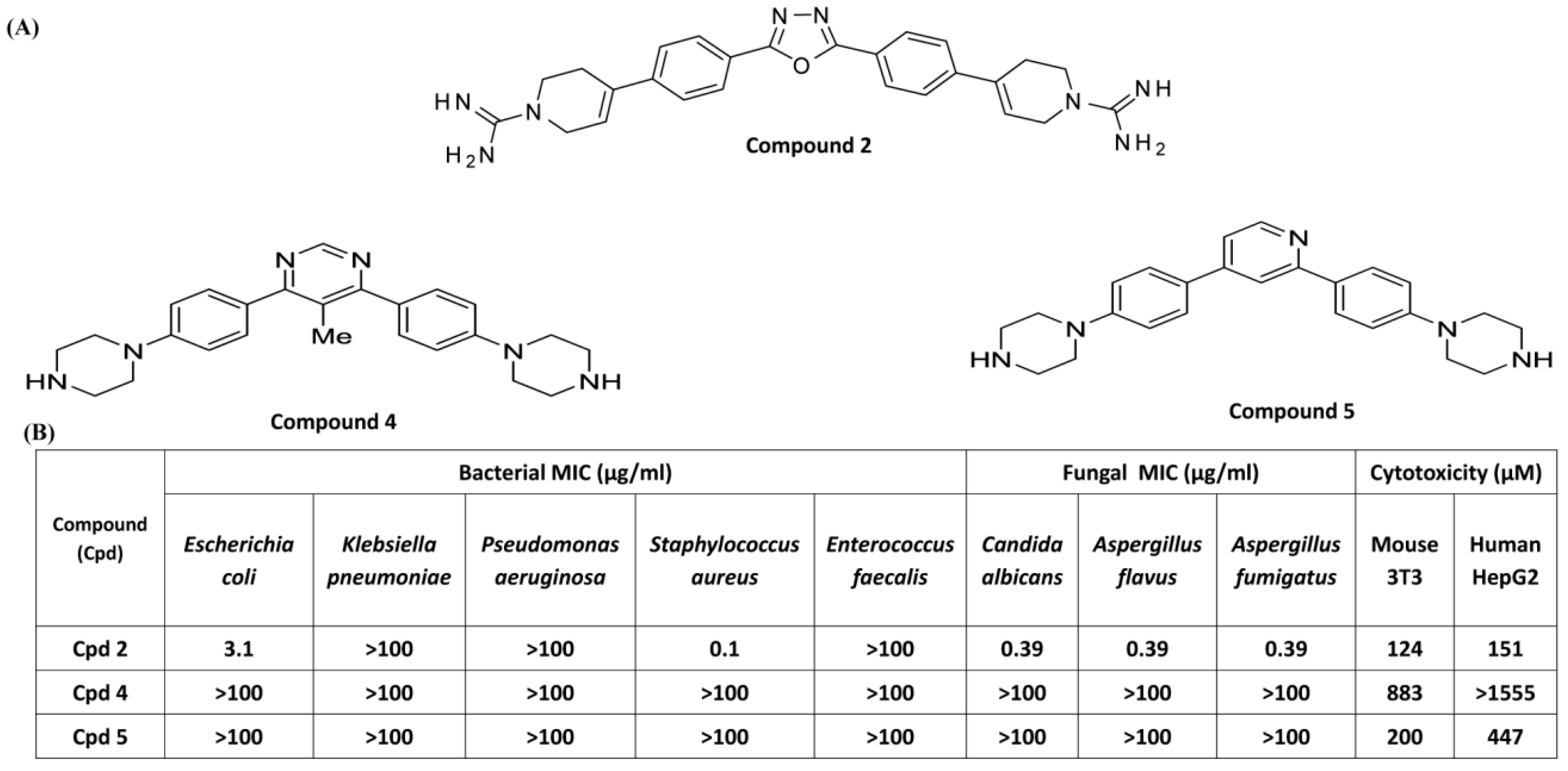
3.3. Effects of Compounds 2, 4, and 5 on Antibacterial Activity and MC Degranulation
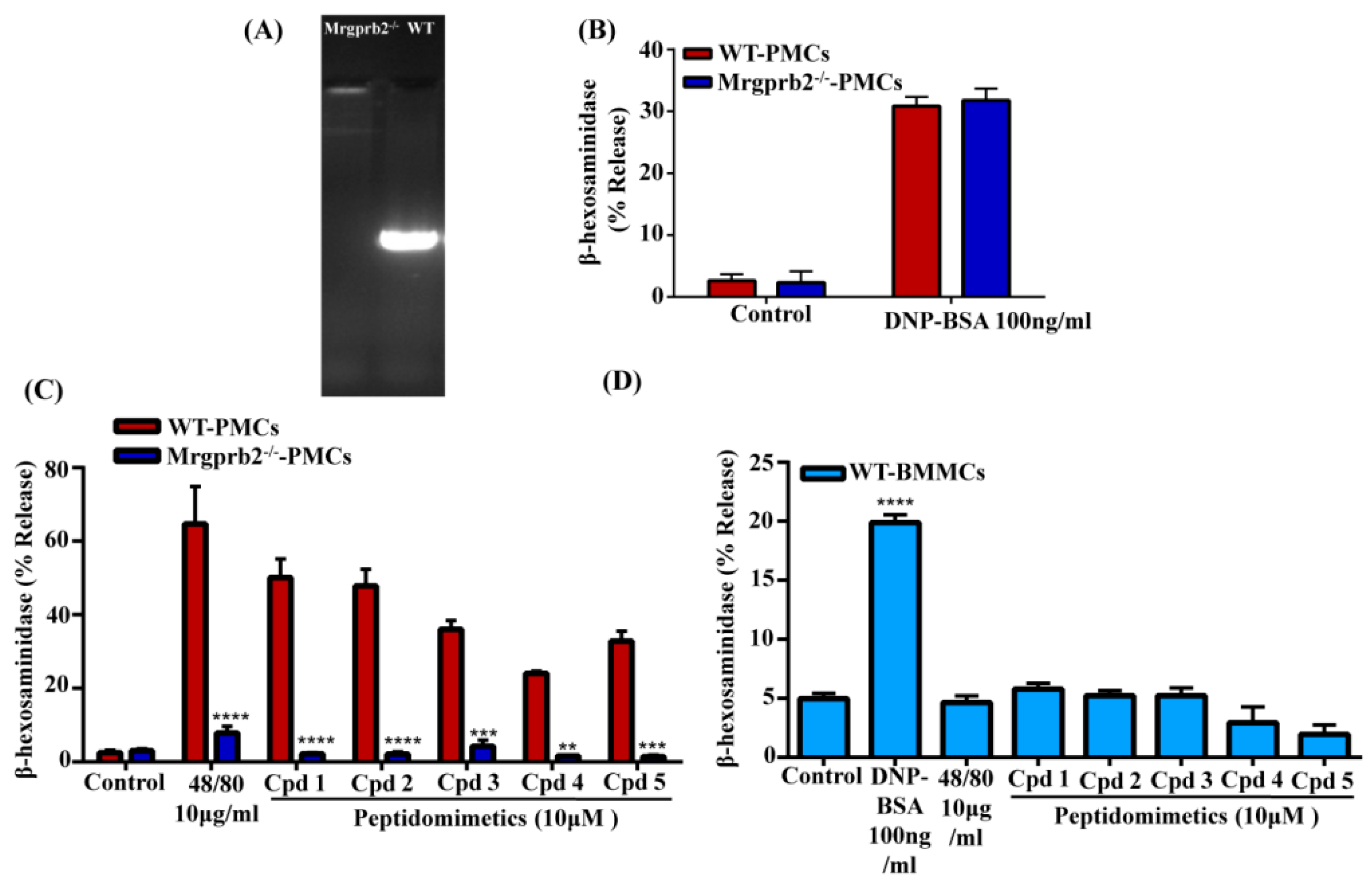
3.4. smHDPMs Activate Murine MCs via Mrgprb2
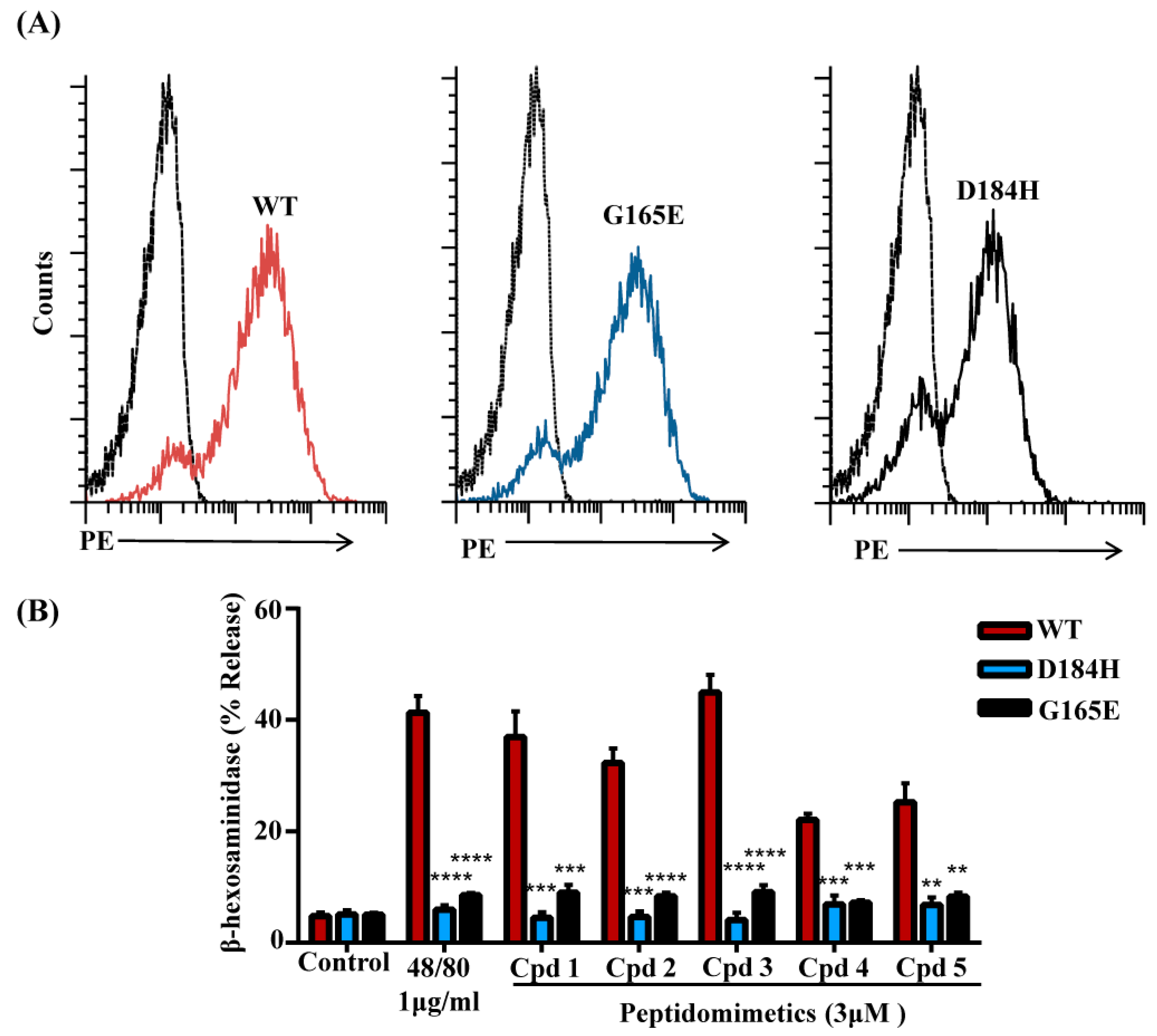
3.5. Naturally Occurring Missense MRGPRX2 Variants D184H, G165E Are Resistant to Activation by smHDPMs
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Frossi, B.; Mion, F.; Tripodo, C.; Colombo, M.P.; Pucillo, C.E. Rheostatic Functions of Mast Cells in the Control of Innate and Adaptive Immune Responses. Trends Immunol. 2017, 38, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Shiota, N.; Nishikori, Y.; Kakizoe, E.; Shimoura, K.; Niibayashi, T.; Shimbori, C.; Tanaka, T.; Okunishi, H. Pathophysiological role of skin mast cells in wound healing after scald injury: Study with mast cell-deficient W/W(V) mice. Int. Arch. Allergy Immunol. 2010, 151, 80–88. [Google Scholar] [CrossRef]
- Groschwitz, K.R.; Ahrens, R.; Osterfeld, H.; Gurish, M.F.; Han, X.; Åbrink, M.; Finkelman, F.D.; Pejler, G.; Hogan, S.P. Mast cells regulate homeostatic intestinal epithelial migration and barrier function by a chymase/Mcpt4-dependent mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 22381–22386. [Google Scholar] [CrossRef]
- Arifuzzaman, M.; Mobley, Y.R.; Choi, H.W.; Bist, P.; Salinas, C.A.; Brown, Z.D.; Chen, S.L.; Staats, H.F.; Abraham, S.N. MRGPR-mediated activation of local mast cells clears cutaneous bacterial infection and protects against reinfection. Sci. Adv. 2019, 5, eaav0216. [Google Scholar] [CrossRef] [PubMed]
- Irani, A.A.; Schechter, N.M.; Craig, S.S.; Deblois, G.; Schwartz, L.B. Two types of human mast cells that have distinct neutral protease compositions. Proc. Natl. Acad. Sci. USA 1986, 83, 4464–4468. [Google Scholar] [CrossRef]
- Bankova, L.G.; Dwyer, D.F.; Liu, A.Y.; Austen, K.F.; Gurish, M.F. Maturation of mast cell progenitors to mucosal mast cells during allergic pulmonary inflammation in mice. Mucosal Immunol. 2015, 8, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, D.F.; Barrett, N.A.; Austen, K.F. Immunological Genome Project Consortium. Expression profiling of constitutive mast cells reveals a unique identity within the immune system. Nat. Immunol. 2016, 17, 878–887. [Google Scholar] [CrossRef] [PubMed]
- McNeil, B.D.; Pundir, P.; Meeker, S.; Han, L.; Undem, B.J.; Kulka, M.; Dong, X. Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature 2015, 519, 237–241. [Google Scholar] [CrossRef]
- Irani, A.M.; Craig, S.S.; Deblois, G.; Elson, C.; Schechter, N.M.; Schwartz, L.B. Deficiency of the tryptase-positive, chymase-negative mast cell type in gastrointestinal mucosa of patients with defective T lymphocyte function. J. Immunol. 1987, 138, 4381–4386. [Google Scholar]
- Fujisawa, D.; Kashiwakura, J.-I.; Kita, H.; Kikukawa, Y.; Fujitani, Y.; Sasaki-Sakamoto, T.; Kuroda, K.; Nunomura, S.; Hayama, K.; Terui, T.; et al. Expression of Mas-related gene X2 on mast cells is upregulated in the skin of patients with severe chronic urticaria. J. Clin. Immunol. 2014, 134, 622–633.e9. [Google Scholar] [CrossRef]
- Thakurdas, S.M.; Melicoff, E.; Sansores-Garcia, L.; Moreira, D.; Petrova, Y.; Stevens, R.L.; Adachi, R. The Mast Cell-restricted Tryptase mMCP-6 Has a Critical Immunoprotective Role in Bacterial Infections. J. Boil. Chem. 2007, 282, 20809–20815. [Google Scholar] [CrossRef] [PubMed]
- Echtenacher, B.; Mannel, D.N.; Hultner, L. Critical protective role of mast cells in a model of acute septic peritonitis. Nature 1996, 381, 75–77. [Google Scholar] [CrossRef] [PubMed]
- Malaviya, R.; Abraham, S.N. Role of mast cell leukotrienes in neutrophil recruitment and bacterial clearance in infectious peritonitis. J. Leukoc. Boil. 2000, 67, 841–846. [Google Scholar] [CrossRef]
- Piliponsky, A.M.; Romani, L. The contribution of mast cells to bacterial and fungal infection immunity. Immunol. Rev. 2018, 282, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Romani, L. Immunity to fungal infections. Nat. Rev. Immunol. 2011, 11, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Bassetti, M.; Taramasso, L.; Nicco, E.; Molinari, M.P.; Mussap, M.; Viscoli, C. Epidemiology, Species Distribution, Antifungal Susceptibility and Outcome of Nosocomial Candidemia in a Tertiary Care Hospital in Italy. PLoS ONE 2011, 6, e24198. [Google Scholar] [CrossRef] [PubMed]
- Guery, B.P.; Arendrup, M.C.; Auzinger, G.; Azoulay, E.; Borges Sa, M.; Johnson, E.M.; Müller, E.; Putensen, C.; Rotstein, C.; Sganga, G.; et al. Management of invasive candidiasis and candidemia in adult non-neutropenic intensive care unit patients: Part I. Epidemiology and diagnosis. Intensive Care Med. 2009, 35, 55–62. [Google Scholar] [CrossRef]
- Guery, B.P.; Arendrup, M.C.; Auzinger, G.; Azoulay, E.; Borges Sa, M.; Johnson, E.M.; Müller, E.; Putensen, C.; Rotstein, C.; Sganga, G.; et al. Management of invasive candidiasis and candidemia in adult non-neutropenic intensive care unit patients: Part II. Treatment. Intensive Care Med. 2009, 35, 206–214. [Google Scholar] [CrossRef]
- Laxminarayan, R.; Duse, A.; Wattal, C.; Zaidi, A.K.M.; Wertheim, H.F.L.; Sumpradit, N.; Vlieghe, E.; Hara, G.L.; Gould, I.M.; Goossens, H.; et al. Antibiotic resistance—The need for global solutions. Infect. Dis. 2013, 13, 1057–1098. [Google Scholar] [CrossRef]
- Diamond, G.; Beckloff, N.; Weinberg, A.; Kisich, K. The Roles of Antimicrobial Peptides in Innate Host Defense. Curr. Pharm. Des. 2009, 15, 2377–2392. [Google Scholar] [CrossRef]
- Hancock, R.E.; Diamond, G. The role of cationic antimicrobial peptides in innate host defences. Trends Microbiol. 2000, 8, 402–410. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Hakansson, J.; Ringstad, L.; Björn, C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef]
- Shai, Y. From Innate Immunity to de-Novo Designed Antimicrobial Peptides. Curr. Pharm. Des. 2002, 8, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Yeaman, M.R.; Hauger, R.L.; Grigoriadis, D.E.; Dallman, M.F.; Plotsky, P.M.; Vale, W.W.; Dautzenberg, F.M. Mechanisms of Antimicrobial Peptide Action and Resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, H.; Gupta, K.; Lee, D.; Bayir, A.K.; Ahn, H.; Ali, H. beta-Defensins activate human mast cells via Mas-related gene X2. J. Immunol. 2013, 191, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, H.; Gupta, K.; Guo, Q.; Price, R.; Ali, H. Mas-related gene X2 (MrgX2) is a novel G protein-coupled receptor for the antimicrobial peptide LL-37 in human mast cells: Resistance to receptor phosphorylation, desensitization, and internalization. J. Biol. Chem. 2011, 286, 44739–44749. [Google Scholar] [CrossRef] [PubMed]
- Koczulla, R.; Von Degenfeld, G.; Kupatt, C.; Krotz, F.; Zahler, S.; Gloe, T.; Issbrücker, K.; Unterberger, P.; Zaiou, M.; Lebherz, C.; et al. An angiogenic role for the human peptide antibiotic LL-37/hCAP-18. J. Clin. Investig. 2003, 111, 1665–1672. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, T.; Spielmann, M.; Zuhaili, B.; Fossum, M.; Metzig, M.; Koehler, T.; Steinau, H.-U.; Yao, F.; Onderdonk, A.B.; Steinstraesser, L.; et al. Human beta-defensin-3 promotes wound healing in infected diabetic wounds. J. Gene Med. 2009, 11, 220–228. [Google Scholar] [CrossRef]
- Gupta, K.; Kotian, A.; Subramanian, H.; Daniell, H.; Ali, H. Activation of human mast cells by retrocyclin and protegrin highlight their immunomodulatory and antimicrobial properties. Oncotarget 2015, 6, 28573–28587. [Google Scholar] [CrossRef]
- Scott, R.W.; Tew, G.N. Mimics of Host Defense Proteins; Strategies for Translation to Therapeutic Applications. Curr. Top. Med. Chem. 2017, 17, 576–589. [Google Scholar] [CrossRef]
- Choi, S.; Isaacs, A.; Clements, D.; Liu, D.; Kim, H.; Scott, R.W.; Winkler, J.D.; DeGrado, W.F. De novo design and in vivo activity of conformationally restrained antimicrobial arylamide foldamers. Proc. Natl. Acad. Sci. USA 2009, 106, 6968–6973. [Google Scholar] [CrossRef]
- Beckloff, N.; Laube, D.; Castro, T.; Furgang, D.; Park, S.; Perlin, D.; Clements, D.; Tang, H.; Scott, R.W.; Tew, G.N.; et al. Activity of an Antimicrobial Peptide Mimetic against Planktonic and Biofilm Cultures of Oral Pathogens. Antimicrob. Agents Chemother. 2007, 51, 4125–4132. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Sato, H.; Sakamaki, K.; Kamada, M.; Okuno, Y.; Fukuishi, N.; Furuta, K.; Tanaka, S. Suppression of IgE-Independent Degranulation of Murine Connective Tissue-Type Mast Cells by Dexamethasone. Cells 2019, 8, 112. [Google Scholar] [CrossRef] [PubMed]
- Alkanfari, I.; Gupta, K.; Jahan, T.; Ali, H. Naturally Occurring Missense MRGPRX2 Variants Display Loss of Function Phenotype for Mast Cell Degranulation in Response to Substance P, Hemokinin-1, Human beta-Defensin-3, and Icatibant. J. Immunol. 2018, 201, 343–349. [Google Scholar] [CrossRef]
- Steinberg, D.; Hurst, M.; Fujii, C.; Kung, A.H.; Ho, J.F.; Cheng, F.C.; Loury, D.J.; Fiddes, J.C. Protegrin-1: A broad-spectrum, rapidly microbicidal peptide with in vivo activity. Antimicrob. Agents Chemother. 1997, 41, 1738–1742. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Hancock, R.E.W. Synergistic Interactions between Mammalian Antimicrobial Defense Peptides. Antimicrob. Agents Chemother. 2001, 45, 1558–1560. [Google Scholar] [CrossRef] [PubMed]
- Ryan, L.K.; Freeman, K.B.; Masso-Silva, J.A.; Falkovsky, K.; Aloyouny, A.; Markowitz, K.; Hise, A.G.; Fatahzadeh, M.; Scott, R.W.; Diamond, G. Activity of Potent and Selective Host Defense Peptide Mimetics in Mouse Models of Oral Candidiasis. Antimicrob. Agents Chemother. 2014, 58, 3820–3827. [Google Scholar] [CrossRef] [PubMed]
- Kirshenbaum, A.S.; Akin, C.; Wu, Y.; Rottem, M.; Goff, J.P.; Beaven, M.; Rao, V.K.; Metcalfe, D.D. Characterization of novel stem cell factor responsive human mast cell lines LAD 1 and 2 established from a patient with mast cell sarcoma/leukemia; activation following aggregation of FcεRI or FcγRI. Leuk. Res. 2003, 27, 677–682. [Google Scholar] [CrossRef]
- Ali, H.; Richardson, R.M.; Tomhave, E.D.; DuBose, R.A.; Haribabu, B.; Snyderman, R. Regulation of stably transfected platelet activating factor receptor in RBL-2H3 cells. Role of multiple G proteins and receptor phosphorylation. J. Biol. Chem. 1994, 269, 24557–24563. [Google Scholar] [PubMed]
- Mrabet-Dahbi, S.; Metz, M.; Dudeck, A.; Zuberbier, T.; Maurer, M. Murine mast cells secrete a unique profile of cytokines and prostaglandins in response to distinct TLR2 ligands. Exp. Dermatol. 2009, 18, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Vukman, K.V.; Visnovitz, T.; Adams, P.N.; Metz, M.; Maurer, M.; O’Neill, S.M. Mast cells cultured from IL-3-treated mice show impaired responses to bacterial antigen stimulation. Inflamm. Res. 2012, 61, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Hua, J.; Scott, R.; Diamond, G. Activity of antimicrobial peptide mimetics in the oral cavity: II. Activity against periopathogenic biofilms and anti-inflammatory activity. Mol. Oral Microbiol. 2010, 25, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Hua, J.; Yamarthy, R.; Felsenstein, S.; Scott, R.W.; Markowitz, K.; Diamond, G. Activity of Antimicrobial Peptide Mimetics in the Oral Cavity: I. Activity Against Biofilms of Candida albicans. Mol. Oral Microbiol. 2010, 25, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Menzel, L.P.; Chowdhury, H.M.; Masso-Silva, J.A.; Ruddick, W.; Falkovsky, K.; Vorona, R.; Malsbary, A.; Cherabuddi, K.; Ryan, L.K.; DiFranco, K.M.; et al. Potent in vitro and in vivo antifungal activity of a small molecule host defense peptide mimic through a membrane-active mechanism. Sci. Rep. 2017, 7, 4353. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.; Weldon, S.; Buchanan, P.J.; Schock, B.; Ernst, R.K.; McAuley, D.F.; Tunney, M.M.; Irwin, C.R.; Elborn, J.S.; Taggart, C.C. Evaluation of the Ability of LL-37 to Neutralise LPS In Vitro and Ex Vivo. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Saluja, R.; Metz, M.; Maurer, M. Role and Relevance of Mast Cells in Fungal Infections. Front. Immunol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Saunte, D.M.; Tarazooie, B.; Arendrup, M.C.; de Hoog, G.S. Black yeast-like fungi in skin and nail: It probably matters. Mycoses 2012, 55, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Sauer, A.; Letscher-Bru, V.; Speeg-Schatz, C.; Touboul, D.; Colin, J.; Bourcier, T.; Candolfi, E. In Vitro Efficacy of Antifungal Treatment Using Riboflavin/UV-A (365 nm) Combination and Amphotericin B. Investig. Opthalmol. Vis. Sci. 2010, 51, 3950–3953. [Google Scholar] [CrossRef]
- Thomas, P.; Kaliamurthy, J. Mycotic keratitis: Epidemiology, diagnosis and management. Clin. Microbiol. Infect. 2013, 19, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhang, H.; Liu, S.; Chen, G.; He, S.; Li, Z.; Wang, L. Mast Cell Activation Protects Cornea by Promoting Neutrophil Infiltration via Stimulating ICAM-1 and Vascular Dilation in Fungal Keratitis. Sci. Rep. 2018, 8, 8365. [Google Scholar] [CrossRef]
- Gupta, K.; Idahosa, C.; Roy, S.; Lee, D.; Subramanian, H.; Dhingra, A.; Boesze-Battaglia, K.; Korostoff, J.; Ali, H. Differential Regulation of Mas-Related G Protein-Coupled Receptor X2-Mediated Mast Cell Degranulation by Antimicrobial Host Defense Peptides and Porphyromonas gingivalis Lipopolysaccharide. Infect. Immun. 2017, 85, e00246-17. [Google Scholar] [CrossRef]
- Manorak, W.; Idahosa, C.; Gupta, K.; Roy, S.; Panettieri, R.; Ali, H. Upregulation of Mas-related G Protein coupled receptor X2 in asthmatic lung mast cells and its activation by the novel neuropeptide hemokinin-1. Respir. Res. 2018, 19, 1. [Google Scholar] [CrossRef] [PubMed]
- Babina, M.; Guhl, S.; Artuc, M.; Zuberbier, T. Allergic FcepsilonRI- and pseudo-allergic MRGPRX2-triggered mast cell activation routes are independent and inversely regulated by SCF. Allergy 2018, 73, 256–560. [Google Scholar] [CrossRef] [PubMed]
- Di Nardo, A.; Vitiello, A.; Gallo, R.L. Cutting edge: Mast cell antimicrobial activity is mediated by expression of cathelicidin antimicrobial peptide. J. Immunol. 2003, 170, 2274–2278. [Google Scholar] [CrossRef] [PubMed]
- Di Nardo, A.; Yamasaki, K.; Dorschner, R.A.; Lai, Y.; Gallo, R.L. Mast cell cathelicidin antimicrobial peptide prevents invasive group A Streptococcus infection of the skin. J. Immunol. 2008, 180, 7565–7573. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Suwanpradid, J.; Sanchez-Lagunes, R.; Choi, H.W.; Hoang, P.; Wang, D.; Abraham, S.N.; MacLeod, A.S.; Information, P.E.K.F.C. IL-27 Facilitates Skin Wound Healing through Induction of Epidermal Proliferation and Host Defense. J. Investig. Dermatol. 2017, 137, 1166–1175. [Google Scholar] [CrossRef]
- Kowalski, R.P.; Romanowski, E.G.; Yates, K.A.; Mah, F.S. An Independent Evaluation of a Novel Peptide Mimetic, Brilacidin (PMX30063), for Ocular Anti-Infective. J. Ocul. Pharmacol. Ther. 2016, 32, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, N.; Endo, M.; Kanno, H.; Matsukawa, N.; Tsutsumi, R.; Takeshita, R.; Sato, S. Polymyxins as Novel and Safe Mucosal Adjuvants to Induce Humoral Immune Responses in Mice. PLoS ONE 2013, 8, e61643. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alkanfari, I.; Freeman, K.B.; Roy, S.; Jahan, T.; Scott, R.W.; Ali, H. Small-Molecule Host-Defense Peptide Mimetic Antibacterial and Antifungal Agents Activate Human and Mouse Mast Cells via Mas-Related GPCRs. Cells 2019, 8, 311. https://doi.org/10.3390/cells8040311
Alkanfari I, Freeman KB, Roy S, Jahan T, Scott RW, Ali H. Small-Molecule Host-Defense Peptide Mimetic Antibacterial and Antifungal Agents Activate Human and Mouse Mast Cells via Mas-Related GPCRs. Cells. 2019; 8(4):311. https://doi.org/10.3390/cells8040311
Chicago/Turabian StyleAlkanfari, Ibrahim, Katie B. Freeman, Saptarshi Roy, Tahsin Jahan, Richard W. Scott, and Hydar Ali. 2019. "Small-Molecule Host-Defense Peptide Mimetic Antibacterial and Antifungal Agents Activate Human and Mouse Mast Cells via Mas-Related GPCRs" Cells 8, no. 4: 311. https://doi.org/10.3390/cells8040311
APA StyleAlkanfari, I., Freeman, K. B., Roy, S., Jahan, T., Scott, R. W., & Ali, H. (2019). Small-Molecule Host-Defense Peptide Mimetic Antibacterial and Antifungal Agents Activate Human and Mouse Mast Cells via Mas-Related GPCRs. Cells, 8(4), 311. https://doi.org/10.3390/cells8040311