mTOR Signalling in Head and Neck Cancer: Heads Up



Abstract
:1. Background
2. Genomic Alterations in Head and Neck Cancer
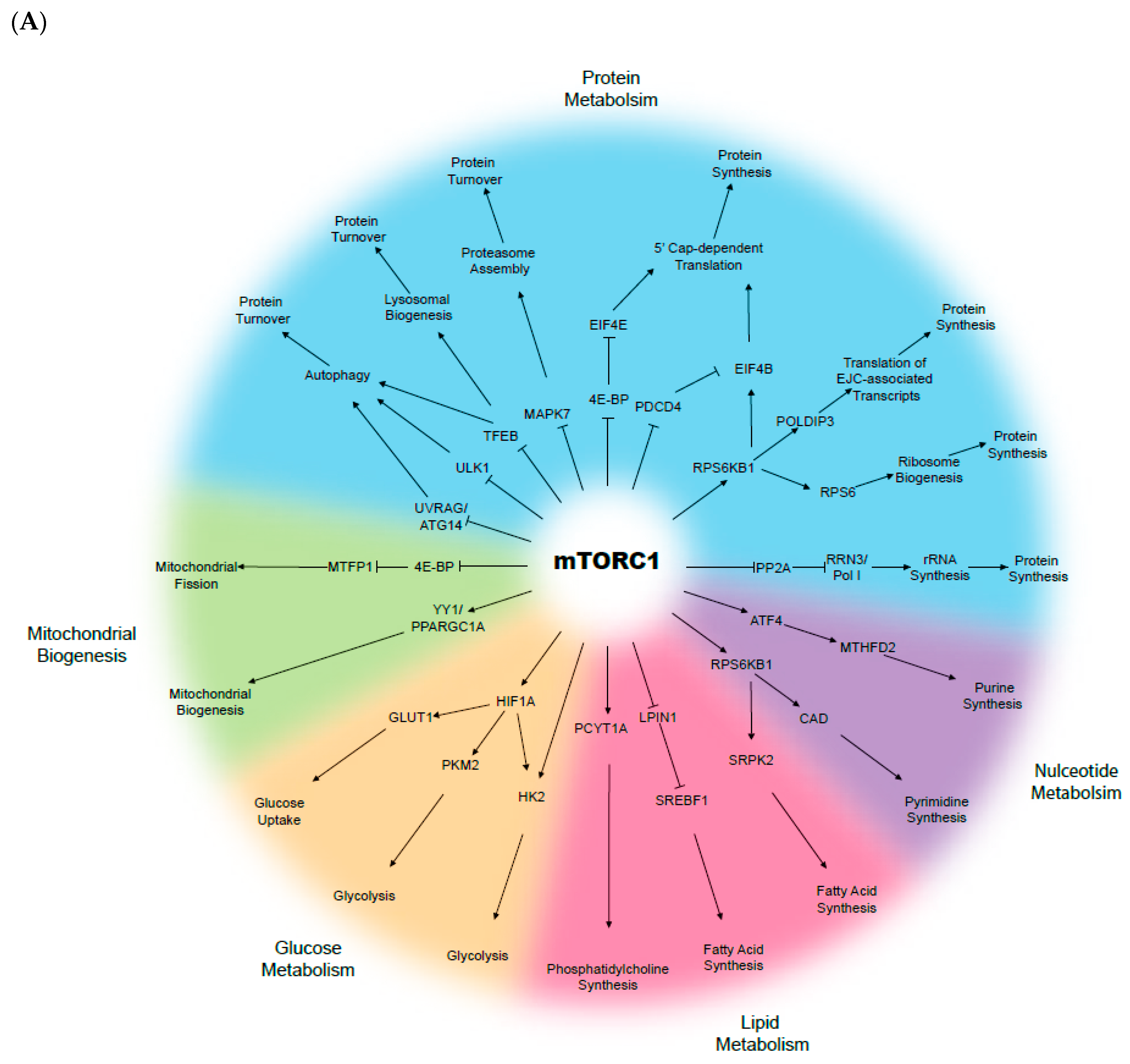
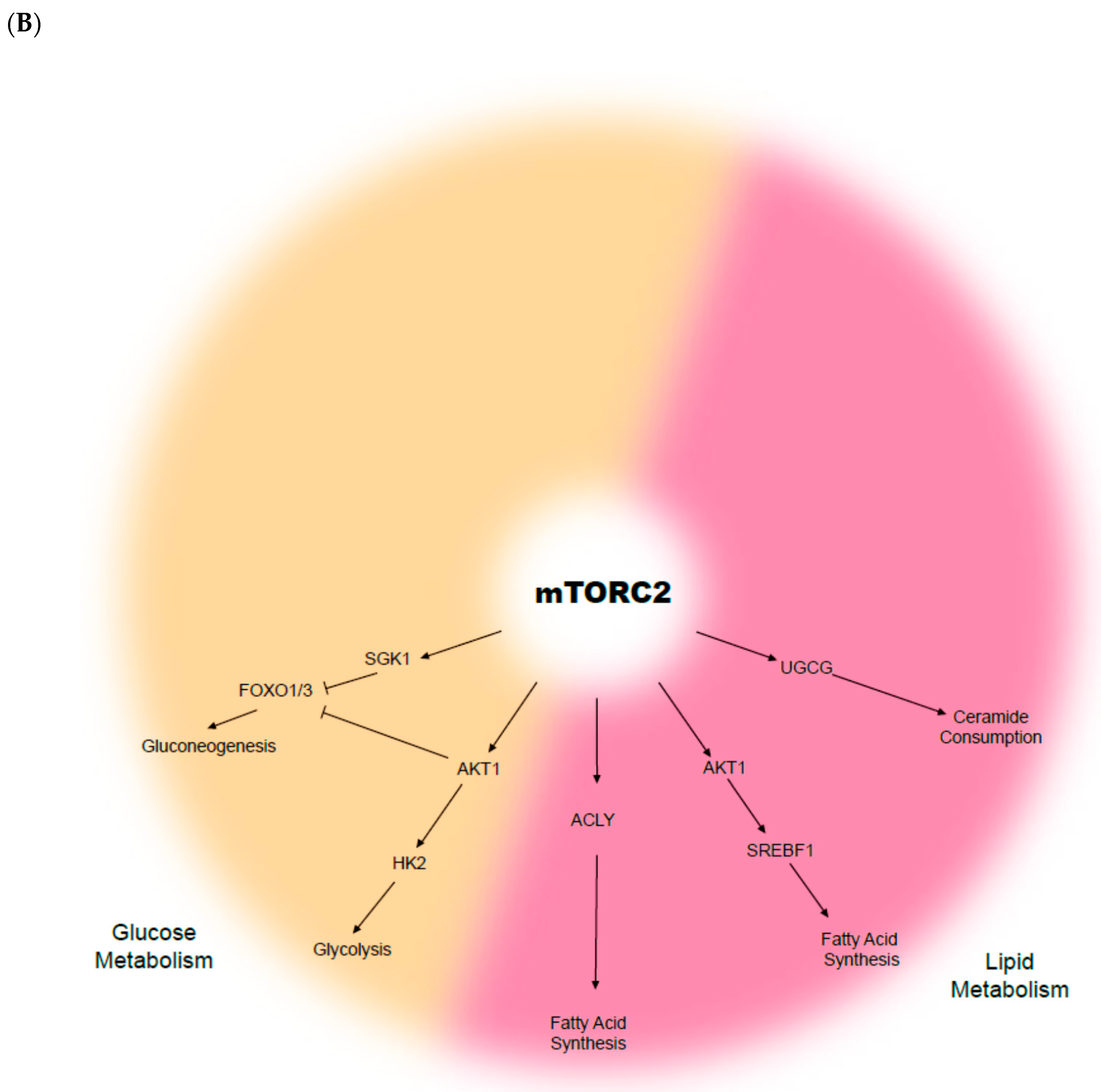
3. The mTOR Complex and the Cellular Metabolism
4. Deregulated mTOR Signalling in HNSCC
5. HPV Status, mTOR Activation and Metabolism in HNSCC
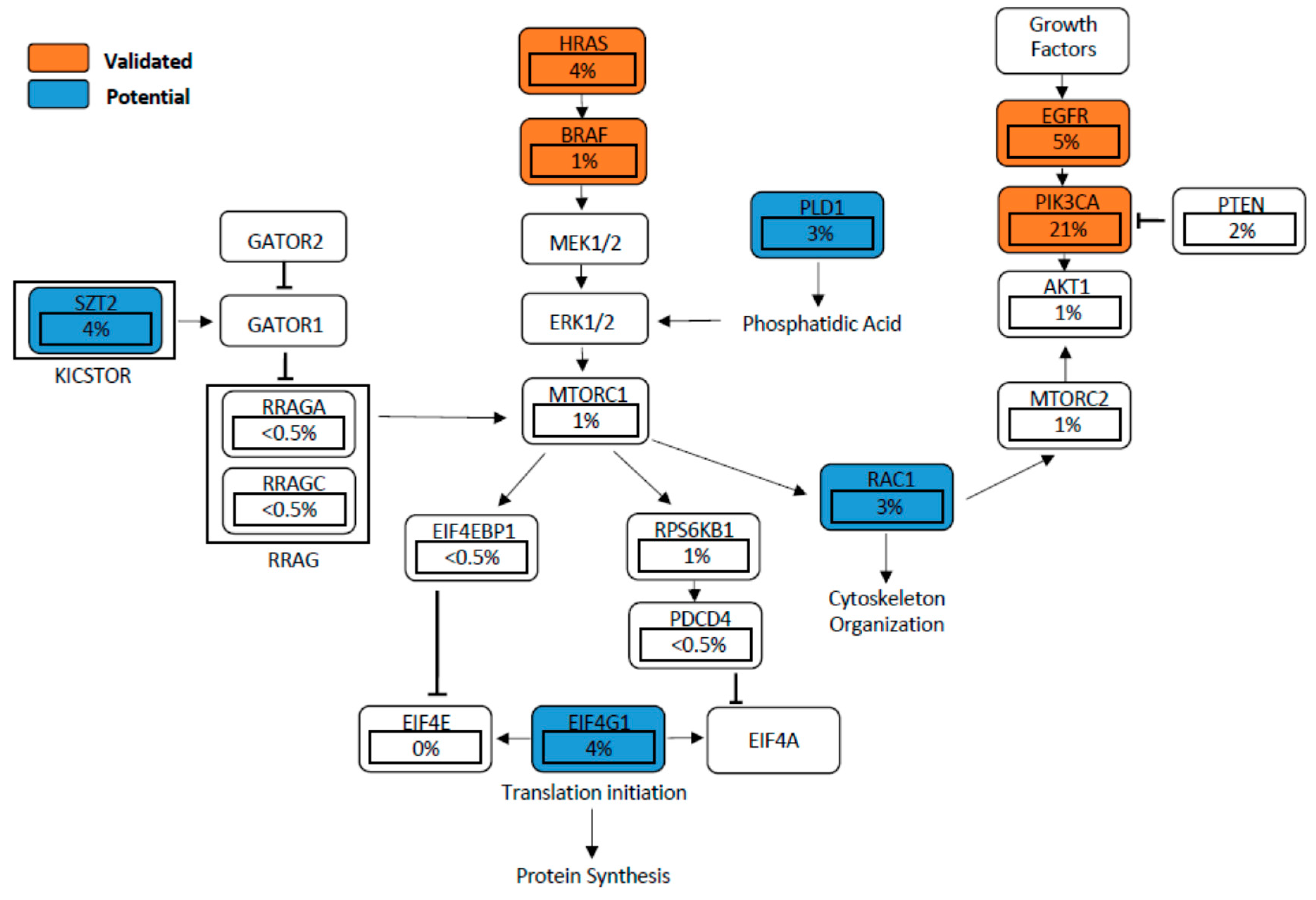
6. Validated Mutant Genes Known to Drive Activation of mTOR Signalling in HNSCC
6.1. EGFR-PI3K-AKT-mTOR Pathway
6.2. PIK3CA Mutation and PTEN Loss
6.3. HRAS
7. Potential Mutant Genes Activating the mTOR Signalling Pathway
7.1. EIF4G1
7.2. RAC1
7.3. SZT2
7.4. PLD1
8. Current Clinical Trials Targeting mTOR in HNSCC
9. Conclusions and Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics 2015. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef]
- Wang, X.; Xu, J.; Wang, L.; Liu, C.; Wang, H. The role of cigarette smoking and alcohol consumption in the differentiation of oral squamous cell carcinoma for the males in China. J. Cancer Res. Ther. 2015, 11, 141–145. [Google Scholar] [CrossRef]
- Lubin, J.H.; Purdue, M.; Kelsey, K.; Zhang, Z.F.; Winn, D.; Wei, Q.; Talamini, R.; Szeszenia-Dabrowska, N.; Sturgis, E.M.; Smith, E.; et al. Total exposure and exposure rate effects for alcohol and smoking and risk of head and neck cancer: A pooled analysis of case-control studies. Am. J. Epidemiol. 2009, 170, 937–947. [Google Scholar] [CrossRef]
- Bagnardi, V.; Rota, M.; Botteri, E.; Tramacere, I.; Islami, F.; Fedirko, V.; Scotti, L.; Jenab, M.; Turati, F.; Pasquali, E.; et al. Alcohol consumption and site-specific cancer risk: A comprehensive dose-response meta-analysis. Br. J. Cancer 2015, 112, 580–593. [Google Scholar] [CrossRef]
- Dalla Torre, D.; Burtscher, D.; Soelder, E.; Offermanns, V.; Rasse, M.; Puelacher, W. HPV prevalence in a Mid-European oral squamous cell cancer population: A cohort study. Oral Dis. 2018, 24, 948–956. [Google Scholar] [CrossRef]
- Young, D.; Xiao, C.C.; Murphy, B.; Moore, M.; Fakhry, C.; Day, T.A. Increase in head and neck cancer in younger patients due to human papillomavirus (HPV). Oral Oncol. 2015, 51, 727–730. [Google Scholar] [CrossRef]
- Kreimer, A.R.; Clifford, G.M.; Boyle, P.; Franceschi, S. Human papillomavirus types in head and neck squamous cell carcinomas worldwide: A systematic review. Cancer Epidemiol. Biomark. Prev. 2005, 14, 467–475. [Google Scholar] [CrossRef]
- Chaturvedi, A.K.; Engels, E.A.; Anderson, W.F.; Gillison, M.L. Incidence trends for human papillomavirus-related and -unrelated oral squamous cell carcinomas in the United States. J. Clin. Oncol. 2008, 26, 612–619. [Google Scholar] [CrossRef]
- Marur, S.; D’Souza, G.; Westra, W.H.; Forastiere, A.A. HPV-associated head and neck cancer: A virus-related cancer epidemic. Lancet Oncol. 2010, 11, 781–789. [Google Scholar] [CrossRef]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tan, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef]
- Griffin, S.; Walker, S.; Sculpher, M.; White, S.; Erhorn, S.; Brent, S.; Dyker, A.; Ferrie, L.; Gilfillan, C.; Horsley, W.; et al. Cetuximab plus radiotherapy for the treatment of locally advanced squamous cell carcinoma of the head and neck. Health Technol. Assess. 2009, 13 Suppl. 1, 49–54. [Google Scholar] [CrossRef]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, J.; et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2006, 354, 567–578. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- Seiwert, T.Y.; Burtness, B.; Mehra, R.; Weiss, J.; Berger, R.; Eder, J.P.; Heath, K.; McClanahan, T.; Lunceford, J.; Gause, C.; et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): An open-label, multicentre, phase 1b trial. Lancet Oncol. 2016, 17, 956–965. [Google Scholar] [CrossRef]
- Chow, L.Q.M.; Haddad, R.; Gupta, S.; Mahipal, A.; Mehra, R.; Tahara, M.; Berger, R.; Eder, J.P.; Burtness, B.; Lee, S.H.; et al. Antitumor Activity of Pembrolizumab in Biomarker-Unselected Patients With Recurrent and/or Metastatic Head and Neck Squamous Cell Carcinoma: Results From the Phase Ib KEYNOTE-012 Expansion Cohort. J. Clin. Oncol. 2016, 34, 3838–3845. [Google Scholar] [CrossRef]
- Champiat, S.; Ferrara, R.; Massard, C.; Besse, B.; Marabelle, A.; Soria, J.C.; Ferte, C. Hyperprogressive disease: Recognizing a novel pattern to improve patient management. Nat. Rev. Clin. Oncol. 2018, 15, 748–762. [Google Scholar] [CrossRef]
- Saada-Bouzid, E.; Defaucheux, C.; Karabajakian, A.; Coloma, V.P.; Servois, V.; Paoletti, X.; Even, C.; Fayette, J.; Guigay, J.; Loirat, D.; et al. Hyperprogression during anti-PD-1/PD-L1 therapy in patients with recurrent and/or metastatic head and neck squamous cell carcinoma. Ann. Oncol. 2017, 28, 1605–1611. [Google Scholar] [CrossRef]
- Pulte, D.; Brenner, H. Changes in survival in head and neck cancers in the late 20th and early 21st century: A period analysis. Oncologist 2010, 15, 994–1001. [Google Scholar] [CrossRef]
- Pickering, C.R.; Zhang, J.; Yoo, S.Y.; Bengtsson, L.; Moorthy, S.; Neskey, D.M.; Zhao, M.; Ortega Alves, M.V.; Chang, K.; Drummond, J.; et al. Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer Discov. 2013, 3, 770–781. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar]
- Lui, V.W.; Hedberg, M.L.; Li, H.; Vangara, B.S.; Pendleton, K.; Zeng, Y.; Lu, Y.; Zhang, Q.; Du, Y.; Gilbert, B.R.; et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013, 3, 761–769. [Google Scholar] [CrossRef]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, J.; Fu, X.; Yang, A. Identification of Key Genes and Pathways in Tongue Squamous Cell Carcinoma Using Bioinformatics Analysis. Med. Sci. Monit. 2017, 23, 5924–5932. [Google Scholar] [CrossRef]
- Cuevas Gonzalez, J.C.; Gaitan Cepeda, L.A.; Borges Yanez, S.A.; Cornejo, A.D.; Mori Estevez, A.D.; Huerta, E.R. p53 and p16 in oral epithelial dysplasia and oral squamous cell carcinoma: A study of 208 cases. Indian J. Pathol. Microbiol. 2016, 59, 153–158. [Google Scholar]
- Karpathiou, G.; Monaya, A.; Forest, F.; Froudarakis, M.; Casteillo, F.; Marc Dumollard, J.; Prades, J.M.; Peoc’h, M. p16 and p53 expression status in head and neck squamous cell carcinoma: A correlation with histological, histoprognostic and clinical parameters. Pathology 2016, 48, 341–348. [Google Scholar] [CrossRef]
- Lin, S.C.; Lin, L.H.; Yu, S.Y.; Kao, S.Y.; Chang, K.W.; Cheng, H.W.; Liu, C.J. FAT1 somatic mutations in head and neck carcinoma are associated with tumor progression and survival. Carcinogenesis 2018, 39, 1320–1330. [Google Scholar] [CrossRef]
- Lee, S.H.; Do, S.I.; Lee, H.J.; Kang, H.J.; Koo, B.S.; Lim, Y.C. Notch1 signaling contributes to stemness in head and neck squamous cell carcinoma. Lab. Investig. 2016, 96, 508–516. [Google Scholar] [CrossRef]
- Plath, M.; Broglie, M.A.; Forbs, D.; Stoeckli, S.J.; Jochum, W. Prognostic significance of cell cycle-associated proteins p16, pRB, cyclin D1 and p53 in resected oropharyngeal carcinoma. J. Otolaryngol. Head Neck Surg. 2018, 47, 53. [Google Scholar] [CrossRef]
- Belobrov, S.; Cornall, A.M.; Young, R.J.; Koo, K.; Angel, C.; Wiesenfeld, D.; Rischin, D.; Garland, S.M.; McCullough, M. The role of human papillomavirus in p16-positive oral cancers. J. Oral Pathol. Med.: Off. Publ. Int. Assoc. Oral Pathol. Am. Acad. Oral Pathol. 2018, 47, 18–24. [Google Scholar] [CrossRef]
- Cheng, H.; Yang, X.; Si, H.; Saleh, A.D.; Xiao, W.; Coupar, J.; Gollin, S.M.; Ferris, R.L.; Issaeva, N.; Yarbrough, W.G.; et al. Genomic and Transcriptomic Characterization Links Cell Lines with Aggressive Head and Neck Cancers. Cell Rep. 2018, 25, 1332–1345.e5. [Google Scholar] [CrossRef]
- Nishikawa, Y.; Miyazaki, T.; Nakashiro, K.; Yamagata, H.; Isokane, M.; Goda, H.; Tanaka, H.; Oka, R.; Hamakawa, H. Human FAT1 cadherin controls cell migration and invasion of oral squamous cell carcinoma through the localization of beta-catenin. Oncol. Rep. 2011, 26, 587–592. [Google Scholar]
- Yoshida, R.; Nagata, M.; Nakayama, H.; Niimori-Kita, K.; Hassan, W.; Tanaka, T.; Shinohara, M.; Ito, T. The pathological significance of Notch1 in oral squamous cell carcinoma. Lab. Investig. 2013, 93, 1068–1081. [Google Scholar] [CrossRef]
- Agrawal, N.; Frederick, M.J.; Pickering, C.R.; Bettegowda, C.; Chang, K.; Li, R.J.; Fakhry, C.; Xie, T.X.; Zhang, J.; Wang, J.; et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011, 333, 1154–1157. [Google Scholar] [CrossRef]
- Grabiner, B.C.; Nardi, V.; Birsoy, K.; Possemato, R.; Shen, K.; Sinha, S.; Jordan, A.; Beck, A.H.; Sabatini, D.M. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov. 2014, 4, 554–563. [Google Scholar] [CrossRef]
- Li, S.; Wang, Z.; Huang, J.; Cheng, S.; Du, H.; Che, G.; Peng, Y. Clinicopathological and prognostic significance of mTOR and phosphorylated mTOR expression in patients with esophageal squamous cell carcinoma: A systematic review and meta-analysis. BMC Cancer 2016, 16, 877. [Google Scholar] [CrossRef]
- Driscoll, D.R.; Karim, S.A.; Sano, M.; Gay, D.M.; Jacob, W.; Yu, J.; Mizukami, Y.; Gopinathan, A.; Jodrell, D.I.; Evans, T.R.; et al. mTORC2 Signaling Drives the Development and Progression of Pancreatic Cancer. Cancer Res. 2016, 76, 6911–6923. [Google Scholar] [CrossRef]
- Vander Haar, E.; Lee, S.I.; Bandhakavi, S.; Griffin, T.J.; Kim, D.H. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 2007, 9, 316–323. [Google Scholar] [CrossRef]
- Liu, P.; Gan, W.; Inuzuka, H.; Lazorchak, A.S.; Gao, D.; Arojo, O.; Liu, D.; Wan, L.; Zhai, B.; Yu, Y.; et al. Sin1 phosphorylation impairs mTORC2 complex integrity and inhibits downstream Akt signalling to suppress tumorigenesis. Nat. Cell Biol. 2013, 15, 1340–1350. [Google Scholar] [CrossRef]
- Tenkerian, C.; Krishnamoorthy, J.; Mounir, Z.; Kazimierczak, U.; Khoutorsky, A.; Staschke, K.A.; Kristof, A.S.; Wang, S.; Hatzoglou, M.; Koromilas, A.E. mTORC2 Balances AKT Activation and eIF2alpha Serine 51 Phosphorylation to Promote Survival under Stress. Mol. Cancer Res. 2015, 13, 1377–1388. [Google Scholar] [CrossRef]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Buller, C.L.; Loberg, R.D.; Fan, M.H.; Zhu, Q.; Park, J.L.; Vesely, E.; Inoki, K.; Guan, K.L.; Brosius, F.C., 3rd. A GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1 glucose transporter expression. Am. J. Physiol. Cell Physiol. 2008, 295, C836–C843. [Google Scholar]
- Li, S.; Yang, X.; Wang, P.; Ran, X. The effects of GLUT1 on the survival of head and neck squamous cell carcinoma. Cell Physiol. Biochem. 2013, 32, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Koukourakis, M.I.; Giatromanolaki, A.; Sivridis, E.; Simopoulos, C.; Turley, H.; Talks, K.; Gatter, K.C.; Harris, A.L. Hypoxia-inducible factor (HIF1A and HIF2A), angiogenesis, and chemoradiotherapy outcome of squamous cell head-and-neck cancer. Int. J. Radiat. Oncol. Biol. Phys. 2002, 53, 1192–1202. [Google Scholar] [CrossRef]
- Kleszcz, R.; Paluszczak, J.; Krajka-Kuzniak, V.; Baer-Dubowska, W. The inhibition of c-MYC transcription factor modulates the expression of glycolytic and glutaminolytic enzymes in FaDu hypopharyngeal carcinoma cells. Adv. Clin. Exp. Med. 2018, 27, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Szablewski, L. Expression of glucose transporters in cancers. Biochim. Biophys. Acta 2013, 1835, 164–169. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, S.; Li, Y.; Tang, Z.; Kong, W. Hexokinase 2 overexpression promotes the proliferation and survival of laryngeal squamous cell carcinoma. Tumour Biol. 2014, 35, 3743–3753. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Pilo, G.M.; Li, X.; Cigliano, A.; Latte, G.; Che, L.; Joseph, C.; Mela, M.; Wang, C.; Jiang, L.; et al. Inactivation of fatty acid synthase impairs hepatocarcinogenesis driven by AKT in mice and humans. J. Hepatol. 2016, 64, 333–341. [Google Scholar] [CrossRef]
- Ricoult, S.J.; Yecies, J.L.; Ben-Sahra, I.; Manning, B.D. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene 2016, 35, 1250–1260. [Google Scholar] [CrossRef]
- Guri, Y.; Colombi, M.; Dazert, E.; Hindupur, S.K.; Roszik, J.; Moes, S.; Jenoe, P.; Heim, M.H.; Riezman, I.; Riezman, H.; et al. mTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell 2017, 32, 807–823 e12. [Google Scholar] [CrossRef]
- Wang, Z.; Martin, D.; Molinolo, A.A.; Patel, V.; Iglesias-Bartolome, R.; Degese, M.S.; Vitale-Cross, L.; Chen, Q.; Gutkind, J.S. mTOR co-targeting in cetuximab resistance in head and neck cancers harboring PIK3CA and RAS mutations. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef]
- Tian, T.; Li, X.; Zhang, J. mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef]
- Amornphimoltham, P.; Leelahavanichkul, K.; Molinolo, A.; Patel, V.; Gutkind, J.S. Inhibition of Mammalian target of rapamycin by rapamycin causes the regression of carcinogen-induced skin tumor lesions. Clin. Cancer Res. 2008, 14, 8094–8101. [Google Scholar] [CrossRef]
- Callejas-Valera, J.L.; Iglesias-Bartolome, R.; Amornphimoltham, P.; Palacios-Garcia, J.; Martin, D.; Califano, J.A.; Molinolo, A.A.; Gutkind, J.S. mTOR inhibition prevents rapid-onset of carcinogen-induced malignancies in a novel inducible HPV-16 E6/E7 mouse model. Carcinogenesis 2016, 37, 1014–1025. [Google Scholar] [CrossRef]
- Czerninski, R.; Amornphimoltham, P.; Patel, V.; Molinolo, A.A.; Gutkind, J.S. Targeting mammalian target of rapamycin by rapamycin prevents tumor progression in an oral-specific chemical carcinogenesis model. Cancer Prev. Res. 2009, 2, 27–36. [Google Scholar] [CrossRef]
- Athar, M.; Kopelovich, L. Rapamycin and mTORC1 inhibition in the mouse: Skin cancer prevention. Cancer Prev. Res. 2011, 4, 957–961. [Google Scholar] [CrossRef]
- Sun, Z.J.; Zhang, L.; Zhang, W.; Hall, B.; Bian, Y.; Kulkarni, A.B. Inhibition of mTOR reduces anal carcinogenesis in transgenic mouse model. PLoS ONE 2013, 8, e74888. [Google Scholar] [CrossRef]
- Zhang, H.; Cohen, A.L.; Krishnakumar, S.; Wapnir, I.L.; Veeriah, S.; Deng, G.; Coram, M.A.; Piskun, C.M.; Longacre, T.A.; Herrler, M.; et al. Patient-derived xenografts of triple-negative breast cancer reproduce molecular features of patient tumors and respond to mTOR inhibition. Breast Cancer Res. 2014, 16, R36. [Google Scholar] [CrossRef]
- Sun, Z.J.; Zhang, L.; Hall, B.; Bian, Y.; Gutkind, J.S.; Kulkarni, A.B. Chemopreventive and chemotherapeutic actions of mTOR inhibitor in genetically defined head and neck squamous cell carcinoma mouse model. Clin. Cancer Res. 2012, 18, 5304–5313. [Google Scholar] [CrossRef]
- Bozec, A.; Ebran, N.; Radosevic-Robin, N.; Sudaka, A.; Monteverde, M.; Toussan, N.; Etienne-Grimaldi, M.C.; Nigro, C.L.; Merlano, M.; Penault-Llorca, F.; et al. Combination of mTOR and EGFR targeting in an orthotopic xenograft model of head and neck cancer. Laryngoscope 2016, 126, E156–E163. [Google Scholar] [CrossRef]
- Moore, E.C.; Cash, H.A.; Caruso, A.M.; Uppaluri, R.; Hodge, J.W.; Van Waes, C.; Allen, C.T. Enhanced Tumor Control with Combination mTOR and PD-L1 Inhibition in Syngeneic Oral Cavity Cancers. Cancer Immunol. Res. 2016, 4, 611–620. [Google Scholar] [CrossRef]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef]
- Abu-Remaileh, M.; Wyant, G.A.; Kim, C.; Laqtom, N.N.; Abbasi, M.; Chan, S.H.; Freinkman, E.; Sabatini, D.M. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 2017, 358, 807–813. [Google Scholar] [CrossRef]
- Bar-Peled, L.; Chantranupong, L.; Cherniack, A.D.; Chen, W.W.; Ottina, K.A.; Grabiner, B.C.; Spear, E.D.; Carter, S.L.; Meyerson, M.; Sabatini, D.M. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 2013, 340, 1100–1106. [Google Scholar] [CrossRef]
- Cosway, B.; Lovat, P. The role of autophagy in squamous cell carcinoma of the head and neck. Oral Oncol. 2016, 54, 1–6. [Google Scholar] [CrossRef]
- Liu, J.L.; Chen, F.F.; Lung, J.; Lo, C.H.; Lee, F.H.; Lu, Y.C.; Hung, C.H. Prognostic significance of p62/SQSTM1 subcellular localization and LC3B in oral squamous cell carcinoma. Br. J. Cancer 2014, 111, 944–954. [Google Scholar] [CrossRef]
- Tang, J.Y.; Hsi, E.; Huang, Y.C.; Hsu, N.C.; Chu, P.Y.; Chai, C.Y. High LC3 expression correlates with poor survival in patients with oral squamous cell carcinoma. Hum. Pathol. 2013, 44, 2558–2562. [Google Scholar] [CrossRef]
- Wright, T.J.; McKee, C.; Birch-Machin, M.A.; Ellis, R.; Armstrong, J.L.; Lovat, P.E. Increasing the therapeutic efficacy of docetaxel for cutaneous squamous cell carcinoma through the combined inhibition of phosphatidylinositol 3-kinase/AKT signalling and autophagy. Clin. Exp. Dermatol. 2013, 38, 421–423. [Google Scholar] [CrossRef]
- Zang, Y.; Thomas, S.M.; Chan, E.T.; Kirk, C.J.; Freilino, M.L.; DeLancey, H.M.; Grandis, J.R.; Li, C.; Johnson, D.E. Carfilzomib and ONX 0912 inhibit cell survival and tumor growth of head and neck cancer and their activities are enhanced by suppression of Mcl-1 or autophagy. Clin. Cancer Res. 2012, 18, 5639–5649. [Google Scholar] [CrossRef]
- Jung, Y.S.; Najy, A.J.; Huang, W.; Sethi, S.; Snyder, M.; Sakr, W.; Dyson, G.; Huttemann, M.; Lee, I.; Ali-Fehmi, R.; et al. HPV-associated differential regulation of tumor metabolism in oropharyngeal head and neck cancer. Oncotarget 2017, 8, 51530–51541. [Google Scholar] [CrossRef]
- Swartz, J.E.; Pothen, A.J.; van Kempen, P.M.; Stegeman, I.; Formsma, F.K.; Cann, E.M.; Willems, S.M.; Grolman, W. Poor prognosis in human papillomavirus-positive oropharyngeal squamous cell carcinomas that overexpress hypoxia inducible factor-1alpha. Head Neck 2016, 38, 1338–1346. [Google Scholar] [CrossRef]
- Rodolico, V.; Arancio, W.; Amato, M.C.; Aragona, F.; Cappello, F.; Di Fede, O.; Pannone, G.; Campisi, G. Hypoxia inducible factor-1 alpha expression is increased in infected positive HPV16 DNA oral squamous cell carcinoma and positively associated with HPV16 E7 oncoprotein. Infect. Agents Cancer 2011, 6, 18. [Google Scholar] [CrossRef]
- Molinolo, A.A.; Marsh, C.; El Dinali, M.; Gangane, N.; Jennison, K.; Hewitt, S.; Patel, V.; Seiwert, T.Y.; Gutkind, J.S. mTOR as a molecular target in HPV-associated oral and cervical squamous carcinomas. Clin. Cancer Res. 2012, 18, 2558–2568. [Google Scholar] [CrossRef]
- Brand, T.M.; Hartmann, S.; Bhola, N.E.; Li, H.; Zeng, Y.; O’Keefe, R.A.; Ranall, M.V.; Bandyopadhyay, S.; Soucheray, M.; Krogan, N.J.; et al. Cross-talk Signaling between HER3 and HPV16 E6 and E7 Mediates Resistance to PI3K Inhibitors in Head and Neck Cancer. Cancer Res. 2018, 78, 2383–2395. [Google Scholar] [CrossRef]
- Madera, D.; Vitale-Cross, L.; Martin, D.; Schneider, A.; Molinolo, A.A.; Gangane, N.; Carey, T.E.; McHugh, J.B.; Komarck, C.M.; Walline, H.M.; et al. Prevention of tumor growth driven by PIK3CA and HPV oncogenes by targeting mTOR signaling with metformin in oral squamous carcinomas expressing OCT3. Cancer Prev. Res. 2015, 8, 197–207. [Google Scholar] [CrossRef]
- Goschzik, T.; Gessi, M.; Denkhaus, D.; Pietsch, T. PTEN mutations and activation of the PI3K/Akt/mTOR signaling pathway in papillary tumors of the pineal region. J. Neuropathol. Exp. Neurol. 2014, 73, 747–751. [Google Scholar] [CrossRef]
- Matsumoto, C.S.; Almeida, L.O.; Guimaraes, D.M.; Martins, M.D.; Papagerakis, P.; Papagerakis, S.; Leopoldino, A.M.; Castilho, R.M.; Squarize, C.H. PI3K-PTEN dysregulation leads to mTOR-driven upregulation of the core clock gene BMAL1 in normal and malignant epithelial cells. Oncotarget 2016, 7, 42393–42407. [Google Scholar] [CrossRef]
- Therkildsen, C.; Bergmann, T.K.; Henrichsen-Schnack, T.; Ladelund, S.; Nilbert, M. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol. 2014, 53, 852–864. [Google Scholar] [CrossRef]
- Nojima, H.; Tokunaga, C.; Eguchi, S.; Oshiro, N.; Hidayat, S.; Yoshino, K.; Hara, K.; Tanaka, N.; Avruch, J.; Yonezawa, K. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J. Biol. Chem. 2003, 278, 15461–15464. [Google Scholar] [CrossRef]
- Schalm, S.S.; Fingar, D.C.; Sabatini, D.M.; Blenis, J. TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr. Biol. 2003, 13, 797–806. [Google Scholar] [CrossRef]
- Wu, C.C.; Hou, S.; Orr, B.A.; Kuo, B.R.; Youn, Y.H.; Ong, T.; Roth, F.; Eberhart, C.G.; Robinson, G.W.; Solecki, D.J.; et al. mTORC1-Mediated Inhibition of 4EBP1 Is Essential for Hedgehog Signaling-Driven Translation and Medulloblastoma. Dev. Cell 2017, 43, 673–688 e5. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar]
- Gkountakos, A.; Pilotto, S.; Mafficini, A.; Vicentini, C.; Simbolo, M.; Milella, M.; Tortora, G.; Scarpa, A.; Bria, E.; Corbo, V. Unmasking the impact of Rictor in cancer: Novel insights of mTORC2 complex. Carcinogenesis 2018. [Google Scholar] [CrossRef]
- Gupta, A.K.; McKenna, W.G.; Weber, C.N.; Feldman, M.D.; Goldsmith, J.D.; Mick, R.; Machtay, M.; Rosenthal, D.I.; Bakanauskas, V.J.; Cerniglia, G.J.; et al. Local recurrence in head and neck cancer: Relationship to radiation resistance and signal transduction. Clin. Cancer Res. 2002, 8, 885–892. [Google Scholar]
- Li, Z.; Yang, Z.; Passaniti, A.; Lapidus, R.G.; Liu, X.; Cullen, K.J.; Dan, H.C. A positive feedback loop involving EGFR/Akt/mTORC1 and IKK/NF-kB regulates head and neck squamous cell carcinoma proliferation. Oncotarget 2016, 7, 31892–31906. [Google Scholar]
- Elkabets, M.; Pazarentzos, E.; Juric, D.; Sheng, Q.; Pelossof, R.A.; Brook, S.; Benzaken, A.O.; Rodon, J.; Morse, N.; Yan, J.J.; et al. AXL mediates resistance to PI3Kalpha inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell 2015, 27, 533–546. [Google Scholar] [CrossRef]
- Bigner, S.H.; Humphrey, P.A.; Wong, A.J.; Vogelstein, B.; Mark, J.; Friedman, H.S.; Bigner, D.D. Characterization of the epidermal growth factor receptor in human glioma cell lines and xenografts. Cancer Res. 1990, 50, 8017–8022. [Google Scholar]
- Sok, J.C.; Coppelli, F.M.; Thomas, S.M.; Lango, M.N.; Xi, S.; Hunt, J.L.; Freilino, M.L.; Graner, M.W.; Wikstrand, C.J.; Bigner, D.D.; et al. Mutant epidermal growth factor receptor (EGFRvIII) contributes to head and neck cancer growth and resistance to EGFR targeting. Clin. Cancer Res. 2006, 12, 5064–5073. [Google Scholar] [CrossRef]
- Khattri, A.; Zuo, Z.; Bragelmann, J.; Keck, M.K.; El Dinali, M.; Brown, C.D.; Stricker, T.; Munagala, A.; Cohen, E.E.; Lingen, M.W.; et al. Rare occurrence of EGFRvIII deletion in head and neck squamous cell carcinoma. Oral Oncol. 2015, 51, 53–58. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwinska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. (Pozn) 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- Hashmi, A.A.; Hussain, Z.F.; Aijaz, S.; Irfan, M.; Khan, E.Y.; Naz, S.; Faridi, N.; Khan, A.; Edhi, M.M. Immunohistochemical expression of epidermal growth factor receptor (EGFR) in South Asian head and neck squamous cell carcinoma: Association with various risk factors and clinico-pathologic and prognostic parameters. World J. Surg. Oncol. 2018, 16, 118. [Google Scholar] [CrossRef]
- Makinoshima, H.; Takita, M.; Saruwatari, K.; Umemura, S.; Obata, Y.; Ishii, G.; Matsumoto, S.; Sugiyama, E.; Ochiai, A.; Abe, R.; et al. Signaling through the Phosphatidylinositol 3-Kinase (PI3K)/Mammalian Target of Rapamycin (mTOR) Axis Is Responsible for Aerobic Glycolysis mediated by Glucose Transporter in Epidermal Growth Factor Receptor (EGFR)-mutated Lung Adenocarcinoma. J. Biol. Chem. 2015, 290, 17495–17504. [Google Scholar] [CrossRef]
- Chiang, C.T.; Demetriou, A.N.; Ung, N.; Choudhury, N.; Ghaffarian, K.; Ruderman, D.L.; Mumenthaler, S.M. mTORC2 contributes to the metabolic reprogramming in EGFR tyrosine-kinase inhibitor resistant cells in non-small cell lung cancer. Cancer Lett. 2018, 434, 152–159. [Google Scholar] [CrossRef]
- Cohen, E.E.; Davis, D.W.; Karrison, T.G.; Seiwert, T.Y.; Wong, S.J.; Nattam, S.; Kozloff, M.F.; Clark, J.I.; Yan, D.H.; Liu, W.; et al. Erlotinib and bevacizumab in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck: A phase I/II study. Lancet Oncol. 2009, 10, 247–257. [Google Scholar] [CrossRef]
- Sharp, H.; Morris, J.C.; Van Waes, C.; Gius, D.; Cooley-Zgela, T.; Singh, A.K. High incidence of oral dysesthesias on a trial of gefitinib, Paclitaxel, and concurrent external beam radiation for locally advanced head and neck cancers. Am. J. Clin. Oncol. 2008, 31, 557–560. [Google Scholar] [CrossRef]
- Xu, M.J.; Johnson, D.E.; Grandis, J.R. EGFR-targeted therapies in the post-genomic era. Cancer Metastasis Rev. 2017, 36, 463–473. [Google Scholar] [CrossRef]
- Porcelli, L.; Quatrale, A.E.; Mantuano, P.; Silvestris, N.; Rolland, J.F.; Biancolillo, L.; Paradiso, A.; Azzariti, A. Synergistic antiproliferative and antiangiogenic effects of EGFR and mTOR inhibitors. Curr. Pharm. Des. 2013, 19, 918–926. [Google Scholar] [CrossRef]
- Schmid, K.; Bago-Horvath, Z.; Berger, W.; Haitel, A.; Cejka, D.; Werzowa, J.; Filipits, M.; Herberger, B.; Hayden, H.; Sieghart, W. Dual inhibition of EGFR and mTOR pathways in small cell lung cancer. Br. J. Cancer 2010, 103, 622–628. [Google Scholar] [CrossRef]
- Boumahdi, S.; Driessens, G.; Lapouge, G.; Rorive, S.; Nassar, D.; Le Mercier, M.; Delatte, B.; Caauwe, A.; Lenglez, S.; Nkusi, E.; et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature 2014, 511, 246–250. [Google Scholar] [CrossRef]
- Keysar, S.B.; Le, P.N.; Miller, B.; Jackson, B.C.; Eagles, J.R.; Nieto, C.; Kim, J.; Tang, B.; Glogowska, M.J.; Morton, J.J.; et al. Regulation of Head and Neck Squamous Cancer Stem Cells by PI3K and SOX2. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef]
- Huang, C.F.; Xu, X.R.; Wu, T.F.; Sun, Z.J.; Zhang, W.F. Correlation of ALDH1, CD44, OCT4 and SOX2 in tongue squamous cell carcinoma and their association with disease progression and prognosis. J. Oral Pathol. Med.: Off. Publ. Int. Assoc. Oral Pathol. Am. Acad. Oral Pathol. 2014, 43, 492–498. [Google Scholar] [CrossRef]
- Suda, T.; Hama, T.; Kondo, S.; Yuza, Y.; Yoshikawa, M.; Urashima, M.; Kato, T.; Moriyama, H. Copy number amplification of the PIK3CA gene is associated with poor prognosis in non-lymph node metastatic head and neck squamous cell carcinoma. BMC Cancer 2012, 12, 416. [Google Scholar] [CrossRef]
- Hao, Y.; Samuels, Y.; Li, Q.; Krokowski, D.; Guan, B.J.; Wang, C.; Jin, Z.; Dong, B.; Cao, B.; Feng, X.; et al. Oncogenic PIK3CA mutations reprogram glutamine metabolism in colorectal cancer. Nat. Commun. 2016, 7, 11971. [Google Scholar] [CrossRef]
- Lau, C.E.; Tredwell, G.D.; Ellis, J.K.; Lam, E.W.; Keun, H.C. Metabolomic characterisation of the effects of oncogenic PIK3CA transformation in a breast epithelial cell line. Sci. Rep. 2017, 7, 46079. [Google Scholar] [CrossRef]
- Pedrero, J.M.; Carracedo, D.G.; Pinto, C.M.; Zapatero, A.H.; Rodrigo, J.P.; Nieto, C.S.; Gonzalez, M.V. Frequent genetic and biochemical alterations of the PI 3-K/AKT/PTEN pathway in head and neck squamous cell carcinoma. Int. J. Cancer 2005, 114, 242–248. [Google Scholar] [CrossRef]
- Henderson, Y.C.; Wang, E.; Clayman, G.L. Genotypic analysis of tumor suppressor genes PTEN/MMAC1 and p53 in head and neck squamous cell carcinomas. Laryngoscope 1998, 108, 1553–1556. [Google Scholar] [CrossRef]
- Shao, X.; Tandon, R.; Samara, G.; Kanki, H.; Yano, H.; Close, L.G.; Parsons, R.; Sato, T. Mutational analysis of the PTEN gene in head and neck squamous cell carcinoma. Int. J. Cancer 1998, 77, 684–688. [Google Scholar] [CrossRef]
- Darido, C.; Georgy, S.R.; Wilanowski, T.; Dworkin, S.; Auden, A.; Zhao, Q.; Rank, G.; Srivastava, S.; Finlay, M.J.; Papenfuss, A.T.; et al. Targeting of the tumor suppressor GRHL3 by a miR-21-dependent proto-oncogenic network results in PTEN loss and tumorigenesis. Cancer Cell 2011, 20, 635–648. [Google Scholar] [CrossRef]
- Cangkrama, M.; Ting, S.B.; Darido, C. Stem cells behind the barrier. Int. J. Mol. Sci. 2013, 14, 13670–13686. [Google Scholar] [CrossRef]
- Cangkrama, M.; Darido, C.; Georgy, S.R.; Partridge, D.; Auden, A.; Srivastava, S.; Wilanowski, T.; Jane, S.M. Two Ancient Gene Families Are Critical for Maintenance of the Mammalian Skin Barrier in Postnatal Life. J. Investig. Dermatol. 2016, 136, 1438–1448. [Google Scholar] [CrossRef]
- Georgy, S.R.; Cangkrama, M.; Srivastava, S.; Partridge, D.; Auden, A.; Dworkin, S.; McLean, C.A.; Jane, S.M.; Darido, C. Identification of a Novel Proto-oncogenic Network in Head and Neck Squamous Cell Carcinoma. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- Miles, L.B.; Dworkin, S.; Darido, C. Review article: Alternative splicing and start sites: Lessons from the Grainyhead-like family. Dev. Biol. 2017, 429, 12–19. [Google Scholar] [CrossRef]
- Darido, C.; Georgy, S.R.; Jane, S.M. The role of barrier genes in epidermal malignancy. Oncogene 2016, 35, 5705–5712. [Google Scholar] [CrossRef]
- Youssef, M.; Cuddihy, A.; Darido, C. Long-Lived Epidermal Cancer-Initiating Cells. Int. J. Mol. Sci. 2017, 18, 1369. [Google Scholar] [CrossRef]
- Darido, C.; Georgy, S.R.; Cullinane, C.; Partridge, D.D.; Walker, R.; Srivastava, S.; Roslan, S.; Carpinelli, M.R.; Dworkin, S.; Pearson, R.B.; et al. Stage-dependent therapeutic efficacy in PI3K/mTOR-driven squamous cell carcinoma of the skin. Cell Death Differ. 2018, 25, 1146–1159. [Google Scholar] [CrossRef]
- Lee, J.I.; Soria, J.C.; Hassan, K.A.; El-Naggar, A.K.; Tang, X.; Liu, D.D.; Hong, W.K.; Mao, L. Loss of PTEN expression as a prognostic marker for tongue cancer. Arch. Otolaryngol. Head Neck Surg. 2001, 127, 1441–1445. [Google Scholar] [CrossRef]
- Squarize, C.H.; Castilho, R.M.; Santos Pinto, D., Jr. Immunohistochemical evidence of PTEN in oral squamous cell carcinoma and its correlation with the histological malignancy grading system. J. Oral Pathol. Med.: Off. Publ. Int. Assoc. Oral Pathol. Am. Acad. Oral Pathol. 2002, 31, 379–384. [Google Scholar] [CrossRef]
- Bian, Y.; Hall, B.; Sun, Z.J.; Molinolo, A.; Chen, W.; Gutkind, J.S.; Waes, C.V.; Kulkarni, A.B. Loss of TGF-beta signaling and PTEN promotes head and neck squamous cell carcinoma through cellular senescence evasion and cancer-related inflammation. Oncogene 2012, 31, 3322–3332. [Google Scholar] [CrossRef]
- Martin, P.L.; Yin, J.J.; Seng, V.; Casey, O.; Corey, E.; Morrissey, C.; Simpson, R.M.; Kelly, K. Androgen deprivation leads to increased carbohydrate metabolism and hexokinase 2-mediated survival in Pten/Tp53-deficient prostate cancer. Oncogene 2017, 36, 525–533. [Google Scholar] [CrossRef]
- Mathur, D.; Stratikopoulos, E.; Ozturk, S.; Steinbach, N.; Pegno, S.; Schoenfeld, S.; Yong, R.; Murty, V.V.; Asara, J.M.; Cantley, L.C.; et al. PTEN Regulates Glutamine Flux to Pyrimidine Synthesis and Sensitivity to Dihydroorotate Dehydrogenase Inhibition. Cancer Discov. 2017, 7, 380–390. [Google Scholar] [CrossRef]
- Garcia-Cao, I.; Song, M.S.; Hobbs, R.M.; Laurent, G.; Giorgi, C.; de Boer, V.C.; Anastasiou, D.; Ito, K.; Sasaki, A.T.; Rameh, L.; et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell 2012, 149, 49–62. [Google Scholar] [CrossRef]
- Kiaris, H.; Spandidos, D.A.; Jones, A.S.; Vaughan, E.D.; Field, J.K. Mutations, expression and genomic instability of the H-ras proto-oncogene in squamous cell carcinomas of the head and neck. Br. J. Cance 1995, 72, 123–128. [Google Scholar] [CrossRef]
- Nakagaki, T.; Tamura, M.; Kobashi, K.; Omori, A.; Koyama, R.; Idogawa, M.; Ogi, K.; Hiratsuka, H.; Tokino, T.; Sasaki, Y. Targeted next-generation sequencing of 50 cancer-related genes in Japanese patients with oral squamous cell carcinoma. Tumour Biol. 2018, 40, 1010428318800180. [Google Scholar] [CrossRef]
- Su, S.C.; Lin, C.W.; Liu, Y.F.; Fan, W.L.; Chen, M.K.; Yu, C.P.; Yang, W.E.; Su, C.W.; Chuang, C.Y.; Li, W.H.; et al. Exome Sequencing of Oral Squamous Cell Carcinoma Reveals Molecular Subgroups and Novel Therapeutic Opportunities. Theranostics 2017, 7, 1088–1099. [Google Scholar] [CrossRef]
- Koumaki, D.; Kostakis, G.; Koumaki, V.; Papadogeorgakis, N.; Makris, M.; Katoulis, A.; Kamakari, S.; Koutsodontis, G.; Perisanidis, C.; Lambadiari, V.; et al. Novel mutations of the HRAS gene and absence of hotspot mutations of the BRAF genes in oral squamous cell carcinoma in a Greek population. Oncol. Rep. 2012, 27, 1555–1560. [Google Scholar] [CrossRef]
- Murugan, A.K.; Hong, N.T.; Cuc, T.T.; Hung, N.C.; Munirajan, A.K.; Ikeda, M.A.; Tsuchida, N. Detection of two novel mutations and relatively high incidence of H-RAS mutations in Vietnamese oral cancer. Oral Oncol. 2009, 45, e161–e166. [Google Scholar] [CrossRef]
- Zheng, W.; Tayyari, F.; Gowda, G.A.; Raftery, D.; McLamore, E.S.; Porterfield, D.M.; Donkin, S.S.; Bequette, B.; Teegarden, D. Altered glucose metabolism in Harvey-ras transformed MCF10A cells. Mol. Carcinog. 2015, 54, 111–120. [Google Scholar] [CrossRef]
- Ruicci, K.M.; Pinto, N.; Khan, M.I.; Yoo, J.; Fung, K.; MacNeil, D.; Mymryk, J.S.; Barrett, J.W.; Nichols, A.C. ERK-TSC2 signalling in constitutively-active HRAS mutant HNSCC cells promotes resistance to PI3K inhibition. Oral Oncol. 2018, 84, 95–103. [Google Scholar] [CrossRef]
- Hah, J.H.; Zhao, M.; Pickering, C.R.; Frederick, M.J.; Andrews, G.A.; Jasser, S.A.; Fooshee, D.R.; Milas, Z.L.; Galer, C.; Sano, D.; et al. HRAS mutations and resistance to the epidermal growth factor receptor tyrosine kinase inhibitor erlotinib in head and neck squamous cell carcinoma cells. Head & Neck 2014, 36, 1547–1554. [Google Scholar]
- Rampias, T.; Giagini, A.; Siolos, S.; Matsuzaki, H.; Sasaki, C.; Scorilas, A.; Psyrri, A. RAS/PI3K crosstalk and cetuximab resistance in head and neck squamous cell carcinoma. Clin. Cancer Res. 2014, 20, 2933–2946. [Google Scholar] [CrossRef] [PubMed]
- Pyronnet, S.; Imataka, H.; Gingras, A.C.; Fukunaga, R.; Hunter, T.; Sonenberg, N. Human eukaryotic translation initiation factor 4G (eIF4G) recruits mnk1 to phosphorylate eIF4E. EMBO J. 1999, 18, 270–279. [Google Scholar] [CrossRef]
- Raught, B.; Gingras, A.C.; Gygi, S.P.; Imataka, H.; Morino, S.; Gradi, A.; Aebersold, R.; Sonenberg, N. Serum-stimulated, rapamycin-sensitive phosphorylation sites in the eukaryotic translation initiation factor 4GI. EMBO J. 2000, 19, 434–444. [Google Scholar] [CrossRef]
- Marcotrigiano, J.; Gingras, A.C.; Sonenberg, N.; Burley, S.K. Cap-dependent translation initiation in eukaryotes is regulated by a molecular mimic of eIF4G. Mol. Cell 1999, 3, 707–716. [Google Scholar] [CrossRef]
- Yang, H.S.; Jansen, A.P.; Komar, A.A.; Zheng, X.; Merrick, W.C.; Costes, S.; Lockett, S.J.; Sonenberg, N.; Colburn, N.H. The transformation suppressor Pdcd4 is a novel eukaryotic translation initiation factor 4A binding protein that inhibits translation. Mol. Cell Biol. 2003, 23, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Comtesse, N.; Keller, A.; Diesinger, I.; Bauer, C.; Kayser, K.; Huwer, H.; Lenhof, H.P.; Meese, E. Frequent overexpression of the genes FXR1, CLAPM1 and EIF4G located on amplicon 3q26-27 in squamous cell carcinoma of the lung. Int. J. Cancer 2007, 120, 2538–2544. [Google Scholar] [CrossRef] [PubMed]
- Silvera, D.; Arju, R.; Darvishian, F.; Levine, P.H.; Zolfaghari, L.; Goldberg, J.; Hochman, T.; Formenti, S.C.; Schneider, R.J. Essential role for eIF4GI overexpression in the pathogenesis of inflammatory breast cancer. Nat. Cell Biol. 2009, 11, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Zhou, Y.; Chen, Y.; Ke, G.; Wen, H.; Wu, X. Decreased expression of EIF4A1 after preoperative brachytherapy predicts better tumor-specific survival in cervical cancer. Int. J. Gynecol. Cancer 2014, 24, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Tu, L.; Liu, Z.; He, X.; He, Y.; Yang, H.; Jiang, Q.; Xie, S.; Xiao, G.; Li, X.; Yao, K.; et al. Over-expression of eukaryotic translation initiation factor 4 gamma 1 correlates with tumor progression and poor prognosis in nasopharyngeal carcinoma. Mol. Cancer 2010, 9, 78. [Google Scholar] [CrossRef]
- Ramirez-Valle, F.; Braunstein, S.; Zavadil, J.; Formenti, S.C.; Schneider, R.J. eIF4GI links nutrient sensing by mTOR to cell proliferation and inhibition of autophagy. J. Cell Biol. 2008, 181, 293–307. [Google Scholar] [CrossRef]
- Moerke, N.J.; Aktas, H.; Chen, H.; Cantel, S.; Reibarkh, M.Y.; Fahmy, A.; Gross, J.D.; Degterev, A.; Yuan, J.; Chorev, M.; et al. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell 2007, 128, 257–267. [Google Scholar] [CrossRef]
- Chen, L.; Aktas, B.H.; Wang, Y.; He, X.; Sahoo, R.; Zhang, N.; Denoyelle, S.; Kabha, E.; Yang, H.; Freedman, R.Y.; et al. Tumor suppression by small molecule inhibitors of translation initiation. Oncotarget 2012, 3, 869–881. [Google Scholar] [CrossRef]
- Papadopoulos, E.; Jenni, S.; Kabha, E.; Takrouri, K.J.; Yi, T.; Salvi, N.; Luna, R.E.; Gavathiotis, E.; Mahalingam, P.; Arthanari, H.; et al. Structure of the eukaryotic translation initiation factor eIF4E in complex with 4EGI-1 reveals an allosteric mechanism for dissociating eIF4G. Proc. Natl. Acad. Sci. USA 2014, 111, E3187–E3195. [Google Scholar] [CrossRef]
- Cencic, R.; Hall, D.R.; Robert, F.; Du, Y.; Min, J.; Li, L.; Qui, M.; Lewis, I.; Kurtkaya, S.; Dingledine, R.; et al. Reversing chemoresistance by small molecule inhibition of the translation initiation complex eIF4F. Proc. Natl. Acad. Sci. USA 2011, 108, 1046–1051. [Google Scholar] [CrossRef]
- Feng, Y.; Pinkerton, A.B.; Hulea, L.; Zhang, T.; Davies, M.A.; Grotegut, S.; Cheli, Y.; Yin, H.; Lau, E.; Kim, H.; et al. SBI-0640756 Attenuates the Growth of Clinically Unresponsive Melanomas by Disrupting the eIF4F Translation Initiation Complex. Cancer Res. 2015, 75, 5211–5218. [Google Scholar] [CrossRef]
- Miyakawa, S.; Oguro, A.; Ohtsu, T.; Imataka, H.; Sonenberg, N.; Nakamura, Y. RNA aptamers to mammalian initiation factor 4G inhibit cap-dependent translation by blocking the formation of initiation factor complexes. RNA 2006, 12, 1825–1834. [Google Scholar] [CrossRef]
- Ridley, A.J.; Paterson, H.F.; Johnston, C.L.; Diekmann, D.; Hall, A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 1992, 70, 401–410. [Google Scholar] [CrossRef]
- Agarwal, N.K.; Chen, C.H.; Cho, H.; Boulbes, D.R.; Spooner, E.; Sarbassov, D.D. Rictor regulates cell migration by suppressing RhoGDI2. Oncogene 2013, 32, 2521–2526. [Google Scholar] [CrossRef]
- Morrison Joly, M.; Williams, M.M.; Hicks, D.J.; Jones, B.; Sanchez, V.; Young, C.D.; Sarbassov, D.D.; Muller, W.J.; Brantley-Sieders, D.; Cook, R.S. Two distinct mTORC2-dependent pathways converge on Rac1 to drive breast cancer metastasis. Breast Cancer Res. 2017, 19, 74. [Google Scholar] [CrossRef]
- Saci, A.; Cantley, L.C.; Carpenter, C.L. Rac1 regulates the activity of mTORC1 and mTORC2 and controls cellular size. Mol. Cell 2011, 42, 50–61. [Google Scholar] [CrossRef]
- Hampsch, R.A.; Shee, K.; Bates, D.; Lewis, L.D.; Desire, L.; Leblond, B.; Demidenko, E.; Stefan, K.; Huang, Y.H.; Miller, T.W. Therapeutic sensitivity to Rac GTPase inhibition requires consequential suppression of mTORC1, AKT, and MEK signaling in breast cancer. Oncotarget 2017, 8, 21806–21817. [Google Scholar] [CrossRef]
- Schnelzer, A.; Prechtel, D.; Knaus, U.; Dehne, K.; Gerhard, M.; Graeff, H.; Harbeck, N.; Schmitt, M.; Lengyel, E. Rac1 in human breast cancer: Overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene 2000, 19, 3013–3020. [Google Scholar] [CrossRef]
- Liu, S.Y.; Yen, C.Y.; Yang, S.C.; Chiang, W.F.; Chang, K.W. Overexpression of Rac-1 small GTPase binding protein in oral squamous cell carcinoma. J. Oral Maxillofac. Surg. 2004, 62, 702–707. [Google Scholar] [CrossRef]
- Kamai, T.; Yamanishi, T.; Shirataki, H.; Takagi, K.; Asami, H.; Ito, Y.; Yoshida, K. Overexpression of RhoA, Rac1, and Cdc42 GTPases is associated with progression in testicular cancer. Clin. Cancer Res. 2004, 10, 4799–4805. [Google Scholar] [CrossRef]
- Engers, R.; Ziegler, S.; Mueller, M.; Walter, A.; Willers, R.; Gabbert, H.E. Prognostic relevance of increased Rac GTPase expression in prostate carcinomas. Endocr. Relat. Cancer 2007, 14, 245–256. [Google Scholar] [CrossRef]
- Gulhati, P.; Bowen, K.A.; Liu, J.; Stevens, P.D.; Rychahou, P.G.; Chen, M.; Lee, E.Y.; Weiss, H.L.; O’Connor, K.L.; Gao, T.; et al. mTORC1 and mTORC2 regulate EMT, motility, and metastasis of colorectal cancer via RhoA and Rac1 signaling pathways. Cancer Res. 2011, 71, 3246–3256. [Google Scholar] [CrossRef]
- Choi, U.J.; Jee, B.K.; Lim, Y.; Lee, K.H. KAI1/CD82 decreases Rac1 expression and cell proliferation through PI3K/Akt/mTOR pathway in H1299 lung carcinoma cells. Cell Biochem. Funct. 2009, 27, 40–47. [Google Scholar] [CrossRef]
- Skvortsov, S.; Jimenez, C.R.; Knol, J.C.; Eichberger, P.; Schiestl, B.; Debbage, P.; Skvortsova, I.; Lukas, P. Radioresistant head and neck squamous cell carcinoma cells: Intracellular signaling, putative biomarkers for tumor recurrences and possible therapeutic targets. Radiother. Oncol. 2011, 101, 177–182. [Google Scholar] [CrossRef]
- Patel, V.; Rosenfeldt, H.M.; Lyons, R.; Servitja, J.M.; Bustelo, X.R.; Siroff, M.; Gutkind, J.S. Persistent activation of Rac1 in squamous carcinomas of the head and neck: Evidence for an EGFR/Vav2 signaling axis involved in cell invasion. Carcinogenesis 2007, 28, 1145–1152. [Google Scholar] [CrossRef]
- Montalvo-Ortiz, B.L.; Castillo-Pichardo, L.; Hernandez, E.; Humphries-Bickley, T.; De la Mota-Peynado, A.; Cubano, L.A.; Vlaar, C.P.; Dharmawardhane, S. Characterization of EHop-016, novel small molecule inhibitor of Rac GTPase. J. Biol. Chem. 2012, 287, 13228–13238. [Google Scholar] [CrossRef]
- Shutes, A.; Onesto, C.; Picard, V.; Leblond, B.; Schweighoffer, F.; Der, C.J. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J. Biol. Chem. 2007, 282, 35666–35678. [Google Scholar] [CrossRef]
- Hagiwara, N.; Watanabe, M.; Iizuka-Ohashi, M.; Yokota, I.; Toriyama, S.; Sukeno, M.; Tomosugi, M.; Sowa, Y.; Hongo, F.; Mikami, K.; et al. Mevalonate pathway blockage enhances the efficacy of mTOR inhibitors with the activation of retinoblastoma protein in renal cell carcinoma. Cancer Lett. 2018, 431, 182–189. [Google Scholar] [CrossRef]
- Frankel, W.N.; Yang, Y.; Mahaffey, C.L.; Beyer, B.J.; O’Brien, T.P. Szt2, a novel gene for seizure threshold in mice. Genes Brain Behav. 2009, 8, 568–576. [Google Scholar] [CrossRef]
- Venkatesan, C.; Angle, B.; Millichap, J.J. Early-life epileptic encephalopathy secondary to SZT2 pathogenic recessive variants. Epileptic Disord. 2016, 18, 195–200. [Google Scholar]
- Pizzino, A.; Whitehead, M.; Sabet Rasekh, P.; Murphy, J.; Helman, G.; Bloom, M.; Evans, S.H.; Murnick, J.G.; Conry, J.; Taft, R.J.; et al. Mutations in SZT2 result in early-onset epileptic encephalopathy and leukoencephalopathy. Am. J. Med. Genet. A 2018, 176, 1443–1448. [Google Scholar] [CrossRef]
- Wolfson, R.L.; Chantranupong, L.; Wyant, G.A.; Gu, X.; Orozco, J.M.; Shen, K.; Condon, K.J.; Petri, S.; Kedir, J.; Scaria, S.M.; et al. KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature 2017, 543, 438–442. [Google Scholar] [CrossRef]
- Peng, M.; Yin, N.; Li, M.O. SZT2 dictates GATOR control of mTORC1 signalling. Nature 2017, 543, 433–437. [Google Scholar] [CrossRef]
- Fang, Y.; Vilella-Bach, M.; Bachmann, R.; Flanigan, A.; Chen, J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 2001, 294, 1942–1945. [Google Scholar] [CrossRef]
- Yoon, M.S.; Sun, Y.; Arauz, E.; Jiang, Y.; Chen, J. Phosphatidic acid activates mammalian target of rapamycin complex 1 (mTORC1) kinase by displacing FK506 binding protein 38 (FKBP38) and exerting an allosteric effect. J. Biol. Chem. 2011, 286, 29568–29574. [Google Scholar] [CrossRef]
- Winter, J.N.; Fox, T.E.; Kester, M.; Jefferson, L.S.; Kimball, S.R. Phosphatidic acid mediates activation of mTORC1 through the ERK signaling pathway. Am. J. Physiol. Cell Physiol. 2010, 299, C335–C344. [Google Scholar] [CrossRef]
- Gozgit, J.M.; Pentecost, B.T.; Marconi, S.A.; Ricketts-Loriaux, R.S.; Otis, C.N.; Arcaro, K.F. PLD1 is overexpressed in an ER-negative MCF-7 cell line variant and a subset of phospho-Akt-negative breast carcinomas. Br. J. Cancer 2007, 97, 809–817. [Google Scholar] [CrossRef]
- Hu, J.; Hu, H.; Hang, J.J.; Yang, H.Y.; Wang, Z.Y.; Wang, L.; Chen, D.H.; Wang, L.W. Simultaneous high expression of PLD1 and Sp1 predicts a poor prognosis for pancreatic ductal adenocarcinoma patients. Oncotarget 2016, 7, 78557–78565. [Google Scholar] [CrossRef]
- Kang, D.W.; Lee, B.H.; Suh, Y.A.; Choi, Y.S.; Jang, S.J.; Kim, Y.M.; Choi, K.Y.; Min, D.S. Phospholipase D1 Inhibition Linked to Upregulation of ICAT Blocks Colorectal Cancer Growth Hyperactivated by Wnt/beta-Catenin and PI3K/Akt Signaling. Clin. Cancer Res. 2017, 23, 7340–7350. [Google Scholar] [CrossRef]
- Ohguchi, K.; Banno, Y.; Nakagawa, Y.; Akao, Y.; Nozawa, Y. Negative regulation of melanogenesis by phospholipase D1 through mTOR/p70 S6 kinase 1 signaling in mouse B16 melanoma cells. J. Cell Physiol. 2005, 205, 444–451. [Google Scholar] [CrossRef]
- Tang, W.; Liang, R.; Duan, Y.; Shi, Q.; Liu, X.; Liao, Y. PLD1 overexpression promotes invasion and migration and function as a risk factor for Chinese glioma patients. Oncotarget 2017, 8, 57039–57046. [Google Scholar] [CrossRef]
- Xiao, J.; Sun, Q.; Bei, Y.; Zhang, L.; Dimitrova-Shumkovska, J.; Lv, D.; Yang, Y.; Cao, Y.; Zhao, Y.; Song, M.; et al. Therapeutic inhibition of phospholipase D1 suppresses hepatocellular carcinoma. Clin. Sci. (Lond.) 2016, 130, 1125–1136. [Google Scholar] [CrossRef]
- Lewis, J.A.; Scott, S.A.; Lavieri, R.; Buck, J.R.; Selvy, P.E.; Stoops, S.L.; Armstrong, M.D.; Brown, H.A.; Lindsley, C.W. Design and synthesis of isoform-selective phospholipase D (PLD) inhibitors. Part I: Impact of alternative halogenated privileged structures for PLD1 specificity. Bioorg. Med. Chem. Lett. 2009, 19, 1916–1920. [Google Scholar]
- Monovich, L.; Mugrage, B.; Quadros, E.; Toscano, K.; Tommasi, R.; LaVoie, S.; Liu, E.; Du, Z.; LaSala, D.; Boyar, W.; et al. Optimization of halopemide for phospholipase D2 inhibition. Bioorg. Med. Chem. Lett. 2007, 17, 2310–2311. [Google Scholar] [CrossRef]
- Grunwald, V.; Keilholz, U.; Boehm, A.; Guntinas-Lichius, O.; Hennemann, B.; Schmoll, H.J.; Ivanyi, P.; Abbas, M.; Lehmann, U.; Koch, A.; et al. TEMHEAD: A single-arm multicentre phase II study of temsirolimus in platin- and cetuximab refractory recurrent and/or metastatic squamous cell carcinoma of the head and neck (SCCHN) of the German SCCHN Group (AIO). Ann. Oncol. 2015, 26, 561–567. [Google Scholar] [CrossRef]
- Dunn, L.A.; Fury, M.G.; Xiao, H.; Baxi, S.S.; Sherman, E.J.; Korte, S.; Pfister, C.; Haque, S.; Katabi, N.; Ho, A.L.; et al. A phase II study of temsirolimus added to low-dose weekly carboplatin and paclitaxel for patients with recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC). Ann. Oncol. 2017, 28, 2533–2538. [Google Scholar] [CrossRef]
- Keam, B.; Kim, S.; Ahn, Y.O.; Kim, T.M.; Lee, S.H.; Kim, D.W.; Heo, D.S. In vitro anticancer activity of PI3K alpha selective inhibitor BYL719 in head and neck cancer. AntiCancer Res. 2015, 35, 175–182. [Google Scholar]
- Mehibel, M.; Ortiz-Martinez, F.; Voelxen, N.; Boyers, A.; Chadwick, A.; Telfer, B.A.; Mueller-Klieser, W.; West, C.M.; Critchlow, S.E.; Williams, K.J.; et al. Statin-induced metabolic reprogramming in head and neck cancer: A biomarker for targeting monocarboxylate transporters. Sci. Rep. 2018, 8, 16804. [Google Scholar] [CrossRef]
- Polanski, R.; Hodgkinson, C.L.; Fusi, A.; Nonaka, D.; Priest, L.; Kelly, P.; Trapani, F.; Bishop, P.W.; White, A.; Critchlow, S.E.; et al. Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clin. Cancer Res. 2014, 20, 926–937. [Google Scholar] [CrossRef]
- Bola, B.M.; Chadwick, A.L.; Michopoulos, F.; Blount, K.G.; Telfer, B.A.; Williams, K.J.; Smith, P.D.; Critchlow, S.E.; Stratford, I.J. Inhibition of monocarboxylate transporter-1 (MCT1) by AZD3965 enhances radiosensitivity by reducing lactate transport. Mol. Cancer Ther. 2014, 13, 2805–2816. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Phase | Status | Targeted Pathway | Targeted Tumour | Reference |
|---|---|---|---|---|---|
| Palbociclib + Gedatolisib | I | Recruiting | CDK4/6 + mTOR | Advanced HNSCC | NCT03065062 |
| BYL719 | II | Recruiting | PI3K | Recurrent or Metastatic HNSCC | NCT02145312 |
| Everolimus + Palbociclib + Trametinib | I | Recruiting | mTOR + CDK4/6 + MEK | Malignant neoplasms of Oral cavity | NCT03065387 |
| CC-115 | I | Active, not recruiting | Dual DNA-PK and TOR kinase | Advanced HNSCC | NCT01353625 |
| PQR 309 | I | Active, not recruiting | PI3K/mTOR/AKT | Advanced HNSCC | NCT02483858 |
| Temsirolimus | II | Completed | mTOR | HNSCSC | NCT01172769 |
| Sirolimus | I/II | Completed | mTOR | Advanced HNSCC | NCT01195922 |
| Temsirolimus + Paclitaxel + Carboplatin | I/II | Completed | mTOR | Recurrent or Metastatic HNSCC | NCT01016769 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, F.H.; Bai, Y.; Saintigny, P.; Darido, C. mTOR Signalling in Head and Neck Cancer: Heads Up. Cells 2019, 8, 333. https://doi.org/10.3390/cells8040333
Tan FH, Bai Y, Saintigny P, Darido C. mTOR Signalling in Head and Neck Cancer: Heads Up. Cells. 2019; 8(4):333. https://doi.org/10.3390/cells8040333
Chicago/Turabian StyleTan, Fiona H., Yuchen Bai, Pierre Saintigny, and Charbel Darido. 2019. "mTOR Signalling in Head and Neck Cancer: Heads Up" Cells 8, no. 4: 333. https://doi.org/10.3390/cells8040333
APA StyleTan, F. H., Bai, Y., Saintigny, P., & Darido, C. (2019). mTOR Signalling in Head and Neck Cancer: Heads Up. Cells, 8(4), 333. https://doi.org/10.3390/cells8040333