Activation of Nrf2/HO-1 Pathway and Human Atherosclerotic Plaque Vulnerability: An In Vitro and In Vivo Study

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Monocyte Isolation and Culture
2.3. Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS) Analysis
2.4. Western Blot Analysis
2.5. Immunofluorescence Analysis
2.6. OCT Image Acquisition and Analysis
2.7. Statistical Analysis
3. Results
3.1. Clinical Features
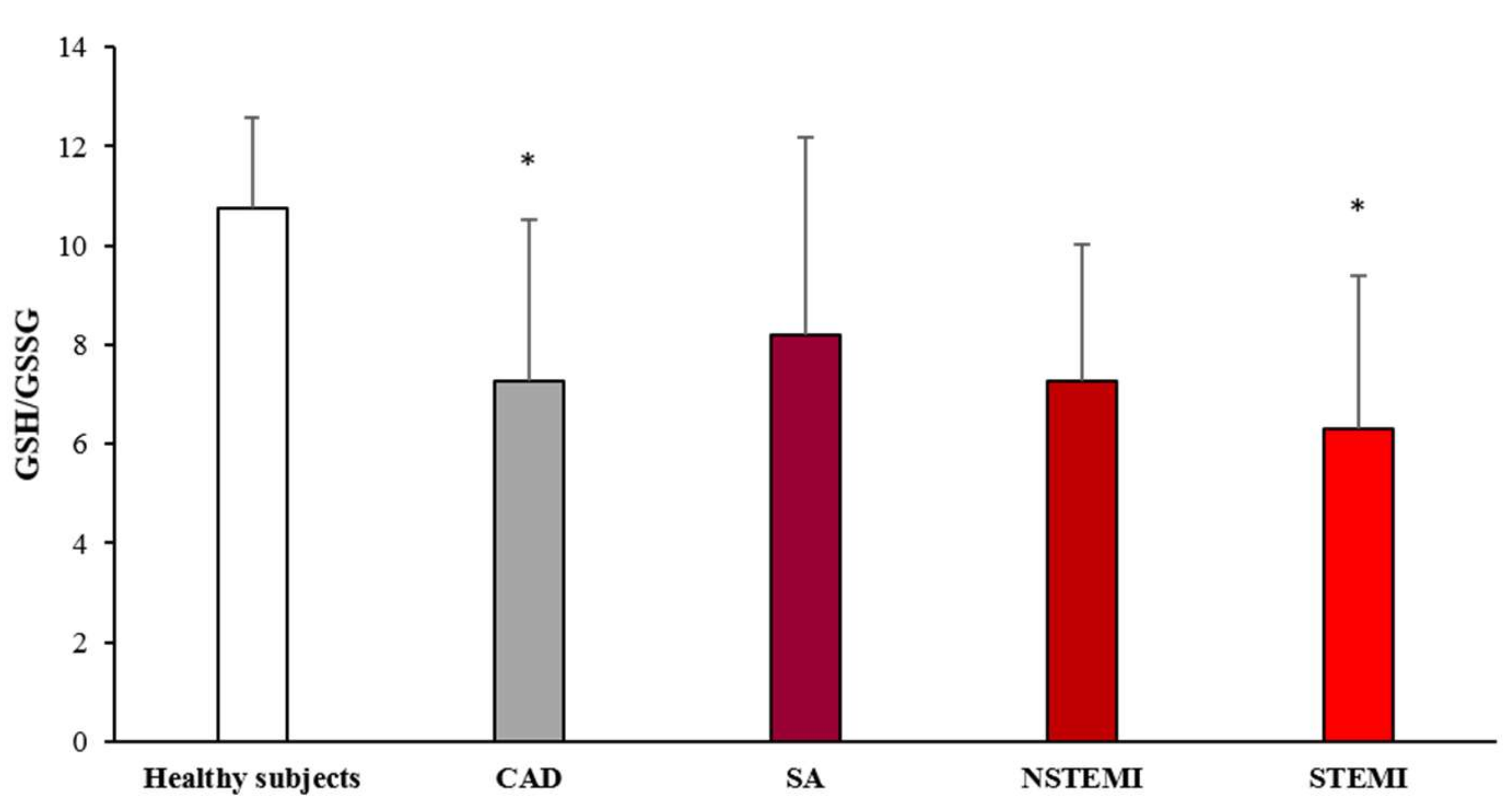
3.2. Oxidative Stress Status
3.3. HO-1 and Nrf2 Expression
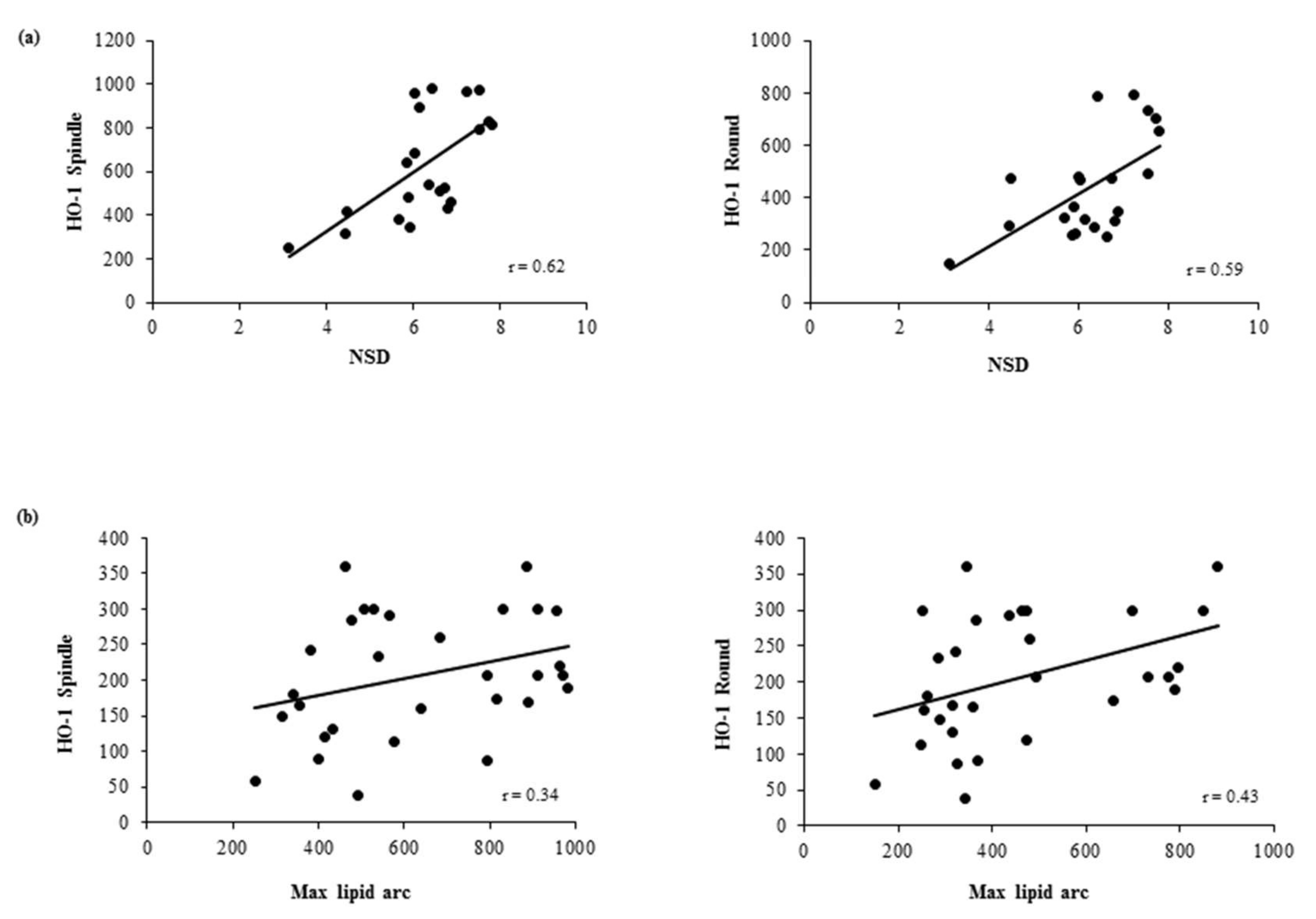
3.4. Association Between In Vivo Plaque Morphology and HO-1 Levels in MDMs
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schaar, J.A.; Muller, J.E.; Falk, E.; Virmani, R.; Fuster, V.; Serruys, P.W.; Colombo, A.; Stefanadis, C.; Ward Casscells, S.; Moreno, P.R.; et al. Terminology for high-risk and vulnerable coronary artery plaques. Report of a meeting on the vulnerable plaque, June 17 and 18, 2003, Santorini, Greece. Eur. Heart J. 2004, 25, 1077–1082. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.M.; Galis, Z.S.; Rosenfeld, M.E.; Falk, E. Plaque rupture in humans and mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 705–713. [Google Scholar] [CrossRef]
- Vancraeynest, D.; Pasquet, A.; Roelants, V.; Gerber, B.L.; Vanoverschelde, J.L. Imaging the vulnerable plaque. J. Am. Coll. Cardiol. 2011, 57, 1961–1979. [Google Scholar] [CrossRef] [PubMed]
- Chinetti-Gbaguidi, G.; Colin, S.; Staels, B. Macrophage subsets in atherosclerosis. Nat. Rev. Cardiol. 2015, 12, 10–17. [Google Scholar] [CrossRef]
- Yokoyama, M. Oxidant stress and atherosclerosis. Curr. Opin. Pharmacol. 2004, 4, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Uno, K.; Nicholls, S.J. Biomarkers of inflammation and oxidative stress in atherosclerosis. Biomark. Med. 2010, 4, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Matsuoka, H.; Umei, H.; Sugano, R.; Fujii, Y.; Soga, J.; Kihara, Y.; Chayama, K.; Imaizumi, T. Endothelial function in subjects with isolated low HDL cholesterol: Role of nitric oxide and circulating progenitor cells. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E202–E209. [Google Scholar] [CrossRef] [PubMed]
- Peluso, I.; Morabito, G.; Urban, L.; Ioannone, F.; Serafini, M. Oxidative stress in atherosclerosis development: The central role of LDL and oxidative burst. Endocr. Metab. Immune Disord. Drug Targets 2012, 12, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.F.; Ullevig, S.; Kim, H.S.; Asmis, R. Regulation of Monocyte Adhesion and Migration by Nox4. PLoS ONE 2013, 8, e66964. [Google Scholar] [CrossRef]
- Wang, L.J.; Lee, T.S.; Lee, F.Y.; Pai, R.C.; Chau, L.Y. Expression of heme oxygenase-1 in atherosclerotic lesions. Am. J. Pathol. 1998, 152, 711–720. [Google Scholar] [PubMed]
- Morita, T. Heme oxygenase and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1786–1795. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Tian, R.; Lu, N. Nitric oxide protected against NADPH oxidase-derived superoxide generation in vascular endothelium: Critical role for heme oxygenase-1. Int. J. Biol. Macromol. 2019, 126, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Ndisang, J.F.; Zhao, W.; Wang, R. Selective regulation of blood pressure by heme oxygenase-1 in hypertension. Hypertension 2002, 40, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Fukano, Y.; Oishi, M.; Chibana, F.; Numazawa, S.; Yoshida, T. Analysis of the expression of heme oxygenase-1 gene in human alveolar epithelial cells exposed to cigarette smoke condensate. J. Toxicol. Sci. 2006, 31, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Itoh, K.; Ruiz, E.; Leake, D.S.; Unoki, H.; Yamamoto, M.; Mann, G.E. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: Activation by oxidatively modified LDL and 4-hydroxynonenal. Circ. Res. 2004, 94, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Noordeloos, A.M.; Jeney, V.; Soares, M.P.; Moll, F.; Pasterkamp, G.; Serruys, P.W.; Duckers, H.J. Heme oxygenase 1 determines atherosclerotic lesion progression into a vulnerable plaque. Circulation 2009, 119, 3017–3027. [Google Scholar] [CrossRef]
- Araujo, J.A.; Zhang, M.; Yin, F. Heme oxygenase-1, oxidation, inflammation, and atherosclerosis. Front. Pharmacol. 2012, 3, 119. [Google Scholar] [CrossRef] [PubMed]
- Durante, W. Protective role of heme oxygenase-1 against inflammation in atherosclerosis. Front. Biosci. (Landmark Ed.) 2011, 16, 2372–2388. [Google Scholar] [CrossRef]
- Naito, Y.; Takagi, T.; Higashimura, Y. Heme oxygenase-1 and anti-inflammatory M2 macrophages. Arch. Biochem. Biophys. 2014, 564, 83–88. [Google Scholar] [CrossRef]
- Zhang, M.; Nakamura, K.; Kageyama, S.; Lawal, A.O.; Gong, K.W.; Bhetraratana, M.; Fujii, T.; Sulaiman, D.; Hirao, H.; Bolisetty, S.; et al. Myeloid HO-1 modulates macrophage polarization and protects against ischemia-reperfusion injury. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Chauveau, C.; Remy, S.; Royer, P.J.; Hill, M.; Tanguy-Royer, S.; Hubert, F.X.; Tesson, L.; Brion, R.; Beriou, G.; Gregoire, M.; et al. Heme oxygenase-1 expression inhibits dendritic cell maturation and proinflammatory function but conserves IL-10 expression. Blood 2005, 106, 1694–1702. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Navab, M.; Leitinger, N.; Fogelman, A.M.; Lusis, A.J. Induction of heme oxygenase-1 inhibits the monocyte transmigration induced by mildly oxidized LDL. J. Clin. Investig. 1997, 100, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Yet, S.F.; Layne, M.D.; Liu, X.; Chen, Y.H.; Ith, B.; Sibinga, N.E.; Perrella, M.A. Absence of heme oxygenase-1 exacerbates atherosclerotic lesion formation and vascular remodeling. FASEB J. 2003, 17, 1759–1761. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Sugawara, D.; Goto, J.; Watanabe, Y.; Kawamura, K.; Shiomi, M.; Itabe, H.; Maruyama, Y. Heme oxygenase-1 inhibits atherogenesis in Watanabe heritable hyperlipidemic rabbits. Circulation 2001, 104, 1831–1836. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Sugawara, D.; Wang, X.; Suzuki, K.; Itabe, H.; Maruyama, Y.; Lusis, A.J. Heme oxygenase-1 inhibits atherosclerotic lesion formation in LDL-receptor knockout mice. Circ. Res. 2001, 88, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.M.; Li, Y.G.; Wang, D.M. Study on changes of heme oxygenase-1 expression in patients with coronary heart disease. Clin. Cardiol. 2005, 28, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Schaer, C.A.; Schoedon, G.; Imhof, A.; Kurrer, M.O.; Schaer, D.J. Constitutive endocytosis of CD163 mediates hemoglobin-heme uptake and determines the noninflammatory and protective transcriptional response of macrophages to hemoglobin. Circ. Res. 2006, 99, 943–950. [Google Scholar] [CrossRef]
- Eligini, S.; Crisci, M.; Bono, E.; Songia, P.; Tremoli, E.; Colombo, G.I.; Colli, S. Human monocyte-derived macrophages spontaneously differentiated in vitro show distinct phenotypes. J. Cell. Physiol. 2013, 228, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Eligini, S.; Cosentino, N.; Fiorelli, S.; Fabbiocchi, F.; Niccoli, G.; Refaat, H.; Camera, M.; Calligaris, G.; De Martini, S.; Bonomi, A.; et al. Biological profile of monocyte-derived macrophages in coronary heart disease patients: Implications for plaque morphology. Sci. Rep. under review.
- Jang, I.K.; Bouma, B.E.; Kang, D.H.; Park, S.J.; Park, S.W.; Seung, K.B.; Choi, K.B.; Shishkov, M.; Schlendorf, K.; Pomerantsev, E.; et al. Visualization of coronary atherosclerotic plaques in patients using optical coherence tomography: comparison with intravascular ultrasound. J. Am. Coll. Cardiol. 2002, 39, 604–609. [Google Scholar] [CrossRef]
- Jang, I.K.; Tearney, G.J.; MacNeill, B.; Takano, M.; Moselewski, F.; Iftima, N.; Shishkov, M.; Houser, S.; Aretz, H.T.; Halpern, E.F.; et al. In vivo characterization of coronary atherosclerotic plaque by use of optical coherence tomography. Circulation 2005, 111, 1551–1555. [Google Scholar] [CrossRef]
- Squellerio, I.; Caruso, D.; Porro, B.; Veglia, F.; Tremoli, E.; Cavalca, V. Direct glutathione quantification in human blood by LC-MS/MS: comparison with HPLC with electrochemical detection. J. Pharm. Biomed. Anal. 2012, 71, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Eligini, S.; Colli, S.; Basso, F.; Sironi, L.; Tremoli, E. Oxidized low density lipoprotein suppresses expression of inducible cyclooxygenase in human macrophages. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1719–1725. [Google Scholar] [CrossRef] [PubMed]
- Eligini, S.; Brambilla, M.; Banfi, C.; Camera, M.; Sironi, L.; Barbieri, S.S.; Auwerx, J.; Tremoli, E.; Colli, S. Oxidized phospholipids inhibit cyclooxygenase-2 in human macrophages via nuclear factor-kappaB/IkappaB- and ERK2-dependent mechanisms. Cardiovasc. Res. 2002, 55, 406–415. [Google Scholar] [CrossRef]
- Eligini, S.; Brioschi, M.; Fiorelli, S.; Tremoli, E.; Banfi, C.; Colli, S. Human monocyte-derived macrophages are heterogenous: Proteomic profile of different phenotypes. J. Proteomics 2015, 124, 112–123. [Google Scholar] [CrossRef]
- Scalone, G.; Niccoli, G.; Refaat, H.; Vergallo, R.; Porto, I.; Leone, A.M.; Burzotta, F.; D’Amario, D.; Liuzzo, G.; Fracassi, F.; et al. Not all plaque ruptures are born equal: An optical coherence tomography study. Eur. Heart J. Cardiovasc. Imaging 2017, 18, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Tearney, G.J.; Regar, E.; Akasaka, T.; Adriaenssens, T.; Barlis, P.; Bezerra, H.G.; Bouma, B.; Bruining, N.; Cho, J.M.; Chowdhary, S.; et al. Consensus standards for acquisition, measurement, and reporting of intravascular optical coherence tomography studies: A report from the International Working Group for Intravascular Optical Coherence Tomography Standardization and Validation. J. Am. Coll. Cardiol. 2012, 59, 1058–1072. [Google Scholar] [CrossRef] [PubMed]
- Tearney, G.J.; Yabushita, H.; Houser, S.L.; Aretz, H.T.; Jang, I.K.; Schlendorf, K.H.; Kauffman, C.R.; Shishkov, M.; Halpern, E.F.; Bouma, B.E. Quantification of macrophage content in atherosclerotic plaques by optical coherence tomography. Circulation 2003, 107, 113–119. [Google Scholar] [CrossRef]
- Di Vito, L.; Agozzino, M.; Marco, V.; Ricciardi, A.; Concardi, M.; Romagnoli, E.; Gatto, L.; Calogero, G.; Tavazzi, L.; Arbustini, E.; et al. Identification and quantification of macrophage presence in coronary atherosclerotic plaques by optical coherence tomography. Eur. Heart J. Cardiovasc. Imaging 2015, 16, 807–813. [Google Scholar] [CrossRef]
- Neubauer, J.A.; Sunderram, J. Heme oxygenase-1 and chronic hypoxia. Respir. Physiol. Neurobiol. 2012, 184, 178–185. [Google Scholar] [CrossRef]
- Ewing, J.F.; Maines, M.D. Glutathione depletion induces heme oxygenase-1 (HSP32) mRNA and protein in rat brain. J. Neurochem. 1993, 60, 1512–1519. [Google Scholar] [CrossRef]
- Biswas, S.K.; Newby, D.E.; Rahman, I.; Megson, I.L. Depressed glutathione synthesis precedes oxidative stress and atherogenesis in Apo-E(-/-) mice. Biochem. Biophys. Res. Commun. 2005, 338, 1368–1373. [Google Scholar] [CrossRef]
- Ozsarlak-Sozer, G.; Sevin, G.; Ozgur, H.H.; Yetik-Anacak, G.; Kerry, Z. Diverse effects of taurine on vascular response and inflammation in GSH depletion model in rabbits. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 1360–1372. [Google Scholar]
- Cavalca, V.; Veglia, F.; Squellerio, I.; Marenzi, G.; Minardi, F.; De Metrio, M.; Cighetti, G.; Boccotti, L.; Ravagnani, P.; Tremoli, E. Glutathione, vitamin E and oxidative stress in coronary artery disease: Relevance of age and gender. Eur. J. Clin. Investig. 2009, 39, 267–272. [Google Scholar] [CrossRef]
- Eligini, S.; Porro, B.; Lualdi, A.; Squellerio, I.; Veglia, F.; Chiorino, E.; Crisci, M.; Garlasche, A.; Giovannardi, M.; Werba, J.P.; et al. Nitric oxide synthetic pathway in red blood cells is impaired in coronary artery disease. PLoS ONE 2013, 8, e66945. [Google Scholar] [CrossRef]
- Kaneda, H.; Ohno, M.; Taguchi, J.; Togo, M.; Hashimoto, H.; Ogasawara, K.; Aizawa, T.; Ishizaka, N.; Nagai, R. Heme oxygenase-1 gene promoter polymorphism is associated with coronary artery disease in Japanese patients with coronary risk factors. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1680–1685. [Google Scholar] [CrossRef]
- Chen, Y.H.; Lin, S.J.; Lin, M.W.; Tsai, H.L.; Kuo, S.S.; Chen, J.W.; Charng, M.J.; Wu, T.C.; Chen, L.C.; Ding, Y.A.; et al. Microsatellite polymorphism in promoter of heme oxygenase-1 gene is associated with susceptibility to coronary artery disease in type 2 diabetic patients. Hum. Genet. 2002, 111, 1–8. [Google Scholar] [CrossRef]
- Schillinger, M.; Exner, M.; Mlekusch, W.; Ahmadi, R.; Rumpold, H.; Mannhalter, C.; Wagner, O.; Minar, E. Heme oxygenase-1 genotype is a vascular anti-inflammatory factor following balloon angioplasty. J. Endovasc. Ther. 2002, 9, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, A.; Oda, Y.; Yachie, A.; Koizumi, S.; Nakanishi, I. Heme oxygenase-1 deficiency: The first autopsy case. Hum. Pathol. 2002, 33, 125–130. [Google Scholar] [CrossRef]
- Wu, M.L.; Ho, Y.C.; Yet, S.F. A central role of heme oxygenase-1 in cardiovascular protection. Antioxid. Redox Signal. 2011, 15, 1835–1846. [Google Scholar] [CrossRef]
- Kawamura, K.; Ishikawa, K.; Wada, Y.; Kimura, S.; Matsumoto, H.; Kohro, T.; Itabe, H.; Kodama, T.; Maruyama, Y. Bilirubin from heme oxygenase-1 attenuates vascular endothelial activation and dysfunction. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 155–160. [Google Scholar] [CrossRef]
- Balla, G.; Jacob, H.S.; Balla, J.; Rosenberg, M.; Nath, K.; Apple, F.; Eaton, J.W.; Vercellotti, G.M. Ferritin: A cytoprotective antioxidant strategem of endothelium. J. Biol. Chem. 1992, 267, 18148–18153. [Google Scholar]
- Zakkar, M.; Van der Heiden, K.; Luong le, A.; Chaudhury, H.; Cuhlmann, S.; Hamdulay, S.S.; Krams, R.; Edirisinghe, I.; Rahman, I.; Carlsen, H.; et al. Activation of Nrf2 in endothelial cells protects arteries from exhibiting a proinflammatory state. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1851–1857. [Google Scholar] [CrossRef] [PubMed]
- Buckley, B.J.; Marshall, Z.M.; Whorton, A.R. Nitric oxide stimulates Nrf2 nuclear translocation in vascular endothelium. Biochem. Biophys. Res. Commun. 2003, 307, 973–979. [Google Scholar] [CrossRef]
- Ungvari, Z.; Bagi, Z.; Feher, A.; Recchia, F.A.; Sonntag, W.E.; Pearson, K.; de Cabo, R.; Csiszar, A. Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H18–H24. [Google Scholar] [CrossRef] [PubMed]
- Donovan, E.L.; McCord, J.M.; Reuland, D.J.; Miller, B.F.; Hamilton, K.L. Phytochemical activation of Nrf2 protects human coronary artery endothelial cells against an oxidative challenge. Oxid. Med. Cell. Longev. 2012, 2012, 132931. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Sumiyoshi, S.; Nakashima, Y.; Doi, Y.; Iida, M.; Kiyohara, Y.; Sueishi, K. Overexpression of heme oxygenase-1 in coronary atherosclerosis of Japanese autopsies with diabetes mellitus: Hisayama study. Atherosclerosis 2009, 202, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.G.; Wang, D.M.; Chen, S.M.; Tan, X.R.; Fang, X.Y.; Wu, J.W.; Zhang, G.H.; Mai, R.Q. Haem oxygenase-1 expression and coronary heart disease--association between levels of haem oxygenase-1 expression and angiographic morphology as well as the quantity of coronary lesions. Acta Cardiol. 2006, 61, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Higgins, C.L.; Chen, I.Y.; Reardon, M.; Lawrie, G.; Vick, G.W., 3rd; Karmonik, C.; Via, D.P.; Morrisett, J.D. Quantitation and localization of matrix metalloproteinases and their inhibitors in human carotid endarterectomy tissues. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2351–2358. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Healthy Subjects (n = 10) | CAD (n = 30) | p Value Healthy Subjects vs. CAD °° | CAD | |||
|---|---|---|---|---|---|---|---|
| SA (n = 10) | NSTEMI (n = 10) | STEMI (n = 10) | ANOVA p Value ° | ||||
| Demographics | |||||||
| Age (years) | 61.5 ± 10 | 63.8 ± 12.1 | 0.5927 | 70.3 ± 7.2 | 61.0 ± 11.9 | 60.0 ± 14.8 | 0.1660 |
| Male sex, n (%) | 5 (50) | 26 (86.7) | 0.0290‡ | 8 (80) | 9 (90) | 8 (80) | 0.8179 ‡ |
| Body mass index (kg/m2) | 23.5 ± 1.6 | 29.3 ± 4.6 | 0.0004 | 28.0 ± 4.5 * | 28.1 ± 3.7 * | 32.3 ± 4.6 * | 0.0002 |
| Clinical characteristics | |||||||
| Current smoking, n (%) | 0 | 18 (60) | 0.0010‡ | 7 (70) | 6 (60) | 5 (50) | 0.5884 ‡ |
| Diabetes mellitus, n (%) | 0 | 16 (53.3) | 0.0030‡ | 5 (50) | 5 (50) | 6 (60) | 0.6593 ‡ |
| Dyslipidemia, n (%) | 0 | 16 (53.3) | 0.0030‡ | 7 (70) | 5 (50) | 4 (40) | 0.4704 ‡ |
| Hypertension, n (%) | 0 | 14 (46.7) | 0.0070‡ | 4 (40) | 5 (50) | 5 (50) | 0.9004 ‡ |
| Family history of CAD, n (%) | 0 | 17 (56.7) | 0.0020‡ | 4 (40) | 9 (90) # | 4 (40) | 0.0149‡ |
| LVEF (%) | NA | 50.1 ± 8.8 | 48 ± 9.3 | 51.3 ± 9.8 | 51.0 ± 7.7 | 0.7510 | |
| Laboratory data | |||||||
| WBC (× 109/L) | 7.6 ± 2.9 | 9.2 ± 3.9 | 0.2537 | 8.9 ± 2.4 | 9.1 ± 5.1 | 9.7 ± 4.0 | 0.6852 |
| RBC (× 1012/L) | 4.5 ± 0.8 | 5.1 ± 2.0 | 0.3917 | 4.6 ± 0.6 | 5.1 ± 0.6 | 5.8 ± 3.6 | 0.4330 |
| Neutrophil count (× 109/L) | 4.8 ± 2.3 | 6.2 ± 3.5 | 0.2585 | 5.8 ± 2.3 | 6.1 ± 4.7 | 6.9 ± 3.4 | 0.6249 |
| Lymphocyte count (× 109/L) | 2.1 ± 1.2 | 2.0 ± 0.9 | 0.7190 | 2.3 ± 1.2 | 1.9 ± 0.7 | 1.8 ± 0.9 | 0.6889 |
| Eosinophil count (× 109/L) | 0.1 ± 0.1 | 0.2 ± 0.2 | 0.6303 | 0.2 ± 0.1 | 0.1 ± 0.1 | 0.2 ± 0.4 | 0.6123 |
| Monocyte count (× 109/L) | 0.5 ± 0.2 | 0.6 ± 0.3 | 0.1398 | 0.6 ± 0.2 | 0.6 ± 0.4 | 0.7 ± 0.4 | 0.3176 |
| Basophil count (× 109/L) | 0.03 ± 0.02 | 0.01 ± 0.00 | 0.0072 | 0.01 ± 0.02 | 0.01 ± 0.03 * | 0.01 ± 0.02 | 0.0343 |
| Platelets (× 109/L) | 248 ± 61.9 | 230.8 ± 83.4 | 0.5714 | 213.7 ± 49.8 | 252.2 ± 106.0 | 223.7 ± 85.6 | 0.6549 |
| hs-CRP (mg/L) | 1.9 (1.4–2.3) | 4.9 (2.0–21.0) | 0.0141† | 2.1 (1.6–2.1) ǂ # | 6.7 (1.6–17.0) * | 38.6 (6.0–75.0) * | 0.0003§ |
| Creatinine (mg/dL) | 1 ± 0.1 | 1.0 ± 0.5 | 0.9372 | 0.8 ± 0.3 # | 0.8 ± 0.3 # | 1.4 ± 0.5 | 0.0015 |
| Glycaemia (mg/dL) | 93.5 ± 12.2 | 140.2 ± 42.8 | 0.0017 | 116.4 ± 27.1 # | 130.3 ± 33.4 # | 178.8 ± 43.7 * | <0.0001 |
| Total cholesterol (mg/dL) | 187.7 ± 22.1 | 204.4 ± 42.6 | 0.2438 | 181.1 ± 34.1 # | 207.9 ± 42.8 | 226.1 ± 41.7 | 0.0417 |
| LDL (mg/dL) | 112.6 ± 26 | 122.4 ± 41.9 | 0.4924 | 102.0 ± 23.5 | 130.4 ± 47.5 | 135.2 ± 46.2 | 0.1890 |
| HDL (mg/dL) | 41.1 ± 5.3 | 48.83 ± 14.9 | 0.0242 | 61.6 ± 16.7 *,ǂ,# | 44.5 ± 10.5 | 43.3 ± 9.3 | 0.0004 |
| Triglycerides (mg/dL) | 143.8 ± 31.6 | 161.2 ± 55.5 | 0.3333 | 117.9 ± 42.7 ǂ# | 176.7 ± 59.6 | 190.3 ± 32.2 | 0.0034 |
| Peak TnI (μg/dL) | NA | 1 (0.0–29.4) | NA | 1.2 (0.5–1.4) # | 29.7 (25.0–163.0) | <0.0001 § | |
| Peak CK-MB (μg/dL) | NA | 11.1 (2.1–110.0) | 2.1 (1.5–2.1) | 12.3 (2.5–28.0) # | 281 (110.0–521.0) # | <0.0001 § | |
| Angiographic data | |||||||
| Culprit or treated vessel, n (%) | 0.1489 ‡ | ||||||
| LAD | NA | 14 (46.7) | 3 (30) | 8 (80) | 3 (30) | ||
| LCX | NA | 10 (30.3) | 4 (40) | 1 (10) | 5 (50) | ||
| RCA | NA | 6 (20) | 3 (30) | 1 (10) | 2 (20) | ||
| Multivessel disease, n (%) | NA | 17 (56.7) | 8 (80) | 4 (40) | 5 (50) | 0.3276 ‡ | |
| Admission therapy | |||||||
| ASA, n (%) | 0 | 11 (36.7) | 0.0380‡ | 3 (30) | 5 (50) | 3 (30) | 0.3192 ‡ |
| Beta-Blockers, n (%) | 0 | 8 (26.7) | 0.1650 ‡ | 2 (20) | 5 (50) | 1 (10) | 0.2319 ‡ |
| ACE-inhibitors, n (%) | 0 | 9 (30) | 0.0810 ‡ | 5 (50) | 2 (20) | 2 (20) | 0.3192 ‡ |
| Statins, n (%) | 0 | 10 (30.3) | 0.0430‡ | 5 (50) | 2 (20) | 3 (30) | 0.3459 ‡ |
| Variables | CAD (n = 30) |
|---|---|
| Lipid plaque, n (%) | 26 (86.67) |
| Fibrous plaque, n (%) | 1 (3.33) |
| Calcific plaque, n (%) | 3 (10) |
| Plaque rupture, n (%) | 15 (50) |
| MLA, mm2 (IQR) | 1.70 (1.43–2.58) |
| TCFA, n (%) | 15 (50) |
| Thrombus, n (%) | 14 (46.67) |
| Lipid quadrants, n | 2.70 ± 1.02 |
| Lipid arc, degree ° (IQR) | 163 (133.5–280) |
| Max lipid arc, degree ° | 206.37 ± 87.10 |
| Macrophage infiltration detection, n (%) | 21 (70) |
| Macrophage NSD | 6.24 ± 1.16 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fiorelli, S.; Porro, B.; Cosentino, N.; Di Minno, A.; Manega, C.M.; Fabbiocchi, F.; Niccoli, G.; Fracassi, F.; Barbieri, S.; Marenzi, G.; et al. Activation of Nrf2/HO-1 Pathway and Human Atherosclerotic Plaque Vulnerability: An In Vitro and In Vivo Study. Cells 2019, 8, 356. https://doi.org/10.3390/cells8040356
Fiorelli S, Porro B, Cosentino N, Di Minno A, Manega CM, Fabbiocchi F, Niccoli G, Fracassi F, Barbieri S, Marenzi G, et al. Activation of Nrf2/HO-1 Pathway and Human Atherosclerotic Plaque Vulnerability: An In Vitro and In Vivo Study. Cells. 2019; 8(4):356. https://doi.org/10.3390/cells8040356
Chicago/Turabian StyleFiorelli, Susanna, Benedetta Porro, Nicola Cosentino, Alessandro Di Minno, Chiara Maria Manega, Franco Fabbiocchi, Giampaolo Niccoli, Francesco Fracassi, Simone Barbieri, Giancarlo Marenzi, and et al. 2019. "Activation of Nrf2/HO-1 Pathway and Human Atherosclerotic Plaque Vulnerability: An In Vitro and In Vivo Study" Cells 8, no. 4: 356. https://doi.org/10.3390/cells8040356
APA StyleFiorelli, S., Porro, B., Cosentino, N., Di Minno, A., Manega, C. M., Fabbiocchi, F., Niccoli, G., Fracassi, F., Barbieri, S., Marenzi, G., Crea, F., Cavalca, V., Tremoli, E., & Eligini, S. (2019). Activation of Nrf2/HO-1 Pathway and Human Atherosclerotic Plaque Vulnerability: An In Vitro and In Vivo Study. Cells, 8(4), 356. https://doi.org/10.3390/cells8040356