The Cytoskeleton—A Complex Interacting Meshwork


Abstract
:1. Introduction
2. Actin Regulation and Structure
2.1. Actin Filaments
2.2. Profilin
2.3. Dendritic Actin Networks
2.4. Non-Muscle Myosin
2.5. Cross-Linked Actin Networks and Actin Bundles
2.6. ADF/Cofilin Induced Actin Disassembly
2.7. Actin Structures Inside the Cell
2.7.1. The Lamellipodium
2.7.2. Filopodia
2.7.3. Stress Fibers
2.7.4. Actin Cortex and Blebs
2.7.5. Nuclear Actin
3. Microtubules
4. Intermediate Filaments
5. Involvement of Cytoskeleton in Cell Motility and Focal Adhesions
5.1. Actin in Motile Processes
5.2. Microtubules in Motile Processes
5.3. Intermediate Filament Involvement in Motile Processes
6. Cytoskeletal Alterations in Glioma
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit. Rev. Oncog. 2013, 18, 43–73. [Google Scholar] [CrossRef]
- Rotty, J.D.; Bear, J.E. Competition and collaboration between different actin assembly pathways allows for homeostatic control of the actin cytoskeleton. Bioarchitecture 2014, 5, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Wen, Q.; Janmey, P.A. Polymer physics of the cytoskeleton. Curr. Opin. Solid State Mater. Sci. 2011, 15, 177–182. [Google Scholar] [CrossRef]
- Sept, D.; Xu, J.; Pollard, T.D.; McCammon, J.A. Annealing accounts for the length of actin filaments formed by spontaneous polymerization. Biophys. J. 1999, 77, 2911–2919. [Google Scholar] [CrossRef]
- Sept, D.; Elcock, A.H.; McCammon, J.A. Computer simulations of actin polymerization can explain the barbed-pointed end asymmetry. J. Mol. Biol. 1999, 294, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D. Actin and Actin-Binding Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D.; Blanchoin, L.; Mullins, R.D. Molecular Mechanisms Controlling Actin Filament Dynamics in Nonmuscle Cells. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 545–576. [Google Scholar] [CrossRef]
- Kang, F.; Purich, D.L.; Frederick, S.; Southwick, F.S. Profilin Promotes Barbed-end Actin Filament Assembly without Lowering the Critical Concentration. J. Biol. Chem. 1999, 274, 36963–36972. [Google Scholar] [CrossRef] [PubMed]
- Machesky, L.M.; Mullins, R.D.; Higgs, H.N.; Kaiser, D.A.; Blanchoin, L.; May, R.C.; Hall, M.E.; Pollard, T.D. Scar, a WASp-related protein, activates nucleation of actin filaments by the Arp2/3 complex. Proc. Natl. Acad. Sci. USA 1999, 96, 3739–3744. [Google Scholar] [CrossRef] [PubMed]
- Kovar, D.R.; Harris, E.S.; Mahaffy, R.; Higgs, H.N.; Pollard, T.D. Control of the assembly of ATP- and ADP-actin by formins and profilin. Cell 2006, 124, 423–435. [Google Scholar] [CrossRef]
- Romero, S.; Le Clainche, C.; Didry, D.; Egile, C.; Pantaloni, D.; Carlier, M.F. Formin is a processive motor that requires profilin to accelerate actin assembly and associated ATP hydrolysis. Cell 2004, 119, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Swaney, K.F.; Li, R. Function and regulation of the Arp2/3 complex during cell migration in diverse environments. Curr. Opin. Cell Biol. 2016, 42, 63–72. [Google Scholar] [CrossRef]
- Svitkina, T.M.; Borisy, G.G. Arp2/3 complex and actin depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J. Cell Biol. 1999, 145, 1009–1026. [Google Scholar] [CrossRef]
- Achard, V.; Martiel, J.L.; Michelot, A.; Guérin, C.; Reymann, A.C.; Blanchoin, L.; Boujemaa-Paterski, R. A “Primer”-Based Mechanism Underlies Branched Actin Filament Network Formation and Motility. Curr. Biol. 2010, 20, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Molinie, N.; Gautreau, A. The Arp2/3 Regulatory System and Its Deregulation in Cancer. Physiol. Rev. 2018, 98, 215–238. [Google Scholar] [CrossRef] [PubMed]
- Akin, O.; Mullins, R.D. Capping Protein Increases the Rate of Actin-Based Motility by Promoting Filament Nucleation by the Arp2/3 Complex. Cell 2008, 133, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Kawska, A.; Carvalho, K.; Manzi, J.; Boujemaa-Paterski, R.; Blanchoin, L.; Martiel, J.-L.; Sykes, C. How actin network dynamics control the onset of actin-based motility. Proc. Natl. Acad. Sci. USA 2012, 109, 14440–14445. [Google Scholar] [CrossRef] [PubMed]
- Vignjevic, D.; Yarar, D.; Welch, M.D.; Peloquin, J.; Svitkina, T.; Borisy, G.G. Formation of filopodia-like bundles in vitro from a dendritic network. J. Cell Biol. 2003, 160, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Bendix, P.M.; Koenderink, G.H.; Cuvelier, D.; Dogic, Z.; Koeleman, B.N.; Brieher, W.M.; Field, C.M.; Mahadevan, L.; Weitz, D.A. A quantitative analysis of contractility in active cytoskeletal protein networks. Biophys. J. 2008, 94, 3126–3136. [Google Scholar] [CrossRef] [PubMed]
- Koenderink, G.H.; Dogic, Z.; Nakamura, F.; Bendix, P.M.; MacKintosh, F.C.; Hartwig, J.H.; Stossel, T.P.; Weitz, D.A. An active biopolymer network controlled by molecular motors. Proc. Natl. Acad. Sci. USA 2009, 106, 15192–15197. [Google Scholar] [CrossRef]
- Finer, J.T.; Simmons, R.M.; Spudich, J.A. Single myosin molecule mechanics: Piconewton forces and nanometre steps. Nature 1994, 368, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, R.K.; Pardee, J.D. Assembly mechanism of Dictyostelium myosin II: Regulation by K+, Mg2+, and actin filaments. Biochemistry 1996, 35, 15504–15514. [Google Scholar] [CrossRef]
- Matsumura, F. Regulation of myosin II during cytokinesis in higher eukaryotes. Trends Cell Biol. 2005, 15, 371–377. [Google Scholar] [CrossRef]
- Jung, H.S.; Komatsu, S.; Ikebe, M.; Craig, R. Head–Head and Head–Tail Interaction: A General Mechanism for Switching Off Myosin II Activity in Cells. Mol. Biol. Cell 2008, 19, 3234–3242. [Google Scholar] [CrossRef] [PubMed]
- Craig, R.; Smith, R.; Kendrick-Jones, J. Light-chain phosphorylation controls the conformation of vertebrate non-muscle and smooth muscle myosin molecules. Nature 1983, 302. [Google Scholar] [CrossRef]
- Yumura, S.; Yoshida, M.; Betapudi, V.; Licate, L.S.; Iwadate, Y.; Nagasaki, A.; Uyeda, T.Q.P.; Egelhoff, T.T. Multiple Myosin II Heavy Chain Kinases: Roles in Filament Assembly Control and Proper Cytokinesis in Dictyostelium. Mol. Biol. Cell 2005, 16, 4256–4266. [Google Scholar] [CrossRef] [PubMed]
- Murakami, N.; Kotula, L.; Hwang, Y.W. Two distinct mechanisms for regulation of nonmuscle myosin assembly via the heavy chain: Phosphorylation for MIIB and Mts 1 binding for MIIA. Biochemistry 2000, 39, 11441–11451. [Google Scholar] [CrossRef] [PubMed]
- Murakami, N.; Chauhan, V.P.S.; Elzinga, M. Two nonmuscle myosin II heavy chain isoforms expressed in rabbit brains: Filament forming properties, the effects of phosphorylation by protein kinase C and casein kinase II, and location of the phosphorylation sites. Biochemistry 1998, 37, 1989–2003. [Google Scholar] [CrossRef]
- Rosenberg, M.; Ravid, S. Protein Kinase C Regulates Myosin IIB Phosphorylation, Cellular Localization, and Filament Assembly. Mol. Biol. Cell 2006, 17, 1364–1374. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, D.A.; Mullins, R.D. Cell mechanics and the cytoskeleton. Nature 2010, 463, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Köhler, S.; Schaller, V.; Bausch, A.R. Collective dynamics of active cytoskeletal networks. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Wagner, B.; Tharmann, R.; Haase, I.; Fischer, M.; Bausch, A.R. Cytoskeletal polymer networks: The molecular structure of cross-linkers determines macroscopic properties. Proc. Natl. Acad. Sci. USA 2006, 103, 13974–13978. [Google Scholar] [CrossRef] [PubMed]
- Falzone, T.T.; Lenz, M.; Kovar, D.R.; Gardel, M.L. Assembly Kinetics Determine the Architecture of α-actinin Crosslinked F-actin Networks. Nat. Commun. 2013, 3. [Google Scholar] [CrossRef]
- Huang, S. Arabidopsis VILLIN1 Generates Actin Filament Cables That Are Resistant to Depolymerization. Plant Cell Online 2005, 17, 486–501. [Google Scholar] [CrossRef]
- Kasza, K.E.; Broedersz, C.P.; Koenderink, G.H.; Lin, Y.C.; Messner, W.; Millman, E.A.; Nakamura, F.; Stossel, T.P.; MacKintosh, F.C.; Weitz, D.A. Actin filament length tunes elasticity of flexibly cross-linked actin networks. Biophys. J. 2010, 99, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.G.; Shi, W.; Ramagopal, U.; Tseng, Y.; Wirtz, D.; Kovar, D.R.; Staiger, C.J.; Almo, S.C. Structure of the actin crosslinking core of fimbrin. Structure 2004, 12, 999–1013. [Google Scholar] [CrossRef] [PubMed]
- Stossel, T.P.; Condeelis, J.; Cooley, L.; Hartwig, J.H.; Noegel, A.; Schleicher, M.; Shapiro, S.S. Filamins as integrattors of cell mechanics and signalling. Nat. Rev. Mol. Cell Biol. 2001, 2, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Schwarz, W.H.; Käs, J.A.; Stossel, T.P.; Janmey, P.A.; Pollard, T.D. Mechanical properties of actin filament networks depend on preparation, polymerization conditions, and storage of actin monomers. Biophys. J. 1998, 74, 2731–2740. [Google Scholar] [CrossRef]
- Bartles, J.R. Parallel actin bundles and their multiple actin-bundling proteins. Curr. Opin. Cell Biol. 2000, 12, 72–78. [Google Scholar] [CrossRef]
- Ferrer, J.M.; Lee, H.; Chen, J.; Pelz, B.; Nakamura, F.; Kamm, R.D.; Lang, M.J. Measuring molecular rupture forces between single actin filaments and actin-binding proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 9221–9226. [Google Scholar] [CrossRef]
- Revenu, C.; Athman, R.; Robine, S.; Louvard, D. The co-workers of actin filaments: From cell structures to signals. Nat. Rev. Mol. Cell Biol. 2004, 5, 635–646. [Google Scholar] [CrossRef]
- Reymann, A.C.; Martiel, J.L.; Cambier, T.; Blanchoin, L.; Boujemaa-Paterski, R.; Théry, M. Nucleation geometry governs ordered actin networks structures. Nat. Mater. 2010, 9, 827–832. [Google Scholar] [CrossRef]
- Chesarone, M.A.; Dupage, A.G.; Goode, B.L. Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat. Rev. Mol. Cell Biol. 2010, 11, 62–74. [Google Scholar] [CrossRef]
- Chen, Q.; Nag, S.; Pollard, T.D. Formins filter modified actin subunits during processive elongation. J. Struct. Biol. 2012, 177, 32–39. [Google Scholar] [CrossRef]
- Goode, B.L.; Eck, M.J. Mechanism and Function of Formins in the Control of Actin Assembly. Annu. Rev. Biochem. 2007, 76, 593–627. [Google Scholar] [CrossRef]
- Michelot, A.; Derivery, E.; Paterski-Boujemaa, R.; Guérin, C.; Huang, S.; Parcy, F.; Staiger, C.J.; Blanchoin, L. A Novel Mechanism for the Formation of Actin-Filament Bundles by a Nonprocessive Formin. Curr. Biol. 2006, 16, 1924–1930. [Google Scholar] [CrossRef]
- Esue, O.; Harris, E.S.; Higgs, H.N.; Wirtz, D. The Filamentous Actin Cross-Linking/Bundling Activity of Mammalian Formins. J. Mol. Biol. 2008, 384, 324–334. [Google Scholar] [CrossRef]
- Edwards, M.; Zwolak, A.; Schafer, D.A.; Sept, D.; Dominguez, R.; Cooper, J.A. Capping protein regulators fine-tune actin assembly dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 677–689. [Google Scholar] [CrossRef]
- Laporte, D.; Ojkic, N.; Vavylonis, D.; Wu, J.-Q. a-Actinin and fimbrin cooperate with myosin II to organize actomyosin bundles during contractile-ring assembly. Mol. Biol. Cell 2012, 23, 3094–3110. [Google Scholar] [CrossRef]
- Köhler, S.; Bausch, A.R. Contraction mechanisms in composite active actin networks. PLoS ONE 2012, 7, 1–8. [Google Scholar] [CrossRef]
- Murrell, M.; Thoresen, T.; Gardel, M. Reconstitution of contractile actomyosin arrays. Methods Enzymol. 2014, 540, 265–282. [Google Scholar] [CrossRef] [PubMed]
- Haviv, L.; Gillo, D.; Backouche, F.; Bernheim-Groswasser, A. A Cytoskeletal Demolition Worker: Myosin II Acts as an Actin Depolymerization Agent. J. Mol. Biol. 2008, 375, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.K.; Petrasek, Z.; Heinemann, F.; Schwille, P. Myosin motors fragment and compact membrane-bound actin filaments. Elife 2013, 2013, 1–18. [Google Scholar] [CrossRef]
- Reymann, A.C.; Boujemaa-Paterski, R.; Martiel, J.L.; Guérin, C.; Cao, W.; Chin, H.F.; De La Cruz, E.M.; Théry, M.; Blanchoin, L. Actin network architecture can determine myosin motor activity. Science 2012, 336, 1310–1314. [Google Scholar] [CrossRef] [PubMed]
- Ingerman, E.; Hsiao, J.Y.; Mullins, R.D. Arp2/3 complex ATP hydrolysis promotes lamellipodial actin network disassembly but is dispensable for assembly. J. Cell Biol. 2013, 200, 619–633. [Google Scholar] [CrossRef]
- Reymann, A.-C.; Suarez, C.; Guerin, C.; Martiel, J.-L.; Staiger, C.J.; Blanchoin, L.; Boujemaa-Paterski, R. Turnover of branched actin filament networks by stochastic fragmentation with ADF/cofilin. Mol. Biol. Cell 2011, 22, 2541–2550. [Google Scholar] [CrossRef]
- Andrianantoandro, E.; Pollard, T.D. Mechanism of Actin Filament Turnover by Severing and Nucleation at Different Concentrations of ADF/Cofilin. Mol. Cell 2006, 24, 13–23. [Google Scholar] [CrossRef]
- Ngo, K.X.; Kodera, N.; Katayama, E.; Ando, T.; Uyeda, T.Q. Cofilin-induced unidirectional cooperative conformational changes in actin filaments revealed by high-speed atomic force microscopy. Elife 2015, 4, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Elam, W.A.; Kang, H.; De La Cruz, E.M. Biophysics of actin filament severing by cofilin. FEBS Lett. 2013, 587, 1215–1219. [Google Scholar] [CrossRef]
- Mccullough, B.R.; Blanchoin, L.; Martiel, J.; La, E.M. De Cofilin Increases the Bending Flexibility of Actin Filaments: Implications for Severing and Cell Mechanics. J. Mol. Biol. 2009, 381, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Goodarzi, J.P.; De La Cruz, E.M. Energetics and Kinetics of Cooperative Cofilin-Actin Filament Interactions. J. Mol. Biol. 2006, 361, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Blanchoin, L.; Pollard, T.D. Mechanism of interaction of Acanthamoeba actophorin (ADF/Cofilin) with actin filaments. J. Biol. Chem. 1999, 274, 15538–15546. [Google Scholar] [CrossRef] [PubMed]
- Kueh, H.Y.; Brieher, W.M.; Mitchison, T.J. Quantitative analysis of actin turnover in listeria comet tails: Evidence for catastrophic filament turnover. Biophys. J. 2010, 99, 2153–2162. [Google Scholar] [CrossRef]
- Hao, Y.K.; Charras, G.T.; Mitchison, T.J.; Brieher, W.M. Actin disassembly by cofilin, coronin, and Aip1 occurs in bursts and is inhibited by barbed-end cappers. J. Cell Biol. 2008, 182, 341–353. [Google Scholar] [CrossRef]
- Suarez, C.; Roland, J.; Boujemaa-Paterski, R.; Kang, H.; McCullough, B.R.; Reymann, A.C.; Guérin, C.; Martiel, J.L.; De La Cruz, E.M.; Blanchoin, L. Cofilin tunes the nucleotide state of actin filaments and severs at bare and decorated segment boundaries. Curr. Biol. 2011, 21, 862–868. [Google Scholar] [CrossRef] [PubMed]
- Breitsprecher, D.; Koestler, S.A.; Chizhov, I.; Nemethova, M.; Mueller, J.; Goode, B.L.; Small, J.V.; Rottner, K.; Faix, J. Cofilin cooperates with fascin to disassemble filopodial actin filaments. J. Cell Sci. 2011, 124, 3305–3318. [Google Scholar] [CrossRef]
- Schmoller, K.M.; Semmrich, C.; Bausch, A.R. Slow down of actin depolymerization by cross-linking molecules. J. Struct. Biol. 2011, 173, 350–357. [Google Scholar] [CrossRef]
- Hayakawa, K.; Tatsumi, H.; Sokabe, M. Actin filaments function as a tension sensor by tension-dependent binding of cofilin to the filament. J. Cell Biol. 2011, 195, 721–727. [Google Scholar] [CrossRef]
- Chan, C.; Beltzner, C.C.; Pollard, T.D. Cofilin Dissociates Arp2/3 Complex and Branches from Actin Filaments. Curr. Biol. 2009, 19, 537–545. [Google Scholar] [CrossRef]
- Abercrombie, M.; Heaysman, J.E.M.; Pegrum, S.M. Locomotion of fibroblasts in culture. V. Surface marking with concanavalin A. Exp. Cell Res. 1972, 73, 536–539. [Google Scholar] [CrossRef]
- Abercrombie, M.; Heaysman, J.E.M.; Pegrum, S.M. The locomotion of fibroblasts in culture. IV. Electron microscopy of the leading lamella. Exp. Cell Res. 1971, 67, 359–367. [Google Scholar] [CrossRef]
- Lauffenburger, D.A.; Horwitz, A.F. Cell migration: A physically integrated molecular process. Cell 1996, 84, 359–369. [Google Scholar] [CrossRef]
- Mitchison, T.J.; Cramer, L.P. Actin-based cell motility and cell locomotion. Cell 1996, 84, 371–379. [Google Scholar] [CrossRef]
- Xu, K.; Babcock, H.P.; Zhuang, X. Dual-objective STORM reveals three-dimensional filament organization in the actin cytoskeleton. Nat. Methods 2012, 9, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Ydenberg, C.A.; Padrick, S.B.; Sweeney, M.O.; Gandhi, M.; Sokolova, O.; Goode, B.L. GMF severs actin-Arp2/3 complex branch junctions by a cofilin- like mechanism. Curr. Biol. 2013, 23, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Manzanares, M.; Ma, X.; Adelstein, R.S.; Horwitz, A.R. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell Biol. 2009, 10, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Vallotton, P.; Gupton, S.L.; Waterman-Storer, C.M.; Danuser, G. Simultaneous mapping of filamentous actin flow and turnover in migrating cells by quantitative fluorescent speckle microscopy. Proc. Natl. Acad. Sci. USA 2004, 101, 9660–9665. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.A.; Tsuchida, M.A.; Allen, G.M.; Barnhart, E.L.; Applegate, K.T.; Yam, P.T.; Ji, L.; Keren, K.; Danuser, G.; Theriot, J.A. Myosin II contributes to cell-scale actin network treadmilling via network disassembly. Nature 2010, 465, 373–377. [Google Scholar] [CrossRef]
- Prass, M.; Jacobson, K.; Mogilner, A.; Radmacher, M. Direct measurement of the lamellipodial protrusive force in a migrating cell. J. Cell Biol. 2006, 174, 767–772. [Google Scholar] [CrossRef]
- Krause, M.; Gautreau, A. Steering cell migration: Lamellipodium dynamics and the regulation of directional persistence. Nat. Rev. Mol. Cell Biol. 2014, 15, 577–590. [Google Scholar] [CrossRef]
- Tang, H.; Li, A.; Bi, J.; Veltman, D.M.; Zech, T.; Spence, H.J.; Yu, X.; Timpson, P.; Insall, R.H.; Frame, M.C.; et al. Loss of scar/WAVE complex promotes N-WASP- and FAK-dependent invasion. Curr. Biol. 2013, 23, 107–117. [Google Scholar] [CrossRef]
- Petrie, R.J.; Gavara, N.; Chadwick, R.S.; Yamada, K.M. Nonpolarized signaling reveals two distinct modes of 3D cell migration. J. Cell Biol. 2012, 197, 439–455. [Google Scholar] [CrossRef]
- Breitsprecher, D.; Kiesewetter, A.K.; Linkner, J.; Vinzenz, M.; Stradal, T.E.B.; Small, J.V.; Curth, U.; Dickinson, R.B.; Faix, J. Molecular mechanism of Ena/VASP-mediated actin-filament elongation. EMBO J. 2011, 30, 456–467. [Google Scholar] [CrossRef]
- Hansen, S.D.; Mullins, R.D. VASP is a processive actin polymerase that requires monomeric actin for barbed end association. J. Cell Biol. 2010, 191, 571–584. [Google Scholar] [CrossRef]
- Mejillano, M.R.; Kojima, S.I.; Applewhite, D.A.; Gertler, F.B.; Svitkina, T.M.; Borisy, G.G. Lamellipodial versus filopodial mode of the actin nanomachinery: Pivotal role of the filament barbed end. Cell 2004, 118, 363–373. [Google Scholar] [CrossRef]
- Small, J.V.; Herzog, M.; Anderson, K. Actin filament organization in the fish keratocyte lamellipodium. J. Cell Biol. 1995, 129, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Helgeson, L.A.; Nolen, B.J. Mechanism of synergistic activation of Arp2/3 complex by cortactin and N-WASP. Elife 2013, 2013, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Dang, I.; Gorelik, R.; Sousa-Blin, C.; Derivery, E.; Guérin, C.; Linkner, J.; Nemethova, M.; Dumortier, J.G.; Giger, F.A.; Chipysheva, T.A.; et al. Inhibitory signalling to the Arp2/3 complex steers cell migration. Nature 2013, 503, 281–284. [Google Scholar] [CrossRef]
- Veltman, D. Actin dynamics: Cell migration takes a new turn with arpin. Curr. Biol. 2014, 24, R31–R33. [Google Scholar] [CrossRef]
- Houk, A.R.; Jilkine, A.; Mejean, C.O.; Boltyanskiy, R.; Dufresne, E.R.; Angenent, S.B.; Altschuler, S.J.; Wu, L.F.; Weiner, O.D. Membrane tension maintains cell polarity by confining signals to the leading edge during neutrophil migration. Cell 2012, 148, 175–188. [Google Scholar] [CrossRef]
- Ofer, N.; Mogilner, A.; Keren, K. Actin disassembly clock determines shape and speed of lamellipodial fragments. PNAS 2011, 108, 20394–20399. [Google Scholar] [CrossRef]
- Svitkina, T.M.; Verkhovsky, A.B.; McQuade, K.M.; Borisy, G.G. Analysis of the actin-myosin II system in fish epidermal keratocytes: Mechanism of cell body translocation. J. Cell Biol. 1997, 139, 397–415. [Google Scholar] [CrossRef]
- Weichsel, J.; Urban, E.; Small, J.V.; Schwarz, U.S. Reconstructing the orientation distribution of actin filaments in the lamellipodium of migrating keratocytes from electron microscopy tomography data. Cytom. Part A 2012, 81 A, 496–507. [Google Scholar] [CrossRef]
- Faix, J.; Rottner, K. The making of filopodia. Curr. Opin. Cell Biol. 2006, 18, 18–25. [Google Scholar] [CrossRef]
- Cojoc, D.; Difato, F.; Ferrari, E.; Shahapure, R.B.; Laishram, J.; Righi, M.; Di Fabrizio, E.M.; Torre, V. Properties of the force exerted by filopodia and lamellipodia and the involvement of cytoskeletal components. PLoS ONE 2007, 2. [Google Scholar] [CrossRef]
- Jacquemet, G.; Hamidi, H.; Ivaska, J. Filopodia in cell adhesion, 3D migration and cancer cell invasion. Curr. Opin. Cell Biol. 2015, 36, 23–31. [Google Scholar] [CrossRef]
- Vignjevic, D.; Peloquin, J.; Borisy, G.G. In vitro assembly of filopodia-like bundles. Methods Enzymol. 2006, 406, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Svitkina, T. Filopodia initiation: Focus on the Arp2/3 complex and formins. Cell Adhes. Migr. 2011, 5, 402–408. [Google Scholar] [CrossRef]
- Block, J.; Breitsprecher, D.; Kühn, S.; Winterhoff, M.; Kage, F.; Geffers, R.; Duwe, P.; Rohn, J.L.; Baum, B.; Brakebusch, C.; et al. FMNL2 drives actin-based protrusion and migration downstream of Cdc42. Curr. Biol. 2012, 22, 1005–1012. [Google Scholar] [CrossRef]
- Miki, H.; Sasaki, T.; Takai, Y.; Takenawa, T. Induction of filopodium formation by a WASP-related actin-depolymerizing protein N-WASP. Nature 1998, 391, 93–96. [Google Scholar] [CrossRef]
- Young, L.E.; Heimsath, E.G.; Higgs, H.N. Cell type-dependent mechanisms for formin-mediated assembly of filopodia. Mol. Biol. Cell 2015, 26, 4646–4659. [Google Scholar] [CrossRef]
- Khurana, S.; George, S.P. The role of actin bundling proteins in the assembly of filopodia in epithelial cells. Cell Adh. Migr. 2011, 5, 409–420. [Google Scholar] [CrossRef]
- Chan, C.E.; Odde, D.J. Traction dynamics of filopodia on compliant substrates. Science 2008, 322, 1687–1691. [Google Scholar] [CrossRef]
- Kress, H.; Stelzer, E.H.K.; Holzer, D.; Buss, F.; Griffiths, G.; Rohrbach, A. Filopodia act as phagocytic tentacles and pull with discrete steps and a load-dependent velocity. Proc. Natl. Acad. Sci. USA 2007, 104, 11633–11638. [Google Scholar] [CrossRef]
- Naumanen, P.; Lappalainen, P.; Hotulainen, P. Mechanisms of actin stress fibre assembly. J. Microsc. 2008, 231, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Cramer, L.P.; Siebert, M.; Mitchison, T.J. Identification of novel graded polarity actin filament bundles in locomoting heart fibroblasts: Implications for the generation of motile force. J. Cell Biol. 1997, 136, 1287–1305. [Google Scholar] [CrossRef] [PubMed]
- Sjöblom, B.; Salmazo, A.; Djinović-Carugo, K. α-Actinin structure and regulation. Cell. Mol. Life Sci. 2008, 65, 2688–2701. [Google Scholar] [CrossRef] [PubMed]
- Koenderink, G.H.; Paluch, E.K. Architecture shapes contractility in actomyosin networks. Curr. Opin. Cell Biol. 2018, 50, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Pellegrin, S.; Mellor, H. Actin stress fibres. J. Cell Sci. 2007, 120, 3491–3499. [Google Scholar] [CrossRef]
- Hotulainen, P.; Lappalainen, P. Stress fibers are generated by two distinct actin assembly mechanisms in motile cells. J. Cell Biol. 2006, 173, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Tojkander, S.; Gateva, G.; Schevzov, G.; Hotulainen, P.; Naumanen, P.; Martin, C.; Gunning, P.W.; Lappalainen, P. A molecular pathway for myosin II recruitment to stress fibers. Curr. Biol. 2011, 21, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Endlich, N.; Schordan, E.; Cohen, C.D.; Kretzler, M.; Lewko, B.; Welsch, T.; Kriz, W.; Otey, C.A.; Endlich, K. Palladin is a dynamic actin-associated protein in podocytes. Kidney Int. 2009, 75, 214–226. [Google Scholar] [CrossRef]
- Schmidt, K.; Nichols, B.J. Functional interdependence between septin and actin cytoskeleton. BMC Cell Biol. 2004, 5, 1–13. [Google Scholar] [CrossRef]
- Lazarides, E.; Burridge, K. α-Actinin: Immunofluorescent localization of a muscle structural protein in nonmuscle cells. Cell 1975, 6, 289–298. [Google Scholar] [CrossRef]
- Vallenius, T.; Makela, T.P. Clik1: A novel kinase targeted to actin stress fibers by the CLP-36 PDZ-LIM protein. J. Cell Sci. 2002, 115, 2067–2073. [Google Scholar]
- Vallenius, T.; Luukko, K.; Mäkelä, T.P. CLP-36 PDZ-LIM Protein Associates with Nonmuscle alpha -Actinin-1 and alpha -Actinin-4. J. Biol. Chem. 2000, 275, 11100–11105. [Google Scholar] [CrossRef]
- Wang, K.; Ash, J.F.; Singer, S.J. Filamin, a New High-Molecular-Weight Protein Found in Smooth Muscle and Nonmuscle Cells. Purification and Properties of Chicken Gizzard Filamin. Proc. Natl. Acad. Sci. USA 1975, 72, 4483–4486. [Google Scholar] [CrossRef]
- Dixon, R.D.S.; Arneman, D.K.; Rachlin, A.S.; Sundaresan, N.R.; Costello, M.J.; Campbell, S.L.; Otey, C.A. Palladin is an actin cross-linking protein that uses immunoglobulin-like domains to bind filamentous actin. J. Biol. Chem. 2008, 283, 6222–6231. [Google Scholar] [CrossRef]
- Elkhatib, N.; Neu, M.B.; Zensen, C.; Schmoller, K.M.; Louvard, D.; Bausch, A.R.; Betz, T.; Vignjevic, D.M. Fascin plays a role in stress fiber organization and focal adhesion disassembly. Curr. Biol. 2014, 24, 1492–1499. [Google Scholar] [CrossRef]
- Boukhelifa, M.; Moza, M.; Johansson, T.; Rachlin, A.; Parast, M.; Huttelmaier, S.; Roy, P.; Jockusch, B.M.; Carpen, O.; Karlsson, R.; et al. The proline-rich protein palladin is a binding partner for profilin. FEBS J. 2006, 273, 26–33. [Google Scholar] [CrossRef]
- Boukhelifa, M.; Parast, M.M.; Bear, J.E.; Gertler, F.B.; Otey, C.A. Palladin Is a Novel Binding Partner for Ena/VASP Family Members. Cell Motil. Cytoskeleton 2004, 58, 17–29. [Google Scholar] [CrossRef]
- Strasser, P.; Gimona, M.; Moessler, H.; Herzog, M.; Small, J.V. Mammalian calponin: Identification and expression of genetic variants. FEBS J. 1993, 330, 13–18. [Google Scholar] [CrossRef]
- Weber, K.; Groeschel-Stewart, U. Antibody to myosin: The specific visualization of myosin-containing filaments in nonmuscle cells. Proc. Natl. Acad. Sci. USA 1974, 71, 4561–4564. [Google Scholar] [CrossRef]
- Yamashiro-Matsumura, S.; Matsumura, F. Characterization of 83-kilodalton nonmuscle caldesmon from cultured rat cells: Stimulation of actin binding of nonmuscle tropomyosin and periodic localization along microfilaments like tropomyosin. J. Cell Biol. 1988, 106, 1973–1983. [Google Scholar] [CrossRef] [PubMed]
- Castellino, F.; Ono, S.; Matsumura, F.; Luini, A. Essential role of caldesmon in the actin filament reorganization induced by glucocorticoids. J. Cell Biol. 1995, 131, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Leung, T.; Chen, X.Q.; Manser, E.; Lim, L. The p160 RhoA-binding kinase ROK alpha is a member of a kinase family and is involved in the reorganization of the cytoskeleton. Mol. Cell. Biol. 1996, 16, 5313–5327. [Google Scholar] [CrossRef]
- Watanabe, N.; Madaule, P.; Reid, T.; Ishizaki, T.; Watanabe, G.; Kakizuka, A.; Saito, Y.; Nakao, K.; Jockusch, B.M.; Narumiya, S. p140mDia, a mammalian homolog of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO J. 1997, 16, 3044–3056. [Google Scholar] [CrossRef]
- Tominaga, T.; Sahai, E.; Chardin, P.; McCormick, F.; Courtneidge, S.A.; Alberts, A.S. Diaphanous-related formins bridge Rho GTPase and Src tyrosine kinase signaling. Mol. Cell 2000, 5, 13–25. [Google Scholar] [CrossRef]
- Maekawa, M.; Ishizaki, T.; Boku, S.; Watanabe, N.; Fujita, A.; Iwamatsu, A.; Obinata, T.; Ohashi, K.; Mizuno, K.; Narumiya, S. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 1999, 285, 895–898. [Google Scholar] [CrossRef]
- Chen, Z.; Borek, D.; Padrick, S.B.; Gomez, T.S.; Metlagel, Z.; Ismail, A.M.; Umetani, J.; Billadeau, D.D.; Otwinowski, Z.; Rosen, M.K. Structure and control of the actin regulatory WAVE complex. Nature 2010, 468, 533–538. [Google Scholar] [CrossRef]
- Lebensohn, A.M.; Kirschner, M.W. Activation of the WAVE Complex by Coincident Signals Controls Actin Assembly. Mol. Cell 2009, 36, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Campellone, K.G.; Welch, M.D. A nucleator arms race: Cellular control of actin assembly. Nat. Rev. Mol. Cell Biol. 2010, 11, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.J.; Rajfur, Z.; Maddox, A.S.; Freel, C.D.; Chen, Y.; Edlund, M.; Otey, C.; Burridge, K. Simultaneous Stretching and Contraction of Stress Fibers In Vivo. Mol. Biol. Cell 2004, 15, 3497–3508. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Oswald, S.J.; Ngu, H.; Yin, F.C.P. Mechanical properties of actin stress fibers in living cells. Biophys. J. 2008, 95, 6060–6071. [Google Scholar] [CrossRef]
- Deguchi, S.; Ohashi, T.; Sato, M. Tensile properties of single stress fibers isolated from cultured vascular smooth muscle cells. J. Biomech. 2006, 39, 2603–2610. [Google Scholar] [CrossRef] [PubMed]
- Anderson, T.W.; Vaughan, A.N.; Cramer, L.P. Retrograde Flow and Myosin II Activity within the Leading Cell Edge Deliver F-Actin to the Lamella to Seed the Formation of Graded Polarity Actomyosin II Filament Bundles. Mol. Biol. Cell 2008, 19, 5006–5018. [Google Scholar] [CrossRef]
- Langanger, G.; Moeremans, M.; Daneels, G.; Sobieszek, A.; De Brabander, M.; De Mey, J. The molecular organisation of myosin in stress fibers of cultured cells. J. Cell Biol. 1986, 102, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Burnette, D.T.; Manley, S.; Sengupta, P.; Sougrat, R.; Davidson, M.W.; Kachar, B.; Lippincott-Schwartz, J. A role for actin arcs in the leading-edge advance of migrating cells. Nat. Cell Biol. 2011, 13, 371–382. [Google Scholar] [CrossRef]
- Hu, S.; Dasbiswas, K.; Guo, Z.; Tee, Y.H.; Thiagarajan, V.; Hersen, P.; Chew, T.L.; Safran, S.A.; Zaidel-Bar, R.; Bershadsky, A.D. Long-range self-organization of cytoskeletal myosin II filament stacks. Nat. Cell Biol. 2017, 19, 133–141. [Google Scholar] [CrossRef]
- Dasanayake, N.L.; Michalski, P.J.; Carlsson, A.E. General mechanism of actomyosin contractility. Phys. Rev. Lett. 2011, 107, 1–4. [Google Scholar] [CrossRef]
- Nemethova, M.; Auinger, S.; Small, J.V. Building the actin cytoskeleton: Filopodia contribute to the construction of contractile bundles in the lamella. J. Cell Biol. 2008, 180, 1233–1244. [Google Scholar] [CrossRef]
- Tojkander, S.; Gateva, G.; Husain, A.; Krishnan, R.; Lappalainen, P. Generation of contractile actomyosin bundles depends on mechanosensitive actin filament assembly and disassembly. Elife 2015, 4. [Google Scholar] [CrossRef]
- Small, J.V.; Rottner, K.; Kaverina, I.; Anderson, K.I. Assembling an actin cytoskeleton for cell attachment and movement. Biochim. Biophys. Acta Mol. Cell Res. 1998, 1404, 271–281. [Google Scholar] [CrossRef]
- Chen, W.-T. Mechanism of the trailing edge during fibroblast movement. J.Cell Biol. 1981, 90, 187–200. [Google Scholar] [CrossRef]
- Khatau, S.B.; Hale, C.M.; Stewart-Hutchinson, P.J.; Patel, M.S.; Stewart, C.L.; Searson, P.C.; Hodzic, D.; Wirtz, D. A perinuclear actin cap regulates nuclear shape. Proc. Natl. Acad. Sci. USA 2009, 106, 19017–19022. [Google Scholar] [CrossRef]
- Burridge, K.; Guilluy, C. Focal adhesions, stress fibers and mechanical tension. Exp. Cell Res. 2016, 343, 14–20. [Google Scholar] [CrossRef]
- Gateva, G.; Tojkander, S.; Koho, S.; Carpen, O.; Lappalainen, P. Palladin promotes assembly of non-contractile dorsal stress fibers through VASP recruitment. J. Cell Sci. 2014, 127, 1887–1898. [Google Scholar] [CrossRef]
- Kovac, B.; Teo, J.L.; Mäkelä, T.P.; Vallenius, T. Assembly of non-contractile dorsal stress fibers requires α-actinin-1 and Rac1 in migrating and spreading cells. J. Cell Sci. 2013, 126, 263–273. [Google Scholar] [CrossRef]
- Shemesh, T.; Verkhovsky, A.B.; Svitkina, T.M.; Bershadsky, A.D.; Kozlov, M.M. Role of focal adhesions and mechanical stresses in the formation and progression of the lamellum interface. Biophys. J. 2009, 97, 1254–1264. [Google Scholar] [CrossRef]
- Eghiaian, F.; Rigato, A.; Scheuring, S. Structural, mechanical, and dynamical variability of the actin cortex in living cells. Biophys. J. 2015, 108, 1330–1340. [Google Scholar] [CrossRef]
- Clausen, M.P.; Colin-York, H.; Schneider, F.; Eggeling, C.; Fritzsche, M. Dissecting the actin cortex density and membrane-cortex distance in living cells by super-resolution microscopy. J. Phys. D Appl. Phys. 2017, 50. [Google Scholar] [CrossRef]
- Hanakam, F.; Albrecht, R.; Eckerskorn, C.; Matzner, M.; Gerisch, G. Myristoylated and non-myristoylated forms of the pH sensor protein hisactophilin II: Intracellular shuttling to plasma membrane and nucleus monitored in real time by a fusion with green fluorescent protein. EMBO J. 1996, 15, 2935–2943. [Google Scholar] [CrossRef]
- Morone, N.; Usukura, J.; Kusumi, A. Three-dimensional reconstruction of the membrane skelton at the plama membrane interface by electron tomography. J. Cell Biol. 2006, 174, 851–862. [Google Scholar] [CrossRef]
- Biro, M.; Romeo, Y.; Kroschwald, S.; Bovellan, M.; Boden, A.; Tcherkezian, J.; Roux, P.P.; Charras, G.; Paluch, E.K. Cell cortex composition and homeostasis resolved by integrating proteomics and quantitative imaging. Cytoskeleton 2013, 70, 741–754. [Google Scholar] [CrossRef]
- Bovellan, M.; Romeo, Y.; Biro, M.; Boden, A.; Chugh, P.; Yonis, A.; Vaghela, M.; Fritzsche, M.; Moulding, D.; Thorogate, R.; et al. Cellular control of cortical actin nucleation. Curr. Biol. 2014, 24, 1628–1635. [Google Scholar] [CrossRef]
- Roh-johnson, M.; Shemer, G.; Higgins, C.D.; Mcclellan, J.H.; Werts, A.D.; Tulu, U.S.; Gao, L.; Betzig, E.; Kiehart, D.P.; Goldstein, B. Triggering a cell shape change by exploiting preexisting actomyosin contractions. Science 2012, 335, 1232–1235. [Google Scholar] [CrossRef]
- Charras, G.T.; Hu, C.K.; Coughlin, M.; Mitchison, T.J. Reassembly of contractile actin cortex in cell blebs. J. Cell Biol. 2006, 175, 477–490. [Google Scholar] [CrossRef]
- McClatchey, A.I.; Fehon, R.G. Merlin and the ERM proteins - regulators of receptor distribution and signaling at the cell cortex. Trends Cell Biol. 2009, 19, 198–206. [Google Scholar] [CrossRef]
- Carlier, M.; Laurent, V.; Santolini, J.; Melki, R.; Didry, D.; Xia, G.; Hong, Y.; Chua, N.; Pantaloni, D. Actin depolymerizing factor (ADF/Cofilin) enhances the rate of filament turnover: Implication in actin-based motility. J. Cell Biol. 1997, 136, 1307–1322. [Google Scholar] [CrossRef]
- Chugh, P.; Clark, A.G.; Smith, M.B.; Cassani, D.A.D.; Dierkes, K.; Ragab, A.; Roux, P.P.; Charras, G.; Salbreux, G.; Paluch, E.K. Actin cortex architecture regulates cell surface tension. Nat. Cell Biol. 2017, 19, 689–697. [Google Scholar] [CrossRef]
- Howard, J. Mechanics of Motor Proteins and the Cytoskeleton; Oxford University Press: Oxford, UK, 2005; ISBN 0-87893-334-4. [Google Scholar]
- Ananthakrishnan, R.; Guck, J.; Wottawah, F.; Schinkinger, S.; Lincoln, B.; Romeyke, M.; Moon, T.; Käs, J. Quantifying the contribution of actin networks to the elastic strength of fibroblasts. J. Theor. Biol. 2006, 242, 502–516. [Google Scholar] [CrossRef]
- Tee, S.Y.; Fu, J.; Chen, C.S.; Janmey, P.A. Cell shape and substrate rigidity both regulate cell stiffness. Biophys. J. 2011, 100, L25–L27. [Google Scholar] [CrossRef]
- Guha, M.; Zhou, M.; Wang, Y.L. Cortical actin turnover during cytokinesis requires myosin II. Curr. Biol. 2005, 15, 732–736. [Google Scholar] [CrossRef]
- Murthy, K.; Wadsworth, P. Myosin-II-dependent localization and dynamics of F-actin during cytokinesis. Curr. Biol. 2005, 15, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Bursac, P.; Lenormand, G.; Fabry, B.; Oliver, M.; Weitz, D.A.; Viasnoff, V.; Butler, J.P.; Fredberg, J.J. Cytoskeletal remodelling and slow dynamics in the living cell. Nat. Mater. 2005, 4, 557–561. [Google Scholar] [CrossRef]
- Gardel, M.L.; Nakamura, F.; Hartwig, J.H.; Crocker, J.C.; Stossel, T.P.; Weitz, D.A. Prestressed F-actin networks cross-linked by hinged filamins replicate mechanical properties of cells. Proc. Natl. Acad. Sci. USA 2006, 103, 1762–1767. [Google Scholar] [CrossRef]
- Lecuit, T.; Lenne, P.F. Cell surface mechanics and the control of cell shape, tissue patterns and morphogenesis. Nat. Rev. Mol. Cell Biol. 2007, 8, 633–644. [Google Scholar] [CrossRef]
- Charras, G.T.; Coughlin, M.; Mitchison, T.J.; Mahadevan, L. Life and times of a cellular bleb. Biophys. J. 2008, 94, 1836–1853. [Google Scholar] [CrossRef]
- Kasza, K.E.; Rowat, A.C.; Liu, J.; Angelini, T.E.; Brangwynne, C.P.; Koenderink, G.H.; Weitz, D.A. The cell as a material. Curr. Opin. Cell Biol. 2007, 19, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Paluch, E.K.; Raz, E. The role and regulation of blebs in cell migration. Curr. Opin. Cell Biol. 2013, 25, 582–590. [Google Scholar] [CrossRef]
- Goudarzi, M.; Banisch, T.U.; Mobin, M.B.; Maghelli, N.; Tarbashevich, K.; Strate, I.; van den Berg, J.; Blaser, H.; Bandemer, S.; Paluch, E.; et al. Identification and Regulation of a Molecular Module for Bleb-Based Cell Motility. Dev. Cell 2012, 23, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Tinevez, J.-Y.; Schulze, U.; Salbreux, G.; Roensch, J.; Joanny, J.-F.; Paluch, E. Role of cortical tension in bleb growth. Proc. Natl. Acad. Sci. USA 2009, 106, 18581–18586. [Google Scholar] [CrossRef] [PubMed]
- Paluch, E.; Piel, M.; Prost, J.; Bornens, M.; Sykes, C. Cortical actomyosin breakage triggers shape oscillations in cells and cell fragments. Biophys. J. 2005, 89, 724–733. [Google Scholar] [CrossRef]
- Charras, G.T.; Yarrow, J.C.; Horton, M.A.; Mahadevan, L.; Mitchison, T.J. Non-equilibration of hydrostatic pressure in blebbing cells. Nature 2005, 435, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Paluch, E.; Van Der Gucht, J.; Sykes, C. Cracking up: Symmetry breaking in cellular systems. J. Cell Biol. 2006, 175, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Loitto, V.M.; Karlsson, T.; Magnusson, K.E. Water flux in cell motility: Expanding the mechanisms of membrane protrusion. Cell Motil. Cytoskeleton 2009, 66, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Blaser, H.; Reichman-Fried, M.; Castanon, I.; Dumstrei, K.; Marlow, F.L.; Kawakami, K.; Solnica-Krezel, L.; Heisenberg, C.P.; Raz, E. Migration of Zebrafish Primordial Germ Cells: A Role for Myosin Contraction and Cytoplasmic Flow. Dev. Cell 2006, 11, 613–627. [Google Scholar] [CrossRef]
- Sahai, E.; Marshall, C.J. Differing modes for tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat. Cell Biol. 2003, 5, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Tournaviti, S.; Hannemann, S.; Terjung, S.; Kitzing, T.M.; Stegmayer, C.; Ritzerfeld, J.; Walther, P.; Grosse, R.; Nickel, W.; Fackler, O.T. SH4-domain-induced plasma membrane dynamization promotes bleb-associated cell motility. J. Cell Sci. 2007, 120, 3820–3829. [Google Scholar] [CrossRef] [PubMed]
- Rossy, J.; Gutjahr, M.C.; Blaser, N.; Schlicht, D.; Niggli, V. Ezrin/moesin in motile Walker 256 carcinosarcoma cells: Signal-dependent relocalization and role in migration. Exp. Cell Res. 2007, 313, 1106–1120. [Google Scholar] [CrossRef]
- Brugues, J.; Maugis, B.; Casademunt, J.; Nassoy, P.; Amblard, F.; Sens, P. Dynamical organization of the cytoskeletal cortex probed by micropipette aspiration. Proc. Natl. Acad. Sci. USA 2010, 107, 15415–15420. [Google Scholar] [CrossRef] [PubMed]
- Charras, G.T. A short history of blebbing. J. Microsc. 2008, 231, 466–478. [Google Scholar] [CrossRef]
- Cunningham, C.C. Actin polymerization and intracellular solvent flow in cell surfase blebbing. J. Cell Biol. 1995, 129, 1589–1599. [Google Scholar] [CrossRef]
- Strychalski, W.; Guy, R.D. A computational model of bleb formation. Math. Med. Biol. 2013, 30, 115–130. [Google Scholar] [CrossRef]
- Keller, H.; Rentsch, P.; Hagmann, J. Differences in cortical actin structure and dynamics document that different types of blebs are formed by distinct mechanisms. Exp. Cell Res. 2002, 277, 161–172. [Google Scholar] [CrossRef]
- Kristó, I.; Bajusz, I.; Bajusz, C.; Borkúti, P.; Vilmos, P. Actin, actin-binding proteins, and actin-related proteins in the nucleus. Histochem. Cell Biol. 2016, 145, 373–388. [Google Scholar] [CrossRef] [PubMed]
- Bajusz, C.; Borkúti, P.; Kristó, I.; Kovács, Z.; Abonyi, C.; Vilmos, P. Nuclear actin: Ancient clue to evolution in eukaryotes? Histochem. Cell Biol. 2018, 150, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Le, H.Q.; Ghatak, S.; Yeung, C.Y.C.; Tellkamp, F.; Günschmann, C.; Dieterich, C.; Yeroslaviz, A.; Habermann, B.; Pombo, A.; Niessen, C.M.; et al. Mechanical regulation of transcription controls Polycomb-mediated gene silencing during lineage commitment. Nat. Cell Biol. 2016, 18, 864–875. [Google Scholar] [CrossRef]
- Obrdlik, A.; Percipalle, P. The F-actin severing protein cofilin-1 is required for RNA polymerase II transcription elongation. Nucleus 2011, 2, 72–79. [Google Scholar] [CrossRef]
- Serebryannyy, L.A.; Parilla, M.; Annibale, P.; Cruz, C.M.; Laster, K.; Gratton, E.; Kudryashov, D.; Kosak, S.T.; Gottardi, C.J.; de Lanerolle, P. Persistent nuclear actin filaments inhibit transcription by RNA polymerase II. J. Cell Sci. 2016, 129, 3412–3425. [Google Scholar] [CrossRef] [PubMed]
- Baarlink, C.; Plessner, M.; Sherrard, A.; Morita, K.; Misu, S.; Virant, D.; Kleinschnitz, E.M.; Harniman, R.; Alibhai, D.; Baumeister, S.; et al. A transient pool of nuclear F-actin at mitotic exit controls chromatin organization. Nat. Cell Biol. 2017, 19, 1389–1399. [Google Scholar] [CrossRef] [PubMed]
- de Leeuw, R.; Gruenbaum, Y.; Medalia, O. Nuclear Lamins: Thin Filaments with Major Functions. Trends Cell Biol. 2018, 28, 34–45. [Google Scholar] [CrossRef]
- Baarlink, C.; Wang, H.; Grosse, R. Nuclear Actin Network Assembly by Formins Regulates the SRF Coactivator MAL. Science 2013, 2, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Plessner, M.; Grosse, R. Dynamizing nuclear actin filaments. Curr. Opin. Cell Biol. 2019, 56, 1–6. [Google Scholar] [CrossRef]
- Parisis, N.; Krasinska, L.; Harker, B.; Urbach, S.; Rossignol, M.; Camasses, A.; Dewar, J.; Morin, N.; Fisher, D. Initiation of DNA replication requires actin dynamics and formin activity. EMBO J. 2017, e201796585. [Google Scholar] [CrossRef] [PubMed]
- Belin, B.J.; Lee, T.; Mullins, R.D. DNA damage induces nuclear actin filament assembly by formin-2 and spire-1/2 that promotes efficient DNA repair. Elife 2015, 4, 1–21. [Google Scholar] [CrossRef]
- Caridi, C.P.; D’agostino, C.; Ryu, T.; Zapotoczny, G.; Delabaere, L.; Li, X.; Khodaverdian, V.Y.; Amaral, N.; Lin, E.; Rau, A.R.; et al. Nuclear F-actin and myosins drive relocalization of heterochromatic breaks. Nature 2018, 559, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Schrank, B.R.; Aparicio, T.; Li, Y.; Chang, W.; Chait, B.T.; Gundersen, G.G.; Gottesman, M.E.; Gautier, J. Nuclear ARP2/3 drives DNA break clustering for homology-directed repair. Nature 2018, 559, 61–66. [Google Scholar] [CrossRef]
- Kelpsch, D.J.; Tootle, T.L. Nuclear Actin: From Discovery to Function. Anat. Rec. 2018, 2013, 1999–2013. [Google Scholar] [CrossRef] [PubMed]
- Ohi, R.; Zanic, M. Ahead of the Curve: New Insights into Microtubule Dynamics. F1000Research 2016, 5, 314. [Google Scholar] [CrossRef] [PubMed]
- Doxsey, S. Re-evaluating centrosome function. Nat. Rev. Mol. Cell Biol. 2001, 2, 688–698. [Google Scholar] [CrossRef]
- Vinogradova, T.; Miller, P.M.; Kaverina, I. Microtubule network asymmetry in motile cells: Role of Golgi-derived array. Cell Cycle 2009, 8, 2168–2174. [Google Scholar] [CrossRef]
- Sanchez, A.D.; Feldman, J.L. Microtubule-organizing centers: From the centrosome to non-centrosomal sites. Curr. Opin. Cell Biol. 2017, 44, 93–101. [Google Scholar] [CrossRef]
- Wu, J.; Akhmanova, A. Microtubule-Organizing Centers. Annu. Rev. Cell Dev. Biol. 2017, 33, 51–75. [Google Scholar] [CrossRef]
- Dimitrov, A.; Quesnoit, M.; Moutel, S.; Cantaloube, I.; Poüs, C.; Perez, F. Detection of GTP-tubulin conformation in vivo reveals a role for GTP remnants in microtubule rescues. Science 2008, 322, 1353–1356. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.; Hyman, A.A. Growth, fluctuation and switching at microtubule plus ends. Nat. Rev. Mol. Cell Biol. 2009, 10, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Duellberg, C.; Cade, N.I.; Holmes, D.; Surrey, T. The size of the EB cap determines instantaneous microtubule stability. Elife 2016, 5, e13470. [Google Scholar] [CrossRef] [PubMed]
- Kita, K.; Wittmann, T.; Näthke, I.S.; Waterman-Storer, C.M. Adenomatous polyposis coli on microtubule plus ends in cell extensions can promote microtubule net growth with or without EB1. Mol. Biol. Cell 2006, 17, 2331–2345. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Zhou, X.Z.; Lu, K.P. Critical role for the EB1 and APC interaction in the regulation of microtubule polymerization. Curr. Biol. 2001, 11, 1062–1067. [Google Scholar] [CrossRef]
- Akhmanova, A.; Steinmetz, M.O. Control of microtubule organization and dynamics: Two ends in the limelight. Nat. Rev. Mol. Cell Biol. 2015, 16, 711–726. [Google Scholar] [CrossRef] [PubMed]
- Mimori-Kiyosue, Y.; Grigoriev, I.; Lansbergen, G.; Sasaki, H.; Matsui, C.; Severin, F.; Galjart, N.; Grosveld, F.; Vorobjev, I.; Tsukita, S.; et al. CLASP1 and CLASP2 bind to EB1 and regulate microtubule plus-end dynamics at the cell cortex. J. Cell Biol. 2005, 168, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Suozzi, K.C.; Wu, X.; Fuchs, E. Spectraplakins: Master orchestrators of cytoskeletal dynamics. J. Cell Biol. 2012, 197, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Drabek, K.; van Ham, M.; Stepanova, T.; Draegestein, K.; van Horssen, R.; Sayas, C.L.; Akhmanova, A.; ten Hagen, T.; Smits, R.; Fodde, R.; et al. Role of CLASP2 in Microtubule Stabilization and the Regulation of Persistent Motility. Curr. Biol. 2006, 16, 2259–2264. [Google Scholar] [CrossRef]
- Leung, C.L.; Sun, D.; Zheng, M.; Knowles, D.R.; Liem, R.K. Microtubule actin cross-linking factor (MACF): A hybrid of dystonin and dystrophin that can interact with the actin and microtubule cytoskeletons. J. Cell Biol. 1999, 147, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Komarova, Y.; De Groot, C.O.; Grigoriev, I.; Gouveia, S.M.; Munteanu, E.L.; Schober, J.M.; Honnappa, S.; Buey, R.M.; Hoogenraad, C.C.; Dogterom, M.; et al. Mammalian end binding proteins control persistent microtubule growth. J. Cell Biol. 2009, 184, 691–706. [Google Scholar] [CrossRef]
- Maurer, S.P.; Cade, N.I.; Bohner, G.; Gustafsson, N.; Boutant, E.; Surrey, T. EB1 Accelerates Two Conformational Transitions Important for Microtubule Maturation and Dynamics. Curr. Biol. 2014, 24, 372–384. [Google Scholar] [CrossRef]
- Dong, Z.; Zhou, L.; Han, N.; Zhang, M.; Lyu, X. Wnt/β-catenin pathway involvement in ionizing radiation-induced invasion of U87 glioblastoma cells. Strahlentherapie und Onkol. 2015, 191, 672–680. [Google Scholar] [CrossRef]
- Evans, K.J.; Gomes, E.R.; Reisenweber, S.M.; Gundersen, G.G.; Lauring, B.P. Linking axonal degeneration to microtubule remodeling by Spastin-mediated microtubule severing. J. Cell Biol. 2005, 168, 599–606. [Google Scholar] [CrossRef]
- Sharp, D.J.; Ross, J.L. Microtubule-severing enzymes at the cutting edge. J. Cell Sci. 2012, 125, 2561–2569. [Google Scholar] [CrossRef]
- Gupta, K.K.; Li, C.; Duan, A.; Alberico, E.O.; Kim, O.V.; Alber, M.S.; Goodson, H. V Mechanism for the catastrophe-promoting activity of the microtubule destabilizer Op18/stathmin. Proc. Natl. Acad. Sci. USA 2013, 110, 20449–20454. [Google Scholar] [CrossRef]
- Brouhard, G.J.; Stear, J.H.; Noetzel, T.L.; Al-Bassam, J.; Kinoshita, K.; Harrison, S.C.; Howard, J.; Hyman, A.A. XMAP215 Is a Processive Microtubule Polymerase. Cell 2008, 132, 79–88. [Google Scholar] [CrossRef]
- Moores, C.A.; Perderiset, M.; Kappeler, C.; Kain, S.; Drummond, D.; Perkins, S.J.; Chelly, J.; Cross, R.; Houdusse, A.; Francis, F. Distinct roles of doublecortin modulating the microtubule cytoskeleton. EMBO J. 2006, 25, 4448–4457. [Google Scholar] [CrossRef]
- Dehmelt, L.; Halpain, S. The MAP2/Tau family of microtubule-associated proteins. Genome Biol. 2004, 6, 204. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Yu, W.; Andreadis, A.; Luo, M.; Baas, P.W. Tau Protects Microtubules in the Axon from Severing by Katanin. J. Neurosci. 2006, 26, 3120–3129. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.M.; Folkmann, A.W.; Maia, A.R.R.; Efimova, N.; Efimov, A.; Kaverina, I. Golgi-derived CLASP-dependent microtubules control Golgi organization and polarized trafficking in motile cells. Nat. Cell Biol. 2009, 11, 1069–1080. [Google Scholar] [CrossRef]
- Yadav, S.; Puri, S.; Linstedt, A. A Primary Role for Golgi Positioning in Directed Secretion, Cell Polarity, and Wound Healing. Mol. Biol. Cell 2009, 20, 1728–1736. [Google Scholar] [CrossRef]
- Verhey, K.J.; Kaul, N.; Soppina, V. Kinesin Assembly and Movement in Cells. Annu. Rev. Biophys. 2011, 40, 267–288. [Google Scholar] [CrossRef]
- Cianfrocco, M.A.; DeSantis, M.E.; Leschziner, A.E.; Reck-Peterson, S.L. Mechanism and regulation of cytoplasmic dynein. Annu. Rev. Cell Dev. Biol. 2015, 31, 83–108. [Google Scholar] [CrossRef]
- Osmani, N.; Peglion, F.; Chavrier, P.; Etienne-Manneville, S. Cdc42 localization and cell polarity depend on membrane traffic. J. Cell Biol. 2010, 191, 1261–1269. [Google Scholar] [CrossRef]
- Palamidessi, A.; Frittoli, E.; Garré, M.; Faretta, M.; Mione, M.; Testa, I.; Diaspro, A.; Lanzetti, L.; Scita, G.; Di Fiore, P.P. Endocytic Trafficking of Rac Is Required for the Spatial Restriction of Signaling in Cell Migration. Cell 2008, 134, 135–147. [Google Scholar] [CrossRef]
- Hookway, C.; Ding, L.; Davidson, M.W.; Rappoport, J.Z.; Danuser, G.; Gelfand, V.I. Microtubule-dependent transport and dynamics of vimentin intermediate filaments. Mol. Biol. Cell 2015, 26, 1675–1686. [Google Scholar] [CrossRef] [PubMed]
- Robert, A.; Tian, P.; Adam, S.A.; Kittisopikul, M.; Jaqaman, K.; Goldman, R.D.; Gelfand, V.I. Kinesin-dependent transport of keratin filaments: A unified mechanism for intermediate filament transport. FASEB J. 2018, fj.201800604R. [Google Scholar] [CrossRef] [PubMed]
- Helfand, B.T.; Mikami, A.; Vallee, R.B.; Goldman, R.D. A requirement for cytoplasmic dynein and dynactin in intermediate filament network assembly and organization. J. Cell Biol. 2002, 157, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Mingle, L.A. Localization of all seven messenger RNAs for the actin-polymerization nucleator Arp2/3 complex in the protrusions of fibroblasts. J. Cell Sci. 2005, 118, 2425–2433. [Google Scholar] [CrossRef] [PubMed]
- Oleynikov, Y.; Singer, R.H. RNA localization: Different zipcodes, same postman? Trends Cell Biol. 1998, 8, 381–383. [Google Scholar] [CrossRef]
- Su, X.; Ohi, R.; Pellman, D. Move in for the kill: Motile microtubule regulators. Trends Cell Biol. 2012, 22, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Moores, C.A.; Yu, M.; Guo, J.; Beraud, C.; Sakowicz, R.; Milligan, R.A. A Mechanism for Microtubule Depolymerization by KinI Kinesins. Mol. Cell 2002, 9, 903–909. [Google Scholar] [CrossRef]
- Liu, M.; Nadar, V.C.; Kozielski, F.; Kozlowska, M.; Yu, W.; Baas, P.W. Kinesin-12, a mitotic microtubule-associated motor protein, impacts axonal growth, navigation, and branching. J. Neurosci. 2010, 30, 14896–14906. [Google Scholar] [CrossRef]
- Myers, K.A.; Baas, P.W. Kinesin-5 regulates the growth of the axon by acting as a brake on its microtubule array. J. Cell Biol. 2007, 178, 1081–1091. [Google Scholar] [CrossRef]
- Jolly, A.L.; Kim, H.; Srinivasan, D.; Lakonishok, M.; Larson, A.G.; Gelfand, V.I. Kinesin-1 heavy chain mediates microtubule sliding to drive changes in cell shape. Proc. Natl. Acad. Sci. USA 2010, 107, 12151–12156. [Google Scholar] [CrossRef]
- Song, Y.; Brady, S.T. Posttranslational Modifications of Tubulin: Pathways to Functional Diversity of Microtubules. Trends Cell Biol. 2015, 25, 125–136. [Google Scholar] [CrossRef]
- Portran, D.; Schaedel, L.; Xu, Z.; Théry, M.; Nachury, M.V. Tubulin acetylation protects long-lived microtubules against mechanical ageing. Nat. Cell Biol. 2017, 19, 391–398. [Google Scholar] [CrossRef]
- Sudo, H.; Baas, P.W. Acetylation of microtubules influences their sensitivity to severing by katanin in neurons and fibroblasts. J. Neurosci. 2010, 30, 7215–7226. [Google Scholar] [CrossRef]
- Roll-Mecak, A. Intrinsically disordered tubulin tails: Complex tuners of microtubule functions? Semin. Cell Dev. Biol. 2015, 37, 11–19. [Google Scholar] [CrossRef]
- Konishi, Y.; Setou, M. Tubulin tyrosination navigates the kinesin-1 motor domain to axons. Nat. Neurosci. 2009, 12, 559–567. [Google Scholar] [CrossRef]
- Roll-Mecak, A.; Vale, R.D. Structural basis of microtubule severing by the hereditary spastic paraplegia protein spastin. Nature 2008, 451, 363–367. [Google Scholar] [CrossRef]
- Lacroix, B.; Van Dijk, J.; Gold, N.D.; Guizetti, J.; Aldrian-Herrada, G.; Rogowski, K.; Gerlich, D.W.; Janke, C. Tubulin polyglutamylation stimulates spastin-mediated microtubule severing. J. Cell Biol. 2010, 189, 945–954. [Google Scholar] [CrossRef]
- Valenstein, M.L.; Roll-Mecak, A. Graded Control of Microtubule Severing by Tubulin Glutamylation. Cell 2016, 164, 911–921. [Google Scholar] [CrossRef]
- Morris, E.J.; Nader, G.P.F.; Ramalingam, N.; Bartolini, F.; Gundersen, G.G. Kif4 interacts with EB1 and stabilizes microtubules downstream of Rho-mDia in migrating fibroblasts. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Wen, Y.; Eng, C.H.; Schmoranzer, J.; Cabrera-Poch, N.; Morris, E.J.S.; Chen, M.; Wallar, B.J.; Alberts, A.S.; Gundersen, G.G. EB1 and APC bind to mDia to EB1 and APC bind to mDia to stabilize stabilize microtubules downstream of Rho and promote cell migration. Nat. Cell Biol. 2004, 6, 820–830. [Google Scholar] [CrossRef]
- Ishizaki, T.; Morishima, Y.; Okamoto, M.; Furuyashiki, T.; Kato, T.; Narumiya, S. Coordination of microtubules and the actin cytoskeleton by the Rho effector mDia1. Nat. Cell Biol. 2001, 3, 8–14. [Google Scholar] [CrossRef]
- Fife, C.M.; Sagnella, S.M.; Teo, W.S.; Po’Uha, S.T.; Byrne, F.L.; Yeap, Y.Y.C.; Ng, D.C.H.; Davis, T.P.; McCarroll, J.A.; Kavallaris, M. Stathmin mediates neuroblastoma metastasis in a tubulin-independent manner via RhoA/ROCK signaling and enhanced transendothelial migration. Oncogene 2017, 36, 501–511. [Google Scholar] [CrossRef]
- Daub, H.; Gevaert, K.; Vandekerckhove, J.; Sobel, A.; Hall, A. Rac/Cdc42 and p65PAK regulate the microtubule-destabilizing protein stathmin through phosphorylation at serine 16. J. Biol. Chem. 2001, 276, 1677–1680. [Google Scholar] [CrossRef]
- Wittmann, T.; Bokoch, G.M.; Waterman-Storer, C.M. Regulation of microtubule destabilizing activity of Op18/stathmin downstream of Rac1. J. Biol. Chem. 2004, 279, 6196–6203. [Google Scholar] [CrossRef]
- Wittmann, T.; Bokoch, G.M.; Waterman-Storer, C.M. Regulation of leading edge microtubule and actin dynamics downstream of Rac1. J. Cell Biol. 2003, 161, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Fukata, M.; Watanabe, T.; Noritake, J.; Nakagawa, M.; Yamaga, M.; Kuroda, S.; Matsuura, Y.; Iwamatsu, A.; Perez, F.; Kaibuchi, K. Rac1 and Cdc42 capture microtubules through IQGAP1 and CLIP-170. Cell 2002, 109, 873–885. [Google Scholar] [CrossRef]
- Brown, R.A.; Talas, G.; Porter, R.A.; McGrouther, D.A.; Eastwood, M. Balanced mechanical forces and microtubule contribution to fibroblast contraction. J. Cell. Physiol. 1996, 169, 439–447. [Google Scholar] [CrossRef]
- Rudolph, R.; Woodward, M. Spatial orientation of microtubules in contractile fibroblasts in vivo. Anat. Rec. 1978, 191, 169–181. [Google Scholar] [CrossRef]
- Tomasek, J.J.; Hay, E.D. Analysis of the role of microfilaments and microtubules in acquisition of bipolarity and elongation of fibroblasts in hydrated collagen gels. J. Cell Biol. 1984, 99, 536–549. [Google Scholar] [CrossRef]
- Etienne-Manneville, S. Microtubules in Cell Migration. Annu. Rev. Cell Dev. Biol. 2013, 29, 471–499. [Google Scholar] [CrossRef]
- Hawkins, T.; Mirigian, M.; Selcuk Yasar, M.; Ross, J.L. Mechanics of microtubules. J. Biomech. 2010, 43, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Dogterom, M.; Yurke, B. Measurement of the force-velocity relation for growing microtubules. Science 1997, 278, 856–860. [Google Scholar] [CrossRef]
- Kolomeisky, A.B.; Fisher, M.E. Force-velocity relation for growing microtubules. Biophys. J. 2001, 80, 149–154. [Google Scholar] [CrossRef]
- Janson, M.E.; de Dood, M.E.; Dogterom, M. Dynamic instability of microtubules is regulated by force. J. Cell Biol. 2003, 161, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Robison, P.; Caporizzo, M.A.; Ahmadzadeh, H.; Bogush, A.I.; Chen, C.Y.; Margulies, K.B.; Shenoy, V.B.; Prosser, B.L. Detyrosinated microtubules buckle and bear load in contracting cardiomyocytes. Science 2016, 352, aaf0659. [Google Scholar] [CrossRef]
- Brangwynne, C.P.; MacKintosh, F.C.; Kumar, S.; Geisse, N.A.; Talbot, J.; Mahadevan, L.; Parker, K.K.; Ingber, D.E.; Weitz, D.A. Microtubules can bear enhanced compressive loads in living cells because of lateral reinforcement. J. Cell Biol. 2006, 173, 733–741. [Google Scholar] [CrossRef]
- Laan, L.; Husson, J.; Munteanu, E.L.; Kerssemakers, J.W.J.; Dogterom, M. Force-generation and dynamic instability of microtubule bundles. Proc. Natl. Acad. Sci. USA 2008, 105, 8920–8925. [Google Scholar] [CrossRef]
- Komarova, Y.A.; Vorobjev, I.A.; Borisy, G.G. Life cycle of MTs: Persistent growth in the cell interior, asymmetric transition frequencies and effects of the cell boundary. J. Cell Sci. 2002, 115, 3527–3539. [Google Scholar]
- Drummond, D.R.; Cross, R.A. Dynamics of interphase microtubules in Schizosaccharomyces pombe. Curr. Biol. 2000, 10, 766–775. [Google Scholar] [CrossRef]
- Bicek, A.D.; Tüzel, E.; Demtchouk, A.; Uppalapati, M.; Hancock, W.O.; Kroll, D.M.; Odde, D.J. Anterograde microtubule transport drives microtubule bending in LLC-PK1 epithelial cells. Mol. Biol. Cell 2009, 20, 2943–2953. [Google Scholar] [CrossRef]
- Desai, A.; Mitchison, T.J. Microtubule Polymerization Dynamics. Annu. Rev. Cell Dev. Biol. 1997, 13, 83–117. [Google Scholar] [CrossRef]
- Grishchuk, E.L.; Molodtsov, M.I.; Ataullakhanov, F.I.; McIntosh, J.R. Force production by disassembling microtubules. Nature 2005, 438, 384–388. [Google Scholar] [CrossRef]
- Petry, S. Mechanisms of Mitotic Spindle Assembly. Annu. Rev. Biochem. 2016, 85, 659–683. [Google Scholar] [CrossRef]
- Duro, E.; Marston, A.L. From equator to pole: Splitting chromosomes in mitosis and meiosis. Genes Dev. 2015, 29, 109–122. [Google Scholar] [CrossRef]
- Gadde, S.; Heald, R. Mechanisms and Molecules of the Mitotic Spindle. Curr. Biol. 2004, 14, R797–R805. [Google Scholar] [CrossRef]
- Okada, K.; Bartolini, F.; Deaconescu, A.M.; Moseley, J.B.; Dogic, Z.; Grigorieff, N.; Gundersen, G.G.; Goode, B.L. Adenomatous polyposis coli protein nucleates actin assembly and synergizes with the formin mDia1. J. Cell Biol. 2010, 189, 1087–1096. [Google Scholar] [CrossRef]
- Bartolini, F.; Moseley, J.B.; Schmoranzer, J.; Cassimeris, L.; Goode, B.L.; Gundersen, G.G. The formin mDia2 stabilizes microtubules independently of its actin nucleation activity. J. Cell Biol. 2008, 181, 523–536. [Google Scholar] [CrossRef]
- Wu, X.; Kodama, A.; Fuchs, E. ACF7 Regulates Cytoskeletal-Focal Adhesion Dynamics and Migration and Has ATPase Activity. Cell 2008, 135, 137–148. [Google Scholar] [CrossRef]
- Miller, A.L.; Wang, Y.; Mooseker, M.S.; Koleske, A.J. The Abl-related gene (Arg) requires its F-actin–microtubule cross-linking activity to regulate lamellipodial dynamics during fibroblast adhesion. J. Cell Biol. 2004, 165, 407–420. [Google Scholar] [CrossRef]
- Moll, R.; Divo, M.; Langbein, L. The human keratins: Biology and pathology. Histochem. Cell Biol. 2008, 129, 705–733. [Google Scholar] [CrossRef]
- Eliasson, C.; Sahlgren, C.; Berthold, C.H.; Stakeberg, J.; Celis, J.E.; Betsholtz, C.; Eriksson, J.E.; Pekny, M. Intermediate filament protein partnership in astrocytes. J. Biol. Chem. 1999, 274, 23996–24006. [Google Scholar] [CrossRef]
- Bellin, R.M.; Sernett, S.W.; Becker, B.; Ip, W.; Huiatt, T.W.; Robson, R.M. Molecular characteristics and interactions of the intermediate filament protein synemin. Interactions with alpha-actinin may anchor synemin-containing heterofilaments. J. Biol. Chem. 1999, 274, 29493–29499. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Aebi, U. Intermediate Filaments: Molecular Structure, Assembly Mechanism, and Integration Into Functionally Distinct Intracellular Scaffolds. Annu. Rev. Biochem. 2004, 73, 749–789. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, E. Intermediate Filaments: Structure, Dynamics, Function, and Disease. Annu. Rev. Biochem. 1994, 63, 345–382. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.A.; Strelkov, S.V.; Burkhard, P.; Aebi, U.; Parry, D.A.D. Sequence comparisons of intermediate filament chains: Evidence of a unique functional/structural role for coiled-coil segment 1A and linker L1. J. Struct. Biol. 2002, 137, 128–145. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, A.V.; Kreplak, L.; Wedig, T.; Mucke, N.; Svergun, D.I.; Herrmann, H.; Aebi, U.; Strelkov, S.V. Monitoring intermediate filament assembly by small-angle x-ray scattering reveals the molecular architecture of assembly intermediates. Proc. Natl. Acad. Sci. USA 2006, 103, 16206–16211. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Häner, M.; Brettel, M.; Ku, N.O.; Aebi, U. Characterization of distinct early assembly units of different intermediate filament proteins. J. Mol. Biol. 1999, 286, 1403–1420. [Google Scholar] [CrossRef]
- Mücke, N.; Wedig, T.; Bürer, A.; Marekov, L.N.; Steinert, P.M.; Langowski, J.; Aebi, U.; Herrmann, H. Molecular and biophysical characterization of assembly-starter units of human vimentin. J. Mol. Biol. 2004, 340, 97–114. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulou, S.; Möller, D.; Sachs, N.; Herrmann, H.; Aebi, U. Near-UV Circular Dichroism Reveals Structural Transitions of Vimentin Subunits during Intermediate Filament Assembly. J. Mol. Biol. 2009, 386, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Steinert, P.M.; Marekov, L.N.; Parry, D.A. Diversity of intermediate filament structure. J. Biol. Chem. 1993, 268, 24916–24925. [Google Scholar] [PubMed]
- Herrmann, H.; Kreplak, L.; Aebi, U. Isolation, Characterization, and In Vitro Assembly of Intermediate Filaments. Methods Cell Biol. 2004, 78, 3–24. [Google Scholar] [PubMed]
- Nöding, B.; Herrmann, H.; Köster, S. Direct Observation of Subunit Exchange along Mature Vimentin Intermediate Filaments. Biophys. J. 2014, 107, 2923–2931. [Google Scholar] [CrossRef]
- Chang, L.; Goldman, R.D. Intermediate filaments mediate cytoskeletal crosstalk. Mol. Cell Biol. 2004, 5, 601–613. [Google Scholar] [CrossRef]
- Kayser, J.; Haslbeck, M.; Dempfle, L.; Krause, M.; Grashoff, C.; Buchner, J.; Herrmann, H.; Bausch, A.R. The Small Heat Shock Protein Hsp27 Affects Assembly Dynamics and Structure of Keratin Intermediate Filament Networks. Biophys. J. 2013, 105, 1778–1785. [Google Scholar] [CrossRef] [PubMed]
- Heitlinger, E.; Peter, M.; Lustig, A.; Villiger, W.; Nigg, E.A.; Aebi, U. The role of the head and tail domain in lamin structure and assembly: Analysis of bacterially expressed chicken Lamin A and truncated B2 lamins. J. Struct. Biol. 1992, 108, 74–91. [Google Scholar] [CrossRef]
- Heitlinger, E.; Peter, M.; Häner, M.; Lustig, A.; Aebi, U.; Nigg, E.A. Expression of chicken lamin B2 in Escherichia coli: Characterization of its structure, assembly, and molecular interactions. J. Cell Biol. 1991, 113, 485–495. [Google Scholar] [CrossRef]
- Aebi, U.; Cohn, J.; Buhle, L.; Gerace, L. The nuclear lamina is a meshwork of intermediate-type filaments. Nature 1986, 323, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Kreis, S.; Schönfeld, H.J.; Melchior, C.; Steiner, B.; Kieffer, N. The intermediate filament protein vimentin binds specifically to a recombinant integrin α2/β1 cytoplasmic tail complex and co-localizes with native α2/β1 in endothelial cell focal adhesions. Exp. Cell Res. 2005, 305, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Tsuruta, D. The vimentin cytoskeleton regulates focal contact size and adhesion of endothelial cells subjected to shear stress. J. Cell Sci. 2003, 116, 4977–4984. [Google Scholar] [CrossRef]
- Seltmann, K.; Cheng, F.; Wiche, G.; Eriksson, J.E.; Magin, T.M. Keratins stabilize hemidesmosomes through regulation of β4-integrin turnover. J. Invest. Dermatol. 2015, 135, 1609–1620. [Google Scholar] [CrossRef]
- Loschke, F.; Homberg, M.; Magin, T.M. Keratin isotypes control desmosome stability and dynamics through PKCα. J. Invest. Dermatol. 2016, 136, 202–213. [Google Scholar] [CrossRef]
- Etienne-Manneville, S. Cytoplasmic Intermediate Filaments in Cell Biology. Annu. Rev. Cell Dev. Biol. 2018, 34, 1–28. [Google Scholar] [CrossRef]
- Dupin, I.; Sakamoto, Y.; Etienne-Manneville, S. Cytoplasmic intermediate filaments mediate actin-driven positioning of the nucleus. J. Cell Sci. 2011, 124, 865–872. [Google Scholar] [CrossRef]
- Matveeva, E.A.; Venkova, L.S.; Chernoivanenko, I.S.; Minin, A.A. Vimentin is involved in regulation of mitochondrial motility and membrane potential by Rac1. Biol. Open 2015, 4, 1290–1297. [Google Scholar] [CrossRef]
- Nekrasova, O.E.; Mendez, M.G.; Chernoivanenko, I.S.; Tyurin-Kuzmin, P.A.; Kuczmarski, E.R.; Gelfand, V.I.; Goldman, R.D.; Minin, A.A. Vimentin intermediate filaments modulate the motility of mitochondria. Mol. Biol. Cell 2011, 22, 2282–2289. [Google Scholar] [CrossRef]
- Wang, N.; Stamenović, D. Contribution of intermediate filaments to cell stiffness, stiffening, and growth. Am. J. Physiol. Cell Physiol. 2000, 279, C188–C194. [Google Scholar] [CrossRef]
- Godsel, L.M.; Hobbs, R.P.; Green, K.J. Intermediate filament assembly: Dynamics to disease. Trends Cell Biol. 2008, 18, 28–37. [Google Scholar] [CrossRef]
- Sanghvi-Shah, R.; Weber, G.F. Intermediate Filaments at the Junction of Mechanotransduction, Migration, and Development. Front. Cell Dev. Biol. 2017, 5, 1–19. [Google Scholar] [CrossRef]
- Storm, C.; Pastore, J.J.; Mackintosh, F.C.; Lubensky, T.C.; Janmey, P.A. Nonlinear elasticity in biological gels. Nature 2005, 435, 191–194. [Google Scholar] [CrossRef]
- Guo, M.; Ehrlicher, A.J.; Mahammad, S.; Fabich, H.; Jensen, M.H.; Moore, J.R.; Fredberg, J.J.; Goldman, R.D.; Weitz, D.A. The Role of Vimentin Intermediate Filaments in Cortical and Cytoplasmic Mechanics. Biophys. J. 2013, 105, 1562–1568. [Google Scholar] [CrossRef]
- Mendez, M.G.; Restle, D.; Janmey, P.A. Vimentin enhances cell elastic behavior and protects against compressive stress. Biophys. J. 2014, 107, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-Y.; Lin, H.-H.; Tang, M.-J.; Wang, Y.-K. Vimentin contributes to epithelial-mesenchymal transition cancer cell mechanics by mediating cytoskeletal organization and focal adhesion maturation. Oncotarget 2015, 6, 15966–15983. [Google Scholar] [CrossRef] [PubMed]
- Alberti, C. Cytoskeleton structure and dynamic behaviour: Quick excursus from basic molecular mechanisms to some implications in cancer chemotherapy. Eur. Rev. Med. Pharmacol. Sci. 2009, 13, 13–21. [Google Scholar] [PubMed]
- Izawa, I.; Inagaki, M. Regulatory mechanisms and functions of intermediate filaments: A study using site- and phosphorylation state-specific antibodies. Cancer Sci. 2006, 97, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Hyder, C.L.; Pallari, H.-M.; Kochin, V.; Eriksson, J.E. Providing cellular signposts - Post-translational modifications of intermediate filaments. FEBS Lett. 2008, 582, 2140–2148. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Hobbs, R.P.; Coulombe, P.A. The expanding significance of keratin intermediate filaments in normal and diseased epithelia. Curr. Opin. Cell Biol. 2013, 25, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Rogel, M.R.; Jaitovich, A.; Ridge, K.M. The Role of the Ubiquitin Proteasome Pathway in Keratin Intermediate Filament Protein Degradation. Proc. Am. Thorac. Soc. 2010, 7, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Omary, M.B.; Coulombe, P.A.; McLean, W.H.I. Intermediate Filament Proteins and Their Associated Diseases. N. Engl. J. Med. 2004, 351, 2087–2100. [Google Scholar] [CrossRef]
- Leung, C.L.; Liem, R.K.H.; Parry, D.A.D.; Green, K.J. The plakin family. J. Cell Sci. 2001, 114, 3409–3410. [Google Scholar] [PubMed]
- Leung, C.L.; Green, K.J.; Liem, R.K.H. Plakins: A family of versatile cytolinker proteins. Trends Cell Biol. 2002, 12, 37–45. [Google Scholar] [CrossRef]
- Desai, B.V.; Harmon, R.M.; Green, K.J. Desmosomes at a glance. J. Cell Sci. 2009, 122, 4401–4407. [Google Scholar] [CrossRef]
- Wiche, G.; Osmanagic-Myers, S.; Castañón, M.J. Networking and anchoring through plectin: A key to IF functionality and mechanotransduction. Curr. Opin. Cell Biol. 2015, 32, 21–29. [Google Scholar] [CrossRef]
- Kölsch, A.; Windoffer, R.; Leube, R.E. Actin-dependent dynamics of keratin filament precursors. Cell Motil. Cytoskeleton 2009, 66, 976–985. [Google Scholar] [CrossRef]
- Robert, A.; Herrmann, H.; Davidson, M.W.; Gelfand, V.I. Microtubule-dependent transport of vimentin filament precursors is regulated by actin and by the concerted action of Rho- and p21-activated kinases. FASEB J. 2014, 28, 2879–2890. [Google Scholar] [CrossRef]
- Burgstaller, G.; Gregor, M.; Winter, L.; Wiche, G. Keeping the vimentin network under control: Cell-matrix adhesion-associated plectin 1f affects cell shape and polarity of fibroblasts. Mol. Biol. Cell 2010, 21, 3362–3375. [Google Scholar] [CrossRef]
- Lynch, C.D.; Lazar, A.M.; Iskratsch, T.; Zhang, X.; Sheetz, M.P. Endoplasmic spreading requires coalescence of vimentin intermediate filaments at force-bearing adhesions. Mol. Biol. Cell 2013, 24, 21–30. [Google Scholar] [CrossRef]
- Gregor, M.; Osmanagic-Myers, S.; Burgstaller, G.; Wolfram, M.; Fischer, I.; Walko, G.; Resch, G.P.; Jörgl, A.; Herrmann, H.; Wiche, G. Mechanosensing through focal adhesion-anchored intermediate filaments. FASEB J. 2014, 28, 715–729. [Google Scholar] [CrossRef]
- Esue, O.; Carson, A.A.; Tseng, Y.; Wirtz, D. A direct interaction between actin and vimentin filaments mediated by the tail domain of vimentin. J. Biol. Chem. 2006, 281, 30393–30399. [Google Scholar] [CrossRef]
- Mellad, J.A.; Warren, D.T.; Shanahan, C.M. Nesprins LINC the nucleus and cytoskeleton. Curr. Opin. Cell Biol. 2011, 23, 47–54. [Google Scholar] [CrossRef]
- Wilhelmsen, K.; Litjens, S.H.M.; Kuikman, I.; Tshimbalanga, N.; Janssen, H.; van den Bout, I.; Raymond, K.; Sonnenberg, A. Nesprin-3, a novel outer nuclear membrane protein, associates with the cytoskeletal linker protein plectin. J. Cell Biol. 2005, 171, 799–810. [Google Scholar] [CrossRef]
- Lombardi, M.L.; Jaalouk, D.E.; Shanahan, C.M.; Burke, B.; Roux, K.J.; Lammerding, J. The Interaction between Nesprins and Sun Proteins at the Nuclear Envelope Is Critical for Force Transmission between the Nucleus and Cytoskeleton. J. Biol. Chem. 2011, 286, 26743–26753. [Google Scholar] [CrossRef]
- Morgan, J.T.; Pfeiffer, E.R.; Thirkill, T.L.; Kumar, P.; Peng, G.; Fridolfsson, H.N.; Douglas, G.C.; Starr, D.A.; Barakat, A.I. Nesprin-3 regulates endothelial cell morphology, perinuclear cytoskeletal architecture, and flow-induced polarization. Mol. Biol. Cell 2011, 22, 4324–4334. [Google Scholar] [CrossRef]
- Keeling, M.C.; Flores, L.R.; Dodhy, A.H.; Murray, E.R.; Gavara, N. Actomyosin and vimentin cytoskeletal networks regulate nuclear shape, mechanics and chromatin organization. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Toivola, D.M.; Tao, G.Z.; Habtezion, A.; Liao, J.; Omary, M.B. Cellular integrity plus: Organelle-related and protein-targeting functions of intermediate filaments. Trends Cell Biol. 2005, 15, 608–617. [Google Scholar] [CrossRef]
- Naetar, N.; Ferraioli, S.; Foisner, R. Lamins in the nuclear interior—Life outside the lamina. J. Cell Sci. 2017, 130, 2087–2096. [Google Scholar] [CrossRef]
- Turgay, Y.; Eibauer, M.; Goldman, A.E.; Shimi, T.; Khayat, M.; Ben-Harush, K.; Dubrovsky-Gaupp, A.; Sapra, K.T.; Goldman, R.D.; Medalia, O. The molecular architecture of lamins in somatic cells. Nature 2017, 543, 261–264. [Google Scholar] [CrossRef]
- Buxboim, A.; Swift, J.; Irianto, J.; Spinler, K.R.; Dingal, P.C.D.P.; Athirasala, A.; Kao, Y.C.; Cho, S.; Harada, T.; Shin, J.-W.; et al. Matrix elasticity regulates lamin-A,C phosphorylation and turnover with feedback to actomyosin. Curr. Biol. 2015, 24, 1909–1917. [Google Scholar] [CrossRef]
- Davidson, P.M.; Denais, C.; Bakshi, M.C.; Lammerding, J. Nuclear deformability constitutes a rate-limiting step during cell migration in 3-D environments. Cell. Mol. Bioeng. 2014, 7, 293–306. [Google Scholar] [CrossRef]
- Harada, T.; Swift, J.; Irianto, J.; Shin, J.W.; Spinler, K.R.; Athirasala, A.; Diegmiller, R.; Dingal, P.C.D.P.; Ivanovska, I.L.; Discher, D.E. Nuclear lamin stiffness is a barrier to 3D migration, but softness can limit survival. J. Cell Biol. 2014, 204, 669–682. [Google Scholar] [CrossRef]
- Cho, S.; Irianto, J.; Discher, D.E. Mechanosensing by the nucleus: From pathways to scaling relationships. J. Cell Biol. 2017, 1–11. [Google Scholar] [CrossRef]
- Osmanagic-Myers, S.; Dechat, T.; Foisner, R. Lamins at the crossroads of mechanosignaling. Genes Dev. 2015, 29, 225–237. [Google Scholar] [CrossRef]
- Stephens, A.D.; Banigan, E.J.; Adam, S.A.; Goldman, R.D.; Marko, J.F. Chromatin and lamin A determine two different mechanical response regimes of the cell nucleus. Mol. Biol. Cell 2017, 28, 1984–1996. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Manzanares, M.; Webb, D.J.; Horwitz, A.R. Cell migration at a glance. J. Cell Sci. 2005, 118, 4917–4919. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Wolf, K. Plasticity of cell migration: A multiscale tuning model. J. Cell Biol. 2010, 188, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Parri, M.; Chiarugi, P. Rac and Rho GTPases in cancer cell motility control. Cell Commun. Signal. 2010, 8, 23. [Google Scholar] [CrossRef]
- Nguyen, L.K.; Kholodenko, B.N.; von Kriegsheim, A. Rac1 and RhoA: Networks, loops and bistability. Small GTPases 2016, 0, 1–6. [Google Scholar] [CrossRef]
- Brunner, C.A.; Ehrlicher, A.; Kohlstrunk, B.; Knebel, D.; Käs, J.A.; Goegler, M. Cell migration through small gaps. Eur. Biophys. J. 2006, 35, 713–719. [Google Scholar] [CrossRef]
- Heinemann, F.; Doschke, H.; Radmacher, M. Keratocyte lamellipodial protrusion is characterized by a concave force-velocity relation. Biophys. J. 2011, 100, 1420–1427. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D.; Borisy, G.G. Cellular motility driven by assembly and disassembly of actin filaments. Cell 2003, 112, 453–465. [Google Scholar] [CrossRef]
- Kage, F.; Winterhoff, M.; Dimchev, V.; Mueller, J.; Thalheim, T.; Freise, A.; Brühmann, S.; Kollasser, J.; Block, J.; Dimchev, G.; et al. FMNL formins boost lamellipodial force generation. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, R.; Gautreau, A. The Arp2/3 inhibitory protein arpin induces cell turning by pausing cell migration. Cytoskeleton 2015, 72, 362–371. [Google Scholar] [CrossRef]
- Choi, C.K.; Vicente-Manzanaras, M.; Zareno, J.; Whitmore, L.A.; Mogilner, A.; Horwitz, A.R. Actin and α-actinin orchestrate the assembly and maturation of nascent adhesions in a myosin II motor-independent manner. Nat. Cell Biol. 2008, 9, 1039–1050. [Google Scholar] [CrossRef]
- Ridley, A.J. Life at the Leading Edge. Cell 2011, 145, 1012–1022. [Google Scholar] [CrossRef]
- Pasapera, A.M.; Plotnikov, S.V.; Fischer, R.S.; Case, L.B.; Egelhoff, T.T.; Waterman, C.M. Rac1-Dependent Phosphorylation and Focal Adhesion Recruitment of Myosin IIA Regulates Migration and Mechanosensing. Curr. Biol. 2015, 25, 175–186. [Google Scholar] [CrossRef]
- Kuo, J.-C.; Han, X.; Hsiao, C.-T.; Yates III, J.R.; Waterman, C.M. Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for β-Pix in negative regulation of focal adhesion maturation. Nat. Cell Biol. 2011, 13, 383–393. [Google Scholar] [CrossRef]
- Schiller, H.B.; Friedel, C.C.; Boulegue, C.; Fässler, R. Quantitative proteomics of the integrin adhesome show a myosin II-dependent recruitment of LIM domain proteins. EMBO Rep. 2011, 12, 259–266. [Google Scholar] [CrossRef]
- Peng, J.; Wallar, B.J.; Flanders, A.; Swiatek, P.J.; Alberts, A.S. Disruption of the Diaphanous-related formin Drf1 gene encoding mDia1 reveals a role for Drf3 as an effector for Cdc42. Curr. Biol. 2003, 13, 534–545. [Google Scholar] [CrossRef]
- Iskratsch, T.; Yu, C.H.; Mathur, A.; Liu, S.; Stévenin, V.; Dwyer, J.; Hone, J.; Ehler, E.; Sheetz, M. FHOD1 is needed for directed forces and adhesion maturation during cell spreading and migration. Dev. Cell 2013, 27, 545–559. [Google Scholar] [CrossRef]
- DeMali, K.A.; Barlow, C.A.; Burridge, K. Recruitment of the Arp2/3 complex to vinculin: Coupling membrane protrusion to matrix adhesion. J. Cell Biol. 2002, 159, 881–891. [Google Scholar] [CrossRef]
- Serrels, B.; Serrels, A.; Brunton, V.G.; Holt, M.; McLean, G.W.; Gray, C.H.; Jones, G.E.; Frame, M.C. Focal adhesion kinase controls actin assembly via a FERM-mediated interaction with the Arp2/3 complex. Nat. Cell Biol. 2007, 9, 1046–1056. [Google Scholar] [CrossRef]
- Suraneni, P.; Rubinstein, B.; Unruh, J.R.; Durnin, M.; Hanein, D.; Li, R. The Arp2/3 complex is required for lamellipodia extension and directional fibroblast cell migration. J. Cell Biol. 2012, 197, 239–251. [Google Scholar] [CrossRef]
- Wu, C.; Asokan, S.B.; Berginski, M.E.; Haynes, E.M.; Sharpless, N.E.; Griffith, J.D.; Gomez, S.M.; Bear, J.E. Arp2/3 Is Critical for Lamellipodia and Response to Extracellular Matrix Cues but Is Dispensable for Chemotaxis. Cell 2012, 148, 973–987. [Google Scholar] [CrossRef]
- Li, A.; Ma, Y.; Yu, X.; Mort, R.L.; Lindsay, C.R.; Stevenson, D.; Strathdee, D.; Insall, R.H.; Chernoff, J.; Snapper, S.B.; et al. Rac1 Drives Melanoblast Organization during Mouse Development by Orchestrating Pseudopod- Driven Motility and Cell-Cycle Progression. Dev. Cell 2011, 21, 722–734. [Google Scholar] [CrossRef]
- O’Neill, P.R.; Kalyanaraman, V.; Gautman, N. Subcellular optogenetic activation of Cdc42 controls local and distal signaling to drive immune cell migration. Mol. Biol. Cell 2016, 27, 1442–1450. [Google Scholar] [CrossRef]
- Yang, H.W.; Collins, S.R.; Meyer, T. Locally excitable Cdc42 signals steer cells during chemotaxis. Nat. Cell Biol. 2016, 18, 191–201. [Google Scholar] [CrossRef]
- Harris, K.P.; Tepass, U. Cdc42 and Vesicle Trafficking in Polarized Cells. Traffic 2010, 11, 1272–1279. [Google Scholar] [CrossRef]
- Vega, F.M.; Fruhwirth, G.; Ng, T.; Ridley, A.J. RhoA and RhoC have distinct roles in migration and invasion by acting through different targets. J. Cell Biol. 2011, 193, 655–665. [Google Scholar] [CrossRef]
- Petrie, R.J.; Yamada, K.M. At the leading edge of three-dimensional cell migration. J. Cell Sci. 2012, 125, 5917–5926. [Google Scholar] [CrossRef]
- Paluch, E.K.; Aspalter, I.M.; Sixt, M. Focal Adhesion–Independent Cell Migration. Annu. Rev. Cell Dev. Biol. 2016, 32, 469–490. [Google Scholar] [CrossRef]
- Maugis, B.; Brugues, J.; Nassoy, P.; Guillen, N.; Sens, P.; Amblard, F. Dynamic instability of the intracellular pressure drives bleb-based motility. J. Cell Sci. 2010, 123, 3884–3892. [Google Scholar] [CrossRef]
- Bergert, M.; Chandradoss, S.D.; Desai, R.A.; Paluch, E. Cell mechanics control rapid transitions between blebs and lamellipodia during migration. Proc. Natl. Acad. Sci. USA 2012, 109, 14434–14439. [Google Scholar] [CrossRef]
- Weiser, D.C.; Row, R.H.; Kimelman, D. Rho-regulated Myosin phosphatase establishes the level of protrusive activity required for cell movements during zebrafish gastrulation. Development 2009, 136, 2375–2384. [Google Scholar] [CrossRef] [PubMed]
- Sroka, J.; von Gunten, M.; Dunn, G.A.; Keller, H.U. Phenotype modulation in non-adherent and adherent sublines of Walker carcinosarcoma cells: The role of cell-substratum contacts and microtubules in controlling cell shape, locomotion and cytoskeletal structure. Int. J. Biochem. Cell Biol. 2002, 34, 882–899. [Google Scholar] [CrossRef]
- Blaser, H.; Eisenbeiss, S.; Neumann, M.; Reichman-Fried, M.; Thisse, B.; Thisse, C.; Raz, E. Transition from non-motile behaviour to directed migration during early PGC development in zebrafish. J. Cell Sci. 2005, 118, 4027–4038. [Google Scholar] [CrossRef]
- Malawista, S.E.; de Boisfleury Chevance, A.; Boxer, L.A. Random locomotion and chemotaxis of human blood polymorphonuclear leukocytes from a patient with Leukocyte Adhesion Deficiency-1: Normal displacement in close quarters via chimneying. Cell Motil. Cytoskeleton 2000, 46, 183–189. [Google Scholar] [CrossRef]
- Tozluoǧlu, M.; Tournier, A.L.; Jenkins, R.P.; Hooper, S.; Bates, P.A.; Sahai, E. Matrix geometry determines optimal cancer cell migration strategy and modulates response to interventions. Nat. Cell Biol. 2013, 15, 751–762. [Google Scholar] [CrossRef]
- Cattin, A.L.; Burden, J.J.; Van Emmenis, L.; MacKenzie, F.E.; Hoving, J.J.A.; Garcia Calavia, N.; Guo, Y.; McLaughlin, M.; Rosenberg, L.H.; Quereda, V.; et al. Macrophage-Induced Blood Vessels Guide Schwann Cell-Mediated Regeneration of Peripheral Nerves. Cell 2015, 162, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, S.; Chen, E.J.H.; Clarke, F.; Lyck, R.; Affentranger, S.; Burkhardt, J.K.; Niggli, V. Ezrin/Radixin/Moesin Proteins and Flotillins Cooperate to Promote Uropod Formation in T Cells. Front. Immunol. 2013, 4, 84. [Google Scholar] [CrossRef]
- Niggli, V.; Rossy, J. Ezrin/radixin/moesin: Versatile controllers of signaling molecules and of the cortical cytoskeleton. Int. J. Biochem. Cell Biol. 2008, 40, 344–349. [Google Scholar] [CrossRef]
- Lorentzen, A.; Bamber, J.; Sadok, A.; Elson-Schwab, I.; Marshall, C.J. An ezrin-rich, rigid uropod-like structure directs movement of amoeboid blebbing cells. J. Cell Sci. 2011, 124, 1256–1267. [Google Scholar] [CrossRef] [PubMed]
- Yanase, Y.; Hide, I.; Mihara, S.; Shirai, Y.; Saito, N.; Nakata, Y.; Hide, M.; Sakai, N. A critical role of conventional protein kinase C in morphological changes of rodent mast cells. Immunol. Cell Biol. 2011, 89, 149–159. [Google Scholar] [CrossRef]
- Fehon, R.G.; McClatchey, A.I.; Bretscher, A. Organizing the cell cortex: The role of ERM proteins. Nat. Rev. Mol. Cell Biol. 2010, 11, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Evans, E.A.; Skalak, R. Mechanics and Thermodynamics of Biomembranes: Part 1. CRC Crit. Rev. Bioeng. 1979, 3, 181–330. [Google Scholar] [PubMed]
- Pinner, S.; Sahai, E. PDK1 regulates cancer cell motility by antagonising inhibition of ROCK1 by RhoE. Nat. Cell Biol. 2008, 10, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Trinkaus, J.P. Ingression during Early Gastrulation of Fundulus. Dev. Biol. 1996, 177, 356–370. [Google Scholar] [CrossRef] [PubMed]
- Oakes, P.W.; Beckham, Y.; Stricker, J.; Gardel, M.L. Tension is required but not sufficient for focal adhesion maturation without a stress fiber template. J. Cell Biol. 2012, 196, 363–374. [Google Scholar] [CrossRef]
- Puklin-Faucher, E.; Gao, M.; Schulten, K.; Vogel, V. How the headpiece hinge angle is opened: New insights into the dynamics of integrin activation. J. Cell Biol. 2006, 175, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.R.; Vogel, K.-P.; Steinhoff, H.-J.; Zoegler, W.H.; Patel, B.; Emsley, J.; Critchley, D.R.; Roberts, G.C.K.; Barsukov, I.L. Structural and Dynamic Characterization of a Vinculin Binding Site in the Talin Rod. Biochemistry 2006. [Google Scholar] [CrossRef]
- del Rio, A.; Perez-Jimenez, R.; Liu, R.; Roca-Cusachs, P.; Fernandez, J.M.; Sheetz, M.P. Stretching Single Talin Rod Molecules Activates Vinculin Binding. Science 2009, 323, 638–641. [Google Scholar] [CrossRef]
- Johnson, C.P.; Tang, H.-Y.; Carag, C.; Speicher, D.W.; Discher, D.E. Forced unfolding of proteins within cells. Science 2007, 317, 663–666. [Google Scholar] [CrossRef]
- Vallenius, T. Actin stress fibre subtypes in mesenchymal-migrating cells. Open Biol. 2013, 3, 130001. [Google Scholar] [CrossRef]
- Valerius, N.H.; Stendahl, O.; Hartwig, J.H.; Stossel, T.P. Distribution of actin-binding protein and myosin in polymorphonuclear leukocytes during locomotion and phagocytosis. Cell 1981, 24, 195–202. [Google Scholar] [CrossRef]
- Rubino, S.; Fighetti, M.; Unger, E.; Cappuccinelli, P. Location of actin, myosin, and microtubular structures during directed locomotion of Dictyostelium amebae. J. Cell Biol. 1984, 98, 382–390. [Google Scholar] [CrossRef]
- Cheng, S.; Castillo, V.; Welty, M.; Eliaz, I.; Sliva, D. Honokiol inhibits migration of renal cell carcinoma through activation of RhoA/ROCK/MLC signaling pathway. Int. J. Oncol. 2016, 49, 1525–1530. [Google Scholar] [CrossRef]
- Castella, L.F.; Buscemi, L.; Godbout, C.; Meister, J.-J.; Hinz, B. A new lock-step mechanism of matrix remodelling based on subcellular contractile events. J. Cell Sci. 2010, 123, 1751–1760. [Google Scholar] [CrossRef]
- Kirfel, G.; Rigort, A.; Borm, B.; Herzog, V. Cell migration: Mechanisms of rear detachment and the formation of migration tracks. Eur. J. Cell Biol. 2004, 83, 717–724. [Google Scholar] [CrossRef]
- Vicente-Manzanares, M.; Koach, M.A.; Whitmore, L.; Lamers, M.L.; Horwitz, A.F. Segregation and activation of myosin IIB creates a rear in migrating cells. J. Cell Biol. 2008, 183, 543–554. [Google Scholar] [CrossRef]
- Cuddihy, A.R.; Bristow, R.G. The p53 protein family and radiation sensitivity: Yes or no? Cancer Metastasis Rev. 2004, 23, 237–257. [Google Scholar] [CrossRef]
- Totsukawa, G.; Wu, Y.; Sasaki, Y.; Hartshorne, D.J.; Yamakita, Y.; Yamashiro, S.; Matsumura, F. Distinct roles of MLCK and ROCK in the regulation of membrane protrusions and focal adhesion dynamics during cell migration of fibroblasts. J. Cell Biol. 2004, 164, 427–439. [Google Scholar] [CrossRef]
- Aifuwa, I.; Giri, A.; Longe, N.; Lee, S.H.; An, S.S.; Wirtz, D. Senescent stromal cells induce cancer cell migration via inhibition of RhoA/ROCK/myosin-based cell contractility. Oncotarget 2015, 6, 30516–30531. [Google Scholar] [CrossRef]
- Schmidt, C.E.; Horwitz, A.F.; Lauffenburger, D.A.; Sheetz, M.P. Integrin-cytoskeletal interactions in migrating fibroblasts are dynamic, asymmetric, and regulated. J. Cell Biol. 1993, 123, 977–991. [Google Scholar] [CrossRef] [PubMed]
- Brangwynne, C.P.; MacKintosh, F.C.; Weitz, D.A. Force fluctuations and polymerization dynamics of intracellular microtubules. Proc. Natl. Acad. Sci. USA 2007, 104, 16128–16133. [Google Scholar] [CrossRef] [PubMed]
- Wadsworth, P. Regional regulation of microtubule dynamics in polarized, motile cells. Cell Motil. Cytoskeleton 1999, 42, 48–59. [Google Scholar] [CrossRef]
- Schober, J.M.; Cain, J.M.; Komarova, Y.A.; Borisy, G.G. Migration and actin protrusion in melanoma cells are regulated by EB1 protein. Cancer Lett. 2009, 284, 30–36. [Google Scholar] [CrossRef]
- Al-Bassam, J.; Kim, H.; Brouhard, G.; van Oijen, A.; Harrison, S.C.; Chang, F. CLASP Promotes Microtubule Rescue by Recruiting Tubulin Dimers to the Microtubule. Dev. Cell 2010, 19, 245–258. [Google Scholar] [CrossRef]
- Steinmetz, M.O.; Jahnke, W.; Towbin, H.; Garcia-Echeverria, C.; Voshol, H.; Muller, D.; van Oostrum, J. Phosphorylation disrupts the central helix in Op18/stathmin and suppresses binding to tubulin. EMBO Rep. 2001, 2, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; McEwen, D.P.; Martens, J.R.; Meyhofer, E.; Verhey, K.J. Single Molecule Imaging Reveals Differences in Microtubule Track Selection Between Kinesin Motors. PLoS Biol. 2009, 7, e1000216. [Google Scholar] [CrossRef] [PubMed]
- Fygenson, D.K.; Marko, J.F.; Libchaber, A. Mechanics of microtubule-based membrane extension. Phys. Rev. Lett. 1997, 79, 4497–4500. [Google Scholar] [CrossRef]
- Mogilner, A.; Oster, G. Polymer motors: Pushing out the front and pulling up the back. Curr. Biol. 2003, 13, 721–733. [Google Scholar] [CrossRef]
- Yu, W.; Centonze, V.E.; Ahmad, F.J.; Baas, P.W. Microtubule nucleation and release from the neuronal centrosome. J. Cell Biol. 1993, 122, 349–359. [Google Scholar] [CrossRef]
- Roll-Mecak, A.; McNally, F.J. Microtubule-severing enzymes. Curr. Opin. Cell Biol. 2010, 22, 96–103. [Google Scholar] [CrossRef]
- Marcette, J.D.; Chen, J.J.; Nonet, M.L. The Caenorhabditis elegans microtubule minus-end binding homolog PTRN-1 stabilizes synapses and neurites. Elife 2014, 3, e01637. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.E.; Spilker, K.A.; Cueva, J.G.; Perrino, J.; Goodman, M.B.; Shen, K. PTRN-1, a microtubule minus end-binding CAMSAP homolog, promotes microtubule function in Caenorhabditis elegans neurons. Elife 2014, 3, e01498. [Google Scholar] [CrossRef] [PubMed]
- Yau, K.W.; Schatzle, P.; Tortosa, E.; Pages, S.; Holtmaat, A.; Kapitein, L.C.; Hoogenraad, C.C. Dendrites In Vitro and In Vivo Contain Microtubules of Opposite Polarity and Axon Formation Correlates with Uniform Plus-End-Out Microtubule Orientation. J. Neurosci. 2016, 36, 1071–1085. [Google Scholar] [CrossRef]
- Baas, P.W.; Deitch, J.S.; Black, M.M.; Banker, G.A. Polarity orientation of microtubules in hippocampal neurons: Uniformity in the axon and nonuniformity in the dendrite. Proc. Natl. Acad. Sci. USA 1988, 85, 8335–8339. [Google Scholar] [CrossRef]
- Bradke, F.; Dotti, C.G. The role of local actin instability in axon formation. Science 1999, 283, 1931–1934. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, C.; Faix, J.; Himmel, M.; Bentzien, F.; Linder, S. KIF5B and KIF3A/KIF3B kinesins drive MT1-MMP surface exposure, CD44 shedding, and extracellular matrix degradation in primary macrophages. Blood 2010, 116, 1559–1569. [Google Scholar] [CrossRef]
- Steffen, A.; Le Dez, G.; Poincloux, R.; Recchi, C.; Nassoy, P.; Rottner, K.; Galli, T.; Chavrier, P. MT1-MMP-Dependent Invasion Is Regulated by TI-VAMP/VAMP7. Curr. Biol. 2008, 18, 926–931. [Google Scholar] [CrossRef] [PubMed]
- Bouchet, B.P.; Gough, R.E.; Ammon, Y.C.; van de Willige, D.; Post, H.; Jacquemet, G.; Maarten Altelaar, A.F.; Heck, A.J.R.; Goult, B.T.; Akhmanova, A. Talin-KANK1 interaction controls the recruitment of cortical microtubule stabilizing complexes to focal adhesions. Elife 2016, 5, 1–23. [Google Scholar] [CrossRef]
- Stehbens, S.; Wittmann, T. Targeting and transport: How microtubules control focal adhesion dynamics. J. Cell Biol. 2012, 198, 481–489. [Google Scholar] [CrossRef]
- Rooney, C.; White, G.; Nazgiewicz, A.; Woodcock, S.A.; Anderson, K.I.; Ballestrem, C.; Malliri, A. The Rac activator STEF (Tiam2) regulates cell migration by microtubule-mediated focal adhesion disassembly. EMBO Rep. 2010, 11, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, O.C.; Schaefer, A.W.; Mandato, C.A.; Forscher, P.; Bement, W.M.; Waterman-Storer, C.M. Conserved microtubule–actin interactions in cell movement and morphogenesis. Nat. Cell Biol. 2003, 5, 599–609. [Google Scholar] [CrossRef]
- Krendel, M.; Zenke, F.T.; Bokoch, G.M. Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat. Cell Biol. 2002, 4, 294–301. [Google Scholar] [CrossRef]
- Waterman-Storer, C.M.; Worthylake, R.A.; Liu, B.P.; Burridge, K.; Salmon, E.D. Microtubule growth activates Rac1 to promote lamellipodial protrusion in fibroblasts. Nat. Cell Biol. 1999, 1, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Montenegro-Venegas, C.; Tortosa, E.; Rosso, S.; Peretti, D.; Bollati, F.; Bisbal, M.; Jausoro, I.; Avila, J.; Cáceres, A.; Gonzalez-Billault, C. MAP1B regulates axonal development by modulating Rho-GTPase Rac1 activity. Mol. Biol. Cell 2010, 21, 3518–3528. [Google Scholar] [CrossRef]
- Van Haren, J.; Boudeau, J.; Schmidt, S.; Basu, S.; Liu, Z.; Lammers, D.; Demmers, J.; Benhari, J.; Grosveld, F.; Debant, A.; et al. Dynamic microtubules catalyze formation of navigator-TRIO complexes to regulate neurite extension. Curr. Biol. 2014, 24, 1778–1785. [Google Scholar] [CrossRef] [PubMed]
- Nalbant, P.; Chang, Y.-C.; Birkenfeld, J.; Chang, Z.-F.; Bokoch, G.M. Guanine nucleotide exchange factor-H1 regulates cell migration via localized activation of RhoA at the leading edge. Mol. Biol. Cell 2009, 20, 4070–4082. [Google Scholar] [CrossRef]
- Callow, M.G.; Zozulya, S.; Gishizky, M.L.; Jallal, B.; Smeal, T. PAK4 mediates morphological changes through the regulation of GEF-H1. J. Cell Sci. 2005, 118, 1861–1872. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Li, R.; Zheng, Y.; Busch, H. Cloning and characterization of GEF-H1, a microtubule-associated guanine nucleotide exchange factor for Rac and Rho GTPases. J. Biol. Chem. 1998, 273, 34954–34960. [Google Scholar] [CrossRef]
- Zenke, F.T.; Krendel, M.; DerMardirossian, C.; King, C.C.; Bohl, B.P.; Bokoch, G.M. p21-activated Kinase 1 Phosphorylates and Regulates 14-3-3 Binding to GEF-H1, a Microtubule-localized Rho Exchange Factor. J. Biol. Chem. 2004, 279, 18392–18400. [Google Scholar] [CrossRef]
- Kaverina, I.; Rottner, K.; Small, J.V. Targeting, capture, and stabilization of microtubules at early focal adhesions. J. Cell Biol. 1998, 142, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Noss, E.H.; Hsu, V.W.; Brenner, M.B. Integrins traffic rapidly via circular dorsal ruffles and macropinocytosis during stimulated cell migration. J. Cell Biol. 2011, 193, 61–70. [Google Scholar] [CrossRef] [PubMed]
- López, M.P.; Huber, F.; Grigoriev, I.; Steinmetz, M.O.; Akhmanova, A.; Koenderink, G.H.; Dogterom, M. Actin–microtubule coordination at growing microtubule ends. Nat. Commun. 2014, 5, 4778. [Google Scholar] [CrossRef]
- Wickström, S.A.; Fässler, R. Regulation of membrane traffic by integrin signaling. Trends Cell Biol. 2011, 21, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Etienne-Manneville, S. APC in cell migration. Adv. Exp. Med. Biol. 2009, 656, 30–40. [Google Scholar] [PubMed]
- Matsumoto, S.; Fumoto, K.; Okamoto, T.; Kaibuchi, K.; Kikuchi, A. Binding of APC and dishevelled mediates Wnt5a-regulated focal adhesion dynamics in migrating cells. EMBO J. 2010, 29, 1192–1204. [Google Scholar] [CrossRef]
- Juanes, M.A.; Bouguenina, H.; Eskin, J.A.; Jaiswal, R.; Badache, A.; Goode, B.L. Adenomatous polyposis coli nucleates actin assembly to drive cell migration and microtubuleinduced focal adhesion turnover. J. Cell Biol. 2017, 216, 2859–2875. [Google Scholar] [CrossRef]
- Breitsprecher, D.; Jaiswal, R.; Bombardier, J.P.; Gould, C.J.; Gelles, J.; Goode, B.L. Rocket Launcher Mechanism of Collaborative Actin Assembly Defined by Single-Molecule Imaging. Science 2012, 336, 1164–1168. [Google Scholar] [CrossRef] [PubMed]
- Brandt, D.T.; Grosse, R. Get to grips: Steering local actin dynamics with IQGAPs. EMBO Rep. 2007, 8, 1019–1023. [Google Scholar] [CrossRef]
- Krylyshkina, O.; Anderson, K.I.; Kaverina, I.; Upmann, I.; Manstein, D.J.; Small, J.V.; Toomre, D.K. Nanometer targeting of microtubules to focal adhesions. J. Cell Biol. 2003, 161, 853–859. [Google Scholar] [CrossRef]
- Kaverina, I.; Krylyshkina, O.; Small, J.V. Microtubule targeting of substrate contacts promotes their relaxation and dissociation. J. Cell Biol. 1999, 146, 1033–1043. [Google Scholar] [CrossRef]
- Rid, R.; Schiefermeier, N.; Grigoriev, I.; Small, J.V.; Kaverina, I. The last but not the least: The origin and significance of trailing adhesions in fibroblastic cells. Cell Motil. Cytoskeleton 2005, 61, 161–171. [Google Scholar] [CrossRef]
- Applewhite, D.A.; Grode, K.D.; Keller, D.; Zadeh, A.D.; Zadeh, A.; Slep, K.C.; Rogers, S.L. The spectraplakin Short stop is an actin-microtubule cross-linker that contributes to organization of the microtubule network. Mol. Biol. Cell 2010, 21, 1714–1724. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Shen, Q.-T.; Oristian, D.S.; Lu, C.P.; Zheng, Q.; Wang, H.-W.; Fuchs, E. Skin stem cells orchestrate directional migration by regulating microtubule-ACF7 connections through GSK3β. Cell 2011, 144, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Zaoui, K.; Benseddik, K.; Daou, P.; Salaun, D.; Badache, A. ErbB2 receptor controls microtubule capture by recruiting ACF7 to the plasma membrane of migrating cells. Proc. Natl. Acad. Sci. USA 2010, 107, 18517–18522. [Google Scholar] [CrossRef] [PubMed]
- Higashida, C.; Miyoshi, T.; Fujita, A.; Oceguera-Yanez, F.; Monypenny, J.; Andou, Y.; Narumiya, S.; Watanabe, N. Actin Polymerization-Driven Molecular Movement of mDia1 in Living Cells. Science 2004, 303, 2007–2010. [Google Scholar] [CrossRef] [PubMed]
- Wickström, S.A.; Lange, A.; Hess, M.W.; Polleux, J.; Spatz, J.P.; Krüger, M.; Pfaller, K.; Lambacher, A.; Bloch, W.; Mann, M.; et al. Integrin-Linked Kinase Controls Microtubule Dynamics Required for Plasma Membrane Targeting of Caveolae. Dev. Cell 2010, 19, 574–588. [Google Scholar] [CrossRef] [PubMed]
- Villari, G.; Jayo, A.; Zanet, J.; Fitch, B.; Serrels, B.; Frame, M.; Stramer, B.; Goult, B.; Parsons, M. A direct interaction between fascin and microtubules contributes to adhesion dynamics and cell migration. J. Cell Sci. 2015, 128, 4601–4604. [Google Scholar] [CrossRef]
- Ezratty, E.J.; Partridge, M.A.; Gundersen, G.G. Microtubule-induced focal adhesion disassembly is mediated by dynamin and focal adhesion kinase. Nat. Cell Biol. 2005, 7, 581–590. [Google Scholar] [CrossRef]
- Ezratty, E.J.; Bertaux, C.; Marcantonio, E.E.; Gundersen, G.G. Clathrin mediates integrin endocytosis for focal adhesion disassembly in migrating cells. J. Cell Biol. 2009, 187, 733–747. [Google Scholar] [CrossRef]
- Stehbens, S.J.; Paszek, M.; Pemble, H.; Ettinger, A.; Gierke, S.; Wittmann, T. CLASPs link focal-adhesion-associated microtubule capture to localized exocytosis and adhesion site turnover. Nat. Cell Biol. 2014, 16, 558–570. [Google Scholar] [CrossRef]
- Kenific, C.M.; Stehbens, S.J.; Goldsmith, J.; Leidal, A.M.; Faure, N.; Ye, J.; Wittmann, T.; Debnath, J. NBR 1 enables autophagy-dependent focal adhesion turnover. J. Cell Biol. 2016, 212, 577–590. [Google Scholar] [CrossRef]
- Helfand, B.T.; Mendez, M.G.; Murthy, S.N.P.; Shumaker, D.K.; Grin, B.; Mahammad, S.; Aebi, U.; Wedig, T.; Wu, Y.I.; Hahn, K.M.; et al. Vimentin organization modulates the formation of lamellipodia. Mol. Biol. Cell 2011, 22, 1274–1289. [Google Scholar] [CrossRef]
- Menko, A.S.; Bleaken, B.M.; Libowitz, A.A.; Zhang, L.; Stepp, M.A.; Walker, J.L. A central role for vimentin in regulating repair function during healing of the lens epithelium. Mol. Biol. Cell 2014, 25, 776–790. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Boëda, B.; Etienne-Manneville, S. APC binds intermediate filaments and is required for their reorganization during cell migration. J. Cell Biol. 2013, 200, 249–258. [Google Scholar] [CrossRef]
- Huber, F.; Boire, A.; López, M.P.; Koenderink, G.H. Cytoskeletal crosstalk: When three different personalities team up. Curr. Opin. Cell Biol. 2015, 32, 39–47. [Google Scholar] [CrossRef]
- Shabbir, S.H.; Cleland, M.M.; Goldman, R.D.; Mrksich, M. Geometric control of vimentin intermediate filaments. Biomaterials 2014, 35, 1359–1366. [Google Scholar] [CrossRef]
- Yuan, A.; Rao, M.V.; Veeranna; Nixon, R.A. Neurofilaments and neurofilament proteins in health and disease. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef]
- Mendez, M.G.; Kojima, S.-I.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010, 24, 1838–1851. [Google Scholar] [CrossRef]
- Rogel, M.R.; Soni, P.N.; Troken, J.R.; Sitikov, A.; Trejo, H.E.; Ridge, K.M. Vimentin is sufficient and required for wound repair and remodeling in alveolar epithelial cells. FASEB J. 2011, 25, 3873–3883. [Google Scholar] [CrossRef]
- Chung, B.M.; Rotty, J.D.; Coulombe, P.A. Networking galore: Intermediate filaments and cell migration. Curr. Opin. Cell Biol. 2013, 25, 600–612. [Google Scholar] [CrossRef]
- Battaglia, R.A.; Delic, S.; Herrmann, H.; Snider, N.T. Vimentin on the move: New developments in cell migration. F1000Research 2018, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lepekhin, E.A.; Eliasson, C.; Berthold, C.H.; Berezin, V.; Bock, E.; Pekny, M. Intermediate filaments regulate astrocyte motility. J. Neurochem. 2001, 79, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Rathje, L.-S.Z.; Nordgren, N.; Pettersson, T.; Ronnlund, D.; Widengren, J.; Aspenstrom, P.; Gad, A.K.B. Oncogenes induce a vimentin filament collapse mediated by HDAC6 that is linked to cell stiffness. Proc. Natl. Acad. Sci. USA 2014, 111, 1515–1520. [Google Scholar] [CrossRef]
- Gan, Z.; Ding, L.; Burckhardt, C.J.; Lowery, J.; Zaritsky, A.; Sitterley, K.; Mota, A.; Costigliola, N.; Starker, C.G.; Voytas, D.F.; et al. Vimentin Intermediate Filaments Template Microtubule Networks to Enhance Persistence in Cell Polarity and Directed Migration. Cell Syst. 2016, 3, 252–263. [Google Scholar] [CrossRef]
- Duan, G.; Walther, D. The Roles of Post-translational Modifications in the Context of Protein Interaction Networks. PLoS Comput. Biol. 2015, 11, 1–23. [Google Scholar] [CrossRef]
- Nieuwenhuizen, R.P.J.; Nahidiazar, L.; Manders, E.M.M.; Jalink, K.; Stallinga, S.; Rieger, B. Co-orientation: Quantifying simultaneous co-localization and orientational alignment of filaments in light microscopy. PLoS ONE 2015, 10, 1–21. [Google Scholar] [CrossRef]
- Leduc, C.; Manneville, S.E. Regulation of microtubule-associated intermediate filament network polarization. J. Cell Biol 2017, 216, 1689–1703. [Google Scholar] [CrossRef] [PubMed]
- Havel, L.; Kline, E.; Salgueiro, A.; Marcus, A. Vimentin regulates lung cancer cell adhesion through a VAV2–Rac1 pathway to control focal adhesion kinase activity. Oncogene 2014, 34, 1–12. [Google Scholar] [CrossRef]
- Jiu, Y.; Peränen, J.; Schaible, N.; Cheng, F.; Eriksson, J.E.; Krishnan, R.; Lappalainen, P. Vimentin intermediate filaments control actin stress fiber assembly through GEF-H1 and RhoA. J. Cell Sci. 2017, 130, 892–902. [Google Scholar] [CrossRef]
- De Pascalis, C.; Perez-Gonzalez, C.; Seetharaman, S.; Boeda, B.; Vianay, B.; Burute, M.; Leduc, C.; Borghi, N.; Trepat, X.; Etienne-Manneville, S. Intermediate filaments control collective migration by restricting traction forces and sustaining cell-cell contacts. bioRxiv 2018. [Google Scholar] [CrossRef]
- Kim, J.; Yang, C.; Kim, E.J.; Jang, J.; Kim, S.-J.; Kang, S.M.; Kim, M.G.; Jung, H.; Park, D.; Kim, C. Vimentin filaments regulate integrin–ligand interactions by binding to the cytoplasmic tail of integrin β3. J. Cell Sci. 2016, 129, 2030–2042. [Google Scholar] [CrossRef]
- Costigliola, N.; Ding, L.; Burckhardt, C.J.; Han, S.J.; Gutierrez, E.; Mota, A.; Groisman, A.; Mitchison, T.J.; Danuser, G. Vimentin fibers orient traction stress. Proc. Natl. Acad. Sci. USA 2017, 114, 5195–5200. [Google Scholar] [CrossRef]
- Akhmanova, A.; Stehbens, S.J.; Yap, A.S. Touch, Grasp, Deliver and Control: Functional Cross-Talk Between Microtubules and Cell Adhesions. Traffic 2009, 10, 268–274. [Google Scholar] [CrossRef]
- Janosch, P.; Kieser, A.; Eulitz, M.; Lovric, J.; Sauer, G.; Reichert, M.; Gounari, F.; Büscher, D.; Baccarini, M.; Mischak, H.; et al. The Raf-1 kinase associates with vimentin kinases and regulates the structure of vimentin filaments. FASEB J. 2000, 14, 2008–2021. [Google Scholar] [CrossRef]
- Ehrenreiter, K.; Piazzolla, D.; Velamoor, V.; Sobczak, I.; Small, J.V.; Takeda, J.; Leung, T.; Baccarini, M. Raf-1 regulates Rho signaling and cell migration. J. Cell Biol. 2005, 168, 955–964. [Google Scholar] [CrossRef]
- Amano, M.; Fukata, Y.; Kaibuchi, K. Regulation and Functions of Rho-Associated Kinase. Exp. Cell Res. 2000, 261, 44–51. [Google Scholar] [CrossRef]
- Sin, W.C.; Chen, X.Q.; Leung, T.; Lim, L. RhoA-binding kinase alpha translocation is facilitated by the collapse of the vimentin intermediate filament network. Mol. Cell. Biol. 1998, 18, 6325–6339. [Google Scholar] [CrossRef]
- Ren, X.D.; Kiosses, W.B.; Sieg, D.J.; Otey, C.A.; Schlaepfer, D.D.; Schwartz, M.A. Focal adhesion kinase suppresses Rho activity to promote focal adhesion turnover. J. Cell Sci. 2000, 113, 3673–3678. [Google Scholar]
- Ren, X.D.; Kiosses, W.B.; Schwartz, M.A. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 1999, 18, 578–585. [Google Scholar] [CrossRef]
- Jiu, Y.; Lehtimäki, J.; Tojkander, S.; Cheng, F.; Jäälinoja, H.; Liu, X.; Varjosalo, M.; Eriksson, J.E.; Lappalainen, P. Bidirectional Interplay between Vimentin Intermediate Filaments and Contractile Actin Stress Fibers. Cell Rep. 2015, 11, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Goto, H.; Tanabe, K.; Manser, E.; Lim, L.; Yasui, Y.; Inagaki, M. Phosphorylation and reorganization of vimentin by p21-activated kinase (PAK). Genes Cells 2002, 7, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Petrie, R.J.; Koo, H.; Yamada, K.M. Generation of compartmentalized pressure by a nuclear piston governs cell motility in a 3D matrix. Science 2014, 345, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Huiatt, T.W.; Paulin, D.; Li, Z.; Robson, R.M. Synemin interacts with the LIM domain protein zyxin and is essential for cell adhesion and migration. Exp. Cell Res. 2010, 316, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Jing, R.; Pitre, A.; Williams, B.J.; Skalli, O. Intermediate filament protein synemin contributes to the migratory properties of astrocytoma cells by influencing the dynamics of the actin cytoskeleton. FASEB J. 2008, 22, 3196–3206. [Google Scholar] [CrossRef] [PubMed]
- Uyama, N.; Zhao, L.; Van Rossen, E.; Hirako, Y.; Reynaert, H.; Adams, D.H.; Xue, Z.; Li, Z.; Robson, R.; Pekny, M.; et al. Hepatic stellate cells express synemin, a protein bridging intermediate filaments to focal adhesions. Gut 2006, 55, 1276–1289. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Critchley, D.R.; Paulin, D.; Li, Z.; Robson, R.M. Identification of a repeated domain within mammalian α-synemin that interacts directly with talin. Exp. Cell Res. 2008, 314, 1839–1849. [Google Scholar] [CrossRef]
- Sun, N.; Critchley, D.R.; Paulin, D.; Li, Z.; Robson, R.M. Human alpha-synemin interacts directly with vinculin and metavinculin. Biochem. J. 2008, 409, 657–667. [Google Scholar] [CrossRef]
- Hijikata, T.; Nakamura, A.; Isokawa, K.; Imamura, M.; Yuasa, K.; Ishikawa, R.; Kohama, K.; Takeda, S.; Yorifuji, H. Plectin 1 links intermediate filaments to costameric sarcolemma through -synemin, -dystrobrevin and actin. J. Cell Sci. 2008, 121, 2062–2074. [Google Scholar] [CrossRef]
- Hyder, C.L.; Lazaro, G.; Pylvänäinen, J.W.; Roberts, M.W.G.; Qvarnström, S.M.; Eriksson, J.E. Nestin regulates prostate cancer cell invasion by influencing the localisation and functions of FAK and integrins. J. Cell Sci. 2014, 127, 2161–2173. [Google Scholar] [CrossRef]
- Mathers, C.D.; Loncar, D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006, 3, 2011–2030. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: Globocan 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef] [PubMed]
- Legler, J.M.; Ries, L.A.; Smith, M.A.; Warren, J.L.; Heineman, E.F.; Kaplan, R.S.; Linet, M.S. Cancer surveillance series [corrected]: Brain and other central nervous system cancers: Recent trends in incidence and mortality. J. Natl. Cancer Inst. 1999, 91, 1382–1390. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.G.; McCarthy, B.J.; Freels, S.; Kupelian, V.; Bondy, M.L. The conditional probability of survival of patients with primary malignant brain tumors: Surveillance, epidemiology, and end results (SEER) data. Cancer 1999, 85, 485–491. [Google Scholar] [CrossRef]
- Scherer, H.J. The forms of growth in gliomas and their practical significance. Brain 1940, 63, 1–35. [Google Scholar] [CrossRef]
- Maher, E.A.; Furnari, F.B.; Bachoo, R.M.; Rowitch, D.H.; Louis, D.N.; Cavenee, W.K.; DePinho, R.A. Malignant glioma: Genetics and biology of a grave matter. Genes Dev. 2001, 15, 1311–1333. [Google Scholar] [CrossRef]
- Chamberlain, M.C. Radiographic patterns of relapse in glioblastoma. J. Neurooncol. 2011, 101, 319–323. [Google Scholar] [CrossRef]
- Iwadate, Y.; Sakaida, T.; Saegusa, T.; Hiwasa, T.; Takiguchi, M.; Fujimoto, S.; Yamaura, A. Proteome-based identification of molecular markers predicting chemosensitivity to each category of anticancer agents in human gliomas. Int. J. Oncol. 2005, 26, 993–998. [Google Scholar] [CrossRef]
- Hirata, E.; Yukinaga, H.; Kamioka, Y.; Arakawa, Y.; Miyamoto, S.; Okada, T.; Sahai, E.; Matsuda, M. In vivo fluorescence resonance energy transfer imaging reveals differential activation of Rho-family GTPases in glioblastoma cell invasion. J. Cell Sci. 2012, 125, 858–868. [Google Scholar] [CrossRef]
- Salhia, B.; Tran, N.L.; Chan, A.; Wolf, A.; Nakada, M.; Rutka, F.; Ennis, M.; McDonough, W.S.; Berens, M.E.; Symons, M.; et al. The Guanine Nucleotide Exchange Factors Trio, Ect2, and Vav3 Mediate the Invasive Behavior of Glioblastoma. Am. J. Pathol. 2008, 173, 1828–1838. [Google Scholar] [CrossRef]
- Malchinkhuu, E.; Sato, K.; Maehama, T.; Mogi, C.; Tomura, H.; Ishiuchi, S.; Yoshimoto, Y.; Kurose, H.; Okajima, F. S1P2 receptors mediate inhibition of glioma cell migration through Rho signaling pathways independent of PTEN. Biochem. Biophys. Res. Commun. 2008, 366, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.L.; McDonough, W.S.; Savitch, B.A.; Fortin, S.P.; Winkles, J.A.; Symons, M.; Nakada, M.; Cunliffe, H.E.; Hostetter, G.; Hoelzinger, D.B.; et al. Increased Fibroblast Growth Factor-Inducible 14 Expression Levels Promote Glioma Cell Invasion via Rac1 and Nuclear Factor-κB and Correlate with Poor Patient Outcome. Cancer Res. 2006, 66, 9535–9542. [Google Scholar] [CrossRef]
- Zagzag, D.; Friedlander, D.R.; Margolis, B.; Grumet, M.; Semenza, G.L.; Zhong, H.; Simons, J.W.; Holash, J.; Wiegand, S.J.; Yancopoulos, G.D. Molecular Events Implicated in Brain Tumor Angiogenesis and Invasion. Pediatr. Neurosurg. 2000, 33, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Khalil, B.D.; Hanna, S.; Saykali, B.A.; El-Sitt, S.; Nasrallah, A.; Marston, D.; El-Sabban, M.; Hahn, K.M.; Symons, M.; El-Sibai, M. The regulation of RhoA at focal adhesions by StarD13 is important for astrocytoma cell motility. Exp. Cell Res. 2014, 321, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Seasholtz, T.M.; Radeff-Huang, J.; Sagi, S.A.; Matteo, R.; Weems, J.M.; Cohen, A.S.; Feramisco, J.R.; Brown, J.H. Rho-mediated cytoskeletal rearrangement in response to LPA is functionally antagonized by Rac1 and PIP2. J. Neurochem. 2004, 91, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Salhia, B.; Rutten, F.; Nakada, M.; Beaudry, C.; Berens, M.; Kwan, A.; Rutka, J.T. Inhibition of Rho-kinase affects astrocytoma morphology, motility, and invasion through activation of Rac1. Cancer Res. 2005, 65, 8792–8800. [Google Scholar] [CrossRef]
- Chan, A.Y.; Coniglio, S.J.; Chuang, Y.; Michaelson, D.; Knaus, U.G.; Philips, M.R.; Symons, M. Roles of the Rac1 and Rac3 GTPases in human tumor cell invasion. Oncogene 2005, 24, 7821–7829. [Google Scholar] [CrossRef]
- Chuang, Y.; Tran, N.L.; Rusk, N.; Nakada, M.; Berens, M.E.; Symons, M. Role of Synaptojanin 2 in Glioma Cell Migration and Invasion. Cancer Res. 2004, 64, 8271–8275. [Google Scholar] [CrossRef]
- Yin, M.; Lu, Q.; Liu, X.; Wang, T.; Liu, Y.; Chen, L. Silencing Drp1 inhibits glioma cells proliferation and invasion by RHOA/ROCK1 pathway. Biochem. Biophys. Res. Commun. 2016, 478, 663–668. [Google Scholar] [CrossRef]
- Singh, J.; Sharma, K.; Pillai, P.P. PDGFR inhibition mediated intracellular signalling in C6 glioma growth and migration: Role of ERK and ROCK pathway. Cytotechnology 2018, 70, 465–477. [Google Scholar] [CrossRef]
- Kim, D.-H.; Wirtz, D. Predicting how cells spread and migrate. Cell Adh. Migr. 2013, 7, 293–296. [Google Scholar] [CrossRef]
- Fortin, S.P.; Ennis, M.J.; Schumacher, C.A.; Zylstra-Diegel, C.R.; Williams, B.O.; Ross, J.T.D.; Winkles, J.A.; Loftus, J.C.; Symons, M.H.; Tran, N.L. Cdc42 and the Guanine Nucleotide Exchange Factors Ect2 and Trio Mediate Fn14-Induced Migration and Invasion of Glioblastoma Cells. Mol. Cancer Res. 2012, 10, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Yamana, N.; Arakawa, Y.; Nishino, T.; Kurokawa, K.; Tanji, M.; Itoh, R.E.; Monypenny, J.; Ishizaki, T.; Bito, H.; Nozaki, K.; et al. The Rho-mDia1 Pathway Regulates Cell Polarity and Focal Adhesion Turnover in Migrating Cells through Mobilizing Apc and c-Src. Mol. Cell. Biol. 2006, 26, 6844–6858. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Sun, X.; You, Y.; Liu, N.; Fu, Z. Expression of focal adhesion kinase and phosphorylated focal adhesion kinase in human gliomas is associated with unfavorable overall survival. Transl. Res. 2010, 156, 45–52. [Google Scholar] [CrossRef]
- Jones, G.; Machado, J.; Merlo, A. Loss of focal adhesion kinase (FAK) inhibits epidermal growth factor receptor-dependent migration and induces aggregation of nh(2)-terminal FAK in the nuclei of apoptotic glioblastoma cells. Cancer Res. 2001, 61, 4978–4981. [Google Scholar] [PubMed]
- Obara, S.; Nakata, M.; Takeshima, H.; Kuratsu, J.; Maruyama, I.; Kitajima, I. Inhibition of migration of human glioblastoma cells by cerivastatin in association with focal adhesion kinase (FAK). Cancer Lett. 2002, 185, 153–161. [Google Scholar] [CrossRef]
- Beadle, C.; Assanah, M.; Monzo, P.; Vallee, R.; Rosenfeld, S.; Canoll, P. The Role of Myosin II in Glioma Invasion of the Brain Christopher. Mol. Biol. Cell 2008, 19, 3357–3368. [Google Scholar] [CrossRef] [PubMed]
- Ivkovic, S.; Beadle, C.; Noticewala, S.; Massey, S.C.; Swanson, K.R.; Toro, L.N.; Bresnick, A.R.; Canoll, P.; Rosenfeld, S.S. Direct inhibition of myosin II effectively blocks glioma invasion in the presence of multiple motogens. Mol. Biol. Cell 2012, 23, 533–542. [Google Scholar] [CrossRef]
- Lee, W.; Lim, S.; Kim, Y. The role of myosin II in glioma invasion: A mathematical model. PLoS ONE 2017, 12, 1939–4586. [Google Scholar] [CrossRef]
- Liu, Z.; Yang, X.; Chen, C.; Liu, B.; Ren, B.; Wang, L.; Zhao, K.; Yu, S.; Ming, H. Expression of the Arp2/3 complex in human gliomas and its role in the migration and invasion of glioma cells. Oncol. Rep. 2013, 30, 2127–2136. [Google Scholar] [CrossRef]
- Monzo, P.; Chong, Y.K.; Guetta-Terrier, C.; Krishnasamy, A.; Sathe, S.R.; Yim, E.K.F.; Ng, W.H.; Ang, B.T.; Tang, C.; Ladoux, B.; et al. Mechanical confinement triggers glioma linear migration dependent on formin FHOD3. Mol. Biol. Cell 2016, 27, 1246–1261. [Google Scholar] [CrossRef]
- Zhang, C.; Hai, L.; Zhu, M.; Yu, S.P.; Li, T.; Lin, Y.; Liu, B.; Zhou, X.C.; Chen, L.; Zhao, P.F.; et al. Actin cytoskeleton regulator Arp2/3 complex is required for DLL1 activating Notch1 signaling to maintain the stem cell phenotype of glioma initiating cells. Oncotarget 2017, 8, 33353–33364. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, K.; Ren, B.; Zhu, M.; Zhang, C.; Zhao, P.; Zhou, H.; Chen, L.; Yu, S.; Yang, X. Expression of cortactin in human gliomas and its effect on migration and invasion of glioma cells. Oncol. Rep. 2015, 34, 1815–1824. [Google Scholar] [CrossRef]
- Hwang, J.H.; Smith, C.A.; Salhia, B.; Rutka, J.T. The Role of Fascin in the Migration and Invasiveness of Malignant Glioma Cells. Neoplasia 2008, 10, 149–159. [Google Scholar] [CrossRef]
- Hoa, N.T.; Ge, L.; Erickson, K.L.; Kruse, C.A.; Cornforth, A.N.; Kuznetsov, Y.; McPherson, A.; Martini, F.; Jadus, M.R. Fascin-1 knock-down of human glioma cells reduces their microvilli/filopodia while improving their susceptibility to lymphocyte-mediated cytotoxicity. Am. J. Transl. Res. 2015, 7, 271–284. [Google Scholar]
- Eke, I.; Storch, K.; Kästner, I.; Vehlow, A.; Faethe, C.; Mueller-Klieser, W.; Taucher-Scholz, G.; Temme, A.; Schackert, G.; Cordes, N. Three-dimensional invasion of human glioblastoma cells remains unchanged by X-ray and carbon ion irradiation in vitro. Int. J. Radiat. Oncol. Biol. Phys. 2012, 84. [Google Scholar] [CrossRef]
- Frankel, P.; Pellet-Many, C.; Lehtolainen, P.; D’Abaco, G.M.; Tickner, M.L.; Cheng, L.; Zachary, I.C. Chondroitin sulphate-modified neuropilin 1 is expressed in human tumour cells and modulates 3D invasion in the U87MG human glioblastoma cell line through a p130Cas-mediated pathway. EMBO Rep. 2008, 9, 983–989. [Google Scholar] [CrossRef]
- Oppel, F.; Müller, N.; Schackert, G.; Hendruschk, S.; Martin, D.; Geiger, K.D.; Temme, A. SOX2-RNAi attenuates S-phase entry and induces RhoA-dependent switch to protease-independent amoeboid migration in human glioma cells. Mol. Cancer 2011, 10, 1–16. [Google Scholar] [CrossRef]
- Weeks, A.; Okolowsky, N.; Golbourn, B.; Ivanchuk, S.; Smith, C.; Rutka, J.T. ECT2 and RASAL2 mediate mesenchymal-amoeboid transition in human astrocytoma cells. Am. J. Pathol. 2012, 181, 662–674. [Google Scholar] [CrossRef]
- Quick, Q.; Paul, M.; Skalli, O. Roles and potential clinical applications of intermediate filament proteins in brain tumors. Semin. Pediatr. Neurol. 2015, 22, 40–48. [Google Scholar] [CrossRef]
- Ducray, F.; Mokhtari, K.; Crinire, E.; Idbaih, A.; Marie, Y.; Dehais, C.; Paris, S.; Carpentier, C.; Dieme, M.J.; Adam, C.; et al. Diagnostic and prognostic value of alpha internexin expression in a series of 409 gliomas. Eur. J. Cancer 2011, 47, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Skalli, O.; Wilhelmsson, U.; Örndahl, C.; Fekete, B.; Malmgren, K.; Rydenhag, B.; Pekny, M. Astrocytoma grade IV (glioblastoma multiforme) displays 3 subtypes with unique expression profiles of intermediate filament proteins. Hum. Pathol. 2013, 44, 2081–2088. [Google Scholar] [CrossRef] [PubMed]
- Paetau, A. Glial fibrillary acidic protein, vimentin and fibronectin in primary cultures of human glioma and fetal brain. Acta Neuropathol. 1988, 75, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Yung, W.A.; Luna, M.; Borit, A. Vimentin and Glial Fibrillary Acidic Protein in Human Brain Tumors. J. Neuro-Oncol. 1985, 38, 35–38. [Google Scholar] [CrossRef]
- van Bodegraven, E.J.; van Asperen, J.V.; Robe, P.A.J.; Hol, E.M. Importance of GFAP isoform-specific analyses in astrocytoma. Glia 2019, 1–17. [Google Scholar] [CrossRef]
- Lin, L.; Wang, G.; Ming, J.; Meng, X.; Han, B.; Sun, B.; Cai, J.; Jiang, C. Analysis of expression and prognostic significance of vimentin and the response to temozolomide in glioma patients. Tumor Biol. 2016, 37, 15333–15339. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, L.; Dong, X.; Liu, L.; Huo, L.; Chen, H. High Expression of Vimentin is Associated with Progression and a Poor Outcome in Glioblastoma. Appl. Immunohistochem. Mol. Morphol. 2018, 26, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Mahesparan, R.; Read, T.-A.; Lund-Johansen, M.; Skaftnesmo, K.O.; Bjerkvig, R.; Engebraaten, O. Expression of extracellular matrix components in a highly infiltrative in vivo glioma model. Acta Neuropathol. 2003, 105, 49–57. [Google Scholar] [CrossRef]
- Lv, D.; Lu, L.; Hu, Z.; Fei, Z.; Liu, M.; Wei, L.; Xu, J. Nestin Expression Is Associated with Poor Clinicopathological Features and Prognosis in Glioma Patients: An Association Study and Meta-analysis. Mol. Neurobiol. 2017, 54, 727–735. [Google Scholar] [CrossRef]
- Baumgarten, P.; Quick-Weller, J.; Gessler, F.; Wagner, M.; Tichy, J.; Forster, M.T.; Foerch, C.; Seifert, V.; Mittelbronn, M.; Senft, C. Pre- and early postoperative GFAP serum levels in glioma and brain metastases. J. Neurooncol. 2018, 139, 541–546. [Google Scholar] [CrossRef]
- Elobeid, A.; Bongcam-Rudloff, E.; Westermark, B.; Nistér, M. Effects of inducible glial fibrillary acidic protein on glioma cell motility and proliferation. J. Neurosci. Res. 2000, 60, 245–256. [Google Scholar] [CrossRef]
- Toda, M.; Miura, M.; Asou, H.; Toya, S.; Uyemura, K. Cell growth suppression of astrocytoma C6 cells by glial fibrillary acidic protein cDNA transfection. J. Neurochem. 1994, 63, 1975–1978. [Google Scholar] [CrossRef]
- Kouam, P.N.; Rezniczek, G.A.; Kochanneck, A.; Priesch-Grzeszkowiak, B.; Hero, T.; Adamietz, I.A.; Bühler, H. Robo1 and vimentin regulate radiation-induced motility of human glioblastoma cells. PLoS ONE 2018, 13, 1–18. [Google Scholar] [CrossRef]
- Ishiwata, T.; Teduka, K.; Yamamoto, T.; Kawahara, K.; Matsuda, Y.; Naito, Z. Neuroepithelial stem cell marker nestin regulates the migration, invasion and growth of human gliomas. Oncol. Rep. 2011, 26, 91–99. [Google Scholar] [CrossRef]
- Kitai, R.; Horita, R.; Sato, K.; Yoshida, K.; Arishima, H.; Higashino, Y.; Hashimoto, N.; Takeuchi, H.; Kubota, T.; Kikuta, K.-I. Nestin expression in astrocytic tumors delineates tumor infiltration. Brain Tumor Pathol. 2010, 27, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Jing, R.; Pizzolato, G.; Robson, R.M.; Gabbiani, G.; Skalli, O. Intermediate filament protein synemin is present in human reactive and malignant astrocytes and associates with ruffled membranes in astrocytoma cells. Glia 2005, 50, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Rickman, D.S.; Bobek, M.P.; Misek, D.E.; Kuick, R.; Blaivas, M.; Kurnit, D.M.; Taylor, J.; Hanash, S.M. Distinctive molecular profiles of high-grade and low-grade gliomas based on oligonucleotide microarray analysis. Cancer Res. 2001, 61, 6885–6891. [Google Scholar] [PubMed]
- Katsetos, C.D.; Legido, A.; Perentes, E.; Mörk, S.J. Class III β-Tubulin Isotype: A Key Cytoskeletal Protein at the Crossroads of Developmental Neurobiology and Tumor Neuropathology. J. Child Neurol. 2003, 18, 851–866. [Google Scholar] [CrossRef] [PubMed]
- Katsetos, C.D.; Reddy, G.; Dráberová, E.; Šmejkalová, B.; Del Valle, L.; Ashraf, Q.; Tadevosyan, A.; Yelin, K.; Maraziotis, T.; Mishra, O.P.; et al. Altered Cellular Distribution and Subcellular Sorting of γ-Tubulin in Diffuse Astrocytic Gliomas and Human Glioblastoma Cell Lines. J. Neuropathol. Exp. Neurol. 2006, 65, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Katsetos, C.D.; Reginato, M.J.; Baas, P.W.; D’Agostino, L.; Legido, A.; Tuszyński, J.A.; Dráberová, E.; Dráber, P. Emerging microtubule targets in glioma therapy. Semin. Pediatr. Neurol. 2015, 22, 49–72. [Google Scholar] [CrossRef]
- Orr, G.A.; Verdier-Pinard, P.; McDaid, H.; Horwitz, S.B. Mechanisms of Taxol resistance related to microtubules. Oncogene 2003, 22, 7280–7295. [Google Scholar] [CrossRef] [PubMed]
- Kavallaris, M.; Kuo, D.Y.; Burkhart, C.A.; Regl, D.L.; Norris, M.D.; Haber, M.; Horwitz, S.B. Taxol-resistant epithelial ovarian tumors are associated with altered expression of specific beta-tubulin isotypes. J. Clin. Investig. 1997, 100, 1282–1293. [Google Scholar] [CrossRef]
- Karmakar, S.; Banik, N.L.; Ray, S.K. Combination of all-trans retinoic acid and paclitaxel-induced differentiation and apoptosis in human glioblastoma U87MG xenografts in nude mice. Cancer 2008, 112, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.C.; Kan, D.; Lo, T.H.; Lu, K.S.; Chien, C.-L. Induction of neural differentiation in rat C6 glioma cells with taxol. Brain Behav. 2015, 5, 1–10. [Google Scholar] [CrossRef]
- Godinho, S.A.; Pellman, D. Causes and consequences of centrosome abnormalities in cancer. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369. [Google Scholar] [CrossRef]
- Dráberová, E.; Vinopal, S.; Morfini, G.; Liu, P.S.; Sládková, V.; Sulimenko, T.; Burns, M.R.; Solowska, J.; Kulandaivel, K.; De Chadarévian, J.P.; et al. Microtubule-severing ATPase spastin in glioblastoma: Increased expression in human glioblastoma cell lines and inverse roles in cell motility and proliferation. J. Neuropathol. Exp. Neurol. 2011, 70, 811–826. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Mu, L.; Han, X.; Liu, X.; Fu, S. SiRNA targeting stathmin inhibits invasion and enhances chemotherapy sensitivity of stem cells derived from Glioma cell lines. Acta Biochim. Biophys. Sin. (Shanghai) 2014, 46, 1034–1040. [Google Scholar] [CrossRef]
- Masui, K.; Mawatari, S.; Suzuki, S.O.; Iwaki, T. Evaluation of sensitivity and specificity of doublecortin immunostatining for the detection of infiltrating glioma cells. Brain Tumor Pathol. 2008, 25, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Daou, M.-C.; Smith, T.W.; Litofsky, N.S.; Hsieh, C.C.; Ross, A.H. Doublecortin is preferentially expressed in invasive human brain tumors. Acta Neuropathol. 2005, 110, 472–480. [Google Scholar] [CrossRef]
- Santra, M.; Santra, S.; Roberts, C.; Zhang, R.L.; Chopp, M. Doublecortin induces mitotic microtubule catastrophe and inhibits glioma cell invasion. J. Neurochem. 2009, 108, 231–245. [Google Scholar] [CrossRef]
- Santra, M.; Zhang, X.; Santra, S.; Jiang, F.; Chopp, M. Ectopic Doublecortin Gene Expression Suppresses the Malignant Phenotype in Glioblastoma Cells. Cancer Res. 2006, 66, 11726–11735. [Google Scholar] [CrossRef] [PubMed]
- Pagano, A.; Honoré, S.; Mohan, R.; Berges, R.; Akhmanova, A.; Braguer, D. Epothilone B inhibits migration of glioblastoma cells by inducing microtubule catastrophes and affecting EB1 accumulation at microtubule plus ends. Biochem. Pharmacol. 2012, 84, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Näthke, I.S. THE ADENOMATOUS POLYPOSIS COLI PROTEIN: The Achilles Heel of the Gut Epithelium. Annu. Rev. Cell Dev. Biol. 2004, 20, 337–366. [Google Scholar] [CrossRef] [PubMed]
- Segditsas, S.; Tomlinson, I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene 2006, 25, 7531–7537. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.O.; McKenney, R.J.; Mawatari, S.Y.; Mizuguchi, M.; Mikami, A.; Iwaki, T.; Goldman, J.E.; Canoll, P.; Vallee, R.B. Expression patterns of LIS1, dynein and their interaction partners dynactin, NudE, NudEL and NudC in human gliomas suggest roles in invasion and proliferation. Acta Neuropathol. 2007, 113, 591–599. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, W.-H.; Wang, Y.-L. Microtubules stabilize cell polarity by localizing rear signals. Proc. Natl. Acad. Sci. USA 2014, 111, 16383–16388. [Google Scholar] [CrossRef]
- Bie, L.; Zhao, G.; Wang, Y.P.; Zhang, B. Kinesin family member 2C (KIF2C/MCAK) is a novel marker for prognosis in human gliomas. Clin. Neurol. Neurosurg. 2012, 114, 356–360. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, L.; Li, D.; Deng, J.; Zhao, Z.; He, S.; Zhang, Y.; Tu, Y. Kinesin family member 14 is a candidate prognostic marker for outcome of glioma patients. Cancer Epidemiol. 2013, 37, 79–84. [Google Scholar] [CrossRef]
- Venere, M.; Horbinski, C.; Crish, J.F.; Jin, X.; Vasanji, A.; Major, J.; Burrows, A.C.; Chang, C.; Prokop, J.; Wu, Q.; et al. The mitotic kinesin KIF11 is a driver of invasion, proliferation, and self-renewal in glioblastoma. Sci. Transl. Med. 2015, 7, 304ra143. [Google Scholar] [CrossRef]
- Afghani, N.; Mehta, T.; Wang, J.; Tang, N.; Skalli, O.; Quick, Q.A. Microtubule actin cross-linking factor 1, a novel target in glioblastoma. Int. J. Oncol. 2017, 50, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Arden, J.D.; Lavik, K.I.; Rubinic, K.A.; Chiaia, N.; Khuder, S.A.; Howard, M.J.; Nestor-Kalinoski, A.L.; Alberts, A.S.; Eisenmann, K.M. Small-molecule agonists of mammalian Diaphanous-related (mDia) formins reveal an effective glioblastoma anti-invasion strategy. Mol. Biol. Cell 2015, 26, 3704–3718. [Google Scholar] [CrossRef]
- Hiratsuka, M.; Inoue, T.; Toda, T.; Kimura, N.; Shirayoshi, Y.; Kamitani, H.; Watanabe, T.; Ohama, E.; Tahimic, C.G.T.; Kurimasa, A.; et al. Proteomics-based identification of differentially expressed genes in human gliomas: Down-regulation of SIRT2 gene. Biochem. Biophys. Res. Commun. 2003, 309, 558–566. [Google Scholar] [CrossRef]
- Hoelzinger, D.B.; Mariani, L.; Weis, J.; Woyke, T.; Berens, T.J.; McDonough, W.; Sloan, A.; Coons, S.W.; Berens, M.E. Gene Expression Profile of Glioblastoma Multiforme Invasive Phenotype Points to New Therapeutic Targets. Neoplasia 2005, 7, 7–16. [Google Scholar] [CrossRef]
- Geiger, K.D.; Stoldt, P.; Schlote, W.; Derouiche, A. Ezrin Immunoreactivity Is Associated with Increasing Malignancy of Astrocytic Tumors but Is Absent in Oligodendrogliomas. Am. J. Pathol. 2000, 157, 1785–1793. [Google Scholar] [CrossRef]
- Tynninen, O.; Carpen, O.; Jaaskelainen, J.; Paavonen, T.; Paetau, A. Ezrin expression in tissue microarray of primary and recurrent gliomas. Neuropathol. Appl. Neurobiol. 2004, 30, 472–477. [Google Scholar] [CrossRef]
- Belot, N.; Rorive, S.; Doyen, I.; Lefranc, F.; Bruyneel, E.; Dedecker, R.; Micik, S.; Brotchi, J.; Decaestecker, C.; Salmon, I.; et al. Molecular characterization of cell substratum attachments in human glial tumors relates to prognostic features. Glia 2001, 36, 375–390. [Google Scholar]
- Adams, J.C. Roles of fascin in cell adhesion and motility. Curr. Opin. Cell Biol. 2004, 16, 590–596. [Google Scholar] [CrossRef]
- Peraud, A.; Mondal, S.; Hawkins, C.; Mastronardi, M.; Bailey, K.; Rutka, J.T. Expression of fascin, an actin-bundling protein, in astrocytomas of varying grades. Brain Tumor Pathol. 2003, 20, 53–58. [Google Scholar] [CrossRef]
- Ng, H.K.; Ko, H.C.W.; Tse, C.C.H. Immunohistochemical and Ultrastructural Studies of Oligodendrogliomas Revealed Features of Neuronal Differentiation. Int. J. Surg. Pathol. 1994, 2, 47–55. [Google Scholar] [CrossRef]
- Wharton, S.B.; Chan, K.K.; Whittle, I.R. Microtubule-associated protein 2 (MAP-2) is expressed in low and high grade diffuse astrocytomas. J. Clin. Neurosci. 2002, 9, 165–169. [Google Scholar] [CrossRef]
- Zhang, Y.; Ni, S.; Huang, B.; Wang, L.; Zhang, X.; Li, X.; Wang, H.; Liu, S.; Hao, A.; Li, X. Overexpression of SCLIP promotes growth and motility in glioblastoma cells. Cancer Biol. Ther. 2015, 16, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Jin, X.; Jung, J.E.; Beck, S.; Kim, H. Cell surface Nestin is a biomarker for glioma stem cells. Biochem. Biophys. Res. Commun. 2013, 433, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Strojnik, T.; Røsland, G.V.; Sakariassen, P.O.; Kavalar, R.; Lah, T. Neural stem cell markers, nestin and musashi proteins, in the progression of human glioma: Correlation of nestin with prognosis of patient survival. Surg. Neurol. 2007, 68, 133–143. [Google Scholar] [CrossRef]
- Dehghani, F.; Schachenmayr, W.; Laun, A.; Korf, H.W. Prognostic implication of histopathological, immunohistochemical and clinical features of oligodendrogliomas: A study of 89 cases. Acta Neuropathol. 1998, 95, 493–504. [Google Scholar] [CrossRef]
- Forget, M.-A.; Desrosiers, R.R.; Del, M.; Moumdjian, R.; Shedid, D.; Berthelet, F.; Béliveau, R. The expression of rho proteins decreases with human brain tumor progression: Potential tumor markers. Clin. Exp. Metastasis 2002, 19, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowska, A.; Didier, S.; Fortin, S.; Chuang, Y.; White, T.; Berens, M.E.; Rushing, E.; Eschbacher, J.; Tran, N.L.; Chan, A.; et al. The small GTPase RhoG mediates glioblastoma cell invasion. Mol. Cancer 2012, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Rutka, J.T.; Muller, M.; Hubbard, S.L.; Forsdike, J.; Dirks, P.B.; Jung, S.; Tsugu, A.; Ivanchuk, S.; Costello, P.; Mondal, S.; et al. Astrocytoma Adhesion to Extracellular Matrix: Functional Significance of Integrin and Focal Adhesion Kinase Expression. J. Neuropathol. Exp. Neurol. 1999, 58, 198–209. [Google Scholar] [CrossRef]
- Natarajan, M.; Hecker, T.P.; Gladson, C.L. FAK signaling in anaplastic astrocytoma and glioblastoma tumors. Cancer J. 2003, 9, 126–133. [Google Scholar] [CrossRef]
- Mariani, L.; Beaudry, C.; McDonough, W.S.; Hoelzinger, D.B.; Demuth, T.; Ross, K.R.; Berens, T.; Coons, S.W.; Watts, G.; Trent, J.M.; et al. Glioma cell motility is associated with reduced transcription of proapoptotic and proliferation genes: A cDNA microarray analysis. J. Neurooncol. 2001, 53, 161–176. [Google Scholar] [CrossRef]
- Giese, A.; Loo, A.; Norman, S.A.; Treasurywala, S.; Berens, M.E. Contrasting migratory response of astrocytoma cells to tenascin mediated by different integrins. J. Cell Sci. 1996, 109, 2161–2168. [Google Scholar]
- Plopper, G.E.; McNamee, H.P.; Dike, L.E.; Bojanowski, K.; Ingber, D.E. Convergence of integrin and growth factor receptor signaling pathways within the focal adhesion complex. Mol. Biol. Cell 1995, 6, 1349–1365. [Google Scholar] [CrossRef]
- Wenk, M.B.; Midwood, K.S.; Schwarzbauer, J.E. Tenascin-C suppresses Rho activation. J. Cell Biol. 2000, 150, 913–920. [Google Scholar] [CrossRef]
- Hirata, E.; Arakawa, Y.; Shirahata, M.; Yamaguchi, M.; Kishi, Y.; Okada, T.; Takahashi, J.A.; Matsuda, M.; Hashimoto, N. Endogenous tenascin-C enhances glioblastoma invasion with reactive change of surrounding brain tissue. Cancer Sci. 2009, 100, 1451–1459. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Wick, W.; Wild-Bode, C.; Aulwurm, S.; Dichgans, J.; Weller, M. Transforming Growth Factors β1 (TGF-β1) and TGF-β2 Promote Glioma Cell Migration via Up-Regulation of αVβ3 Integrin Expression. Biochem. Biophys. Res. Commun. 2000, 268, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Tonn, J.C.; Wunderlich, S.; Kerkau, S.; Klein, C.E.; Roosen, K. Invasive behaviour of human gliomas is mediated by interindividually different integrin patterns. Anticancer Res. 1998, 18, 2599–2605. [Google Scholar] [PubMed]
- Rooprai, H.K.; Vanmeter, T.; Panou, C.; Schnüll, S.; Trillo-Pazos, G.; Davies, D.; Pilkington, G.J. The role of integrin receptors in aspects of glioma invasion in vitro. Int. J. Dev. Neurosci. 1999, 17, 613–623. [Google Scholar] [CrossRef]
- Gritsenko, P.G.; Friedl, P. Adaptive adhesion systems mediate glioma cell invasion in complex environments. J. Cell Sci. 2018, 131, jcs216382. [Google Scholar] [CrossRef]
- Serres, E.; Debarbieux, F.; Stanchi, F.; Maggiorella, L.; Grall, D.; Turchi, L.; Burel-Vandenbos, F.; Figarella-Branger, D.; Virolle, T.; Rougon, G.; et al. Fibronectin expression in glioblastomas promotes cell cohesion, collective invasion of basement membrane in vitro and orthotopic tumor growth in mice. Oncogene 2014, 33, 3451–3462. [Google Scholar] [CrossRef]
- Demuth, T.; Berens, M.E. Molecular mechanisms of glioma cell migration and invasion. J. Neurooncol. 2004, 70, 217–228. [Google Scholar] [CrossRef]
- Giese, A.; Kluwe, L.; Laube, B.; Meissner, H.; Berens, M.E.; Westphal, M. Migration of human glioma cells on myelin. Neurosurgery 1996, 38, 755–764. [Google Scholar] [CrossRef]
- Lash, L.L.; Wallar, B.J.; Turner, J.D.; Vroegop, S.M.; Kilkuskie, R.E.; Kitchen-Goosen, S.M.; Xu, H.E.; Alberts, A.S. Small-Molecule Intramimics of Formin Autoinhibition: A New Strategy to Target the Cytoskeletal Remodeling Machinery in Cancer Cells. Cancer Res. 2013, 73, 6793–6803. [Google Scholar] [CrossRef]
- Wiranowska, M.; Ladd, S.; Smith, S.R.; Gottschall, P.E. CD44 adhesion molecule and neuro-glial proteoglycan NG2 as invasive markers of glioma. Brain Cell Biol. 2006, 35, 159–172. [Google Scholar] [CrossRef]
- Akiyama, Y.; Jung, S.; Salhia, B.; Lee, S.; Hubbard, S.; Taylor, M.; Mainprize, T.; Akaishi, K.; van Furth, W.; Rutka, J.T. Hyaluronate Receptors Mediating Glioma Cell Migration and Proliferation. J. Neurooncol. 2001, 53, 115–127. [Google Scholar] [CrossRef]
- Toole, B.P. Hyaluronan: From extracellular glue to pericellular cue. Nat. Rev. Cancer 2004, 4, 528–539. [Google Scholar] [CrossRef]
- Merzak, A.; Koocheckpour, S.; Pilkington, G.J. CD44 mediates human glioma cell adhesion and invasion in vitro. Cancer Res. 1994, 54, 3988–3992. [Google Scholar]
- Park, J.B.; Kwak, H.-J.; Lee, S.-H. Role of hyaluronan in glioma invasion. Cell Adh. Migr. 2008, 2, 202–207. [Google Scholar] [CrossRef]
- Karousou, E.; Misra, S.; Ghatak, S.; Dobra, K.; Götte, M.; Vigetti, D.; Passi, A.; Karamanos, N.K.; Skandalis, S.S. Roles and targeting of the HAS/hyaluronan/CD44 molecular system in cancer. Matrix Biol. 2017, 59, 3–22. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.W. Hyaluronan-mediated CD44 activation of RhoGTPase signaling and cytoskeleton function promotes tumor progression. Semin. Cancer Biol. 2008, 18, 251–259. [Google Scholar] [CrossRef]
- Park, D.; Kim, Y.; Kim, H.; Kim, K.; Lee, Y.-S.; Choe, J.; Hahn, J.-H.; Lee, H.; Jeon, J.; Choi, C.; et al. Hyaluronic acid promotes angiogenesis by inducing RHAMM-TGFβ receptor interaction via CD44-PKCδ. Mol. Cells 2012, 33, 563–574. [Google Scholar] [CrossRef]
- Murai, T.; Miyazaki, Y.; Nishinakamura, H.; Sugahara, K.N.; Miyauchi, T.; Sako, Y.; Yanagida, T.; Miyasaka, M. Engagement of CD44 Promotes Rac Activation and CD44 Cleavage during Tumor Cell Migration. J. Biol. Chem. 2004, 279, 4541–4550. [Google Scholar] [CrossRef]
- Klank, R.L.; Decker Grunke, S.A.; Bangasser, B.L.; Forster, C.L.; Price, M.A.; Odde, T.J.; SantaCruz, K.S.; Rosenfeld, S.S.; Canoll, P.; Turley, E.A.; et al. Biphasic Dependence of Glioma Survival and Cell Migration on CD44 Expression Level. Cell Rep. 2017, 18, 23–31. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
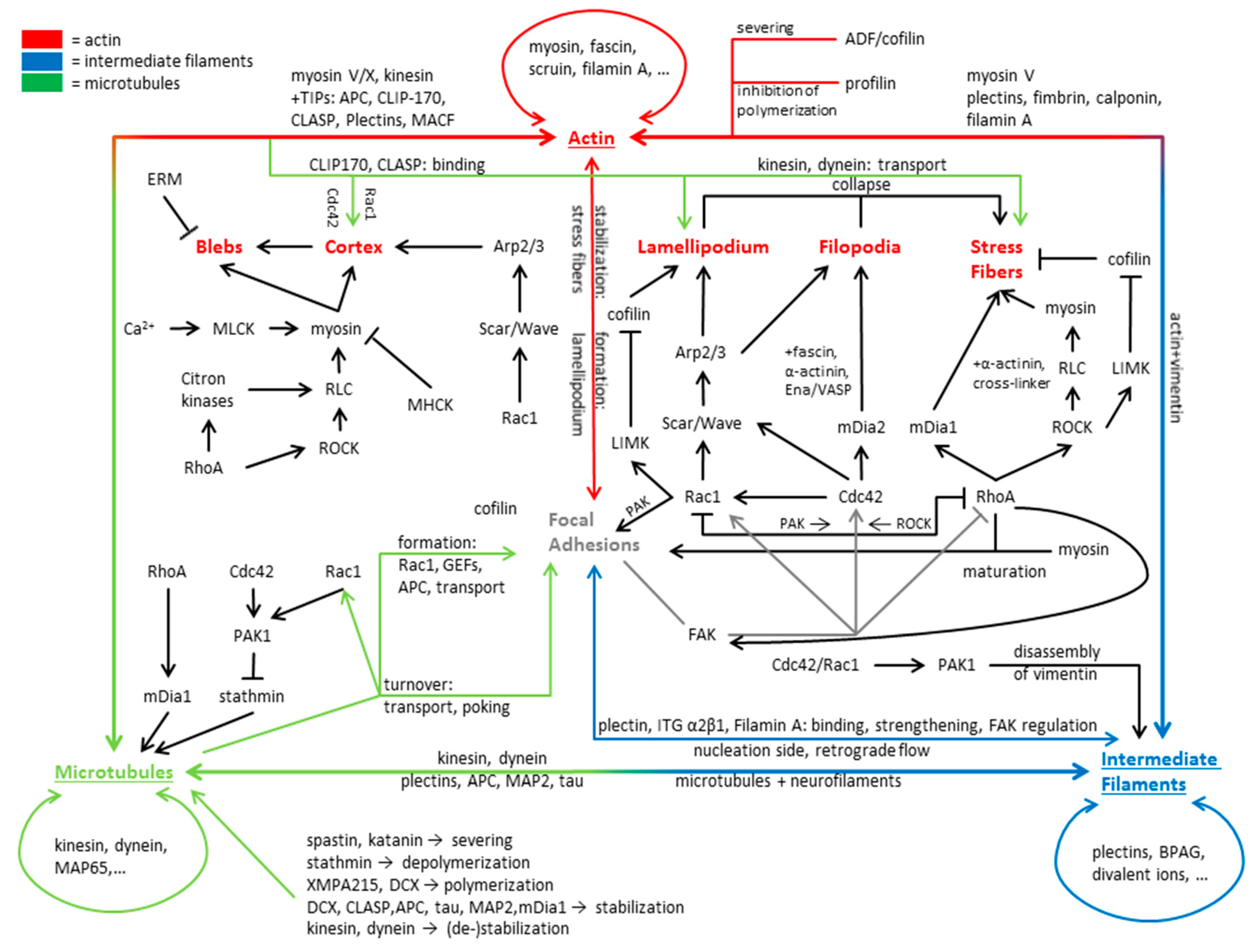{kind=link}
{kind=link}
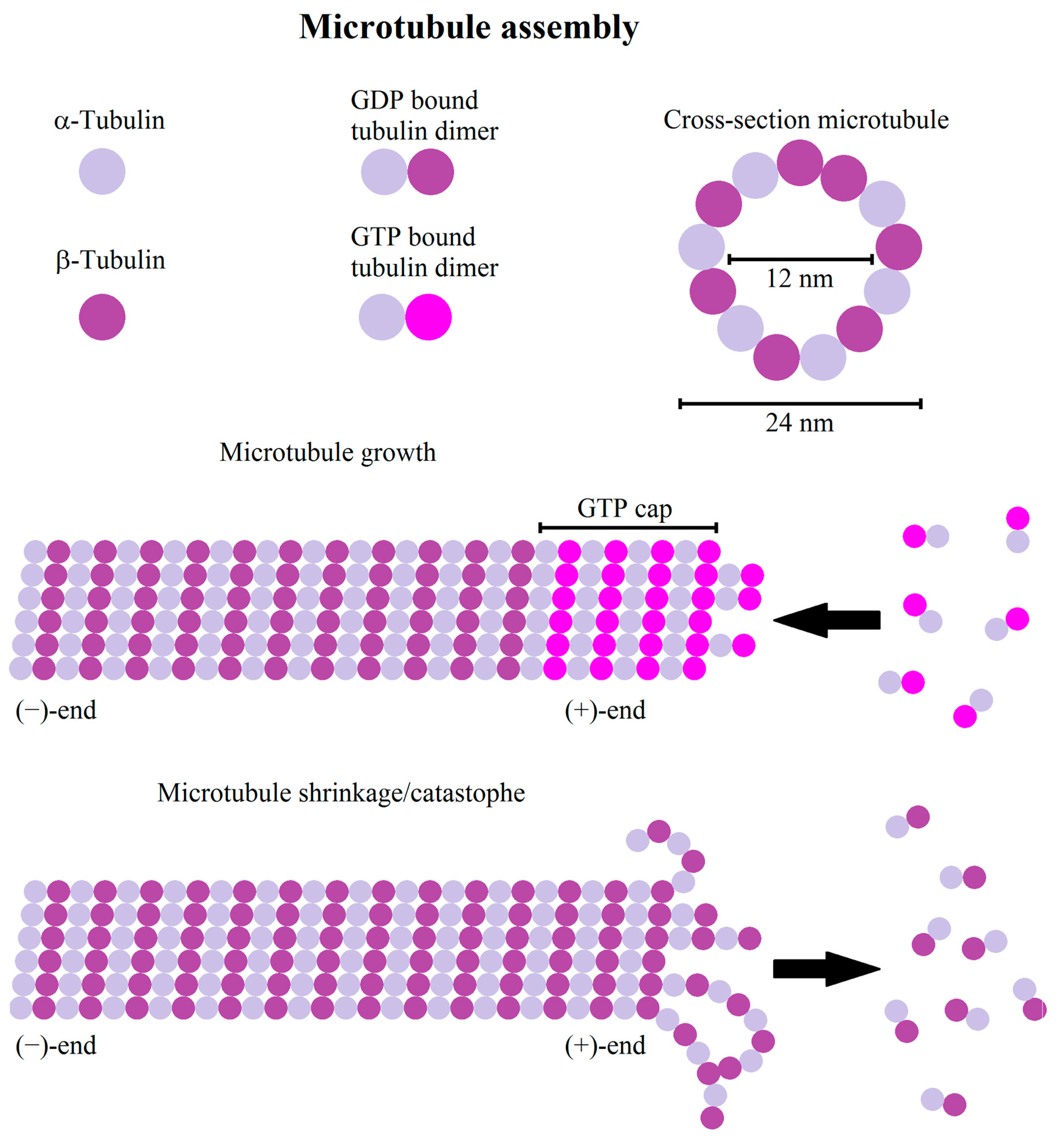{kind=link}
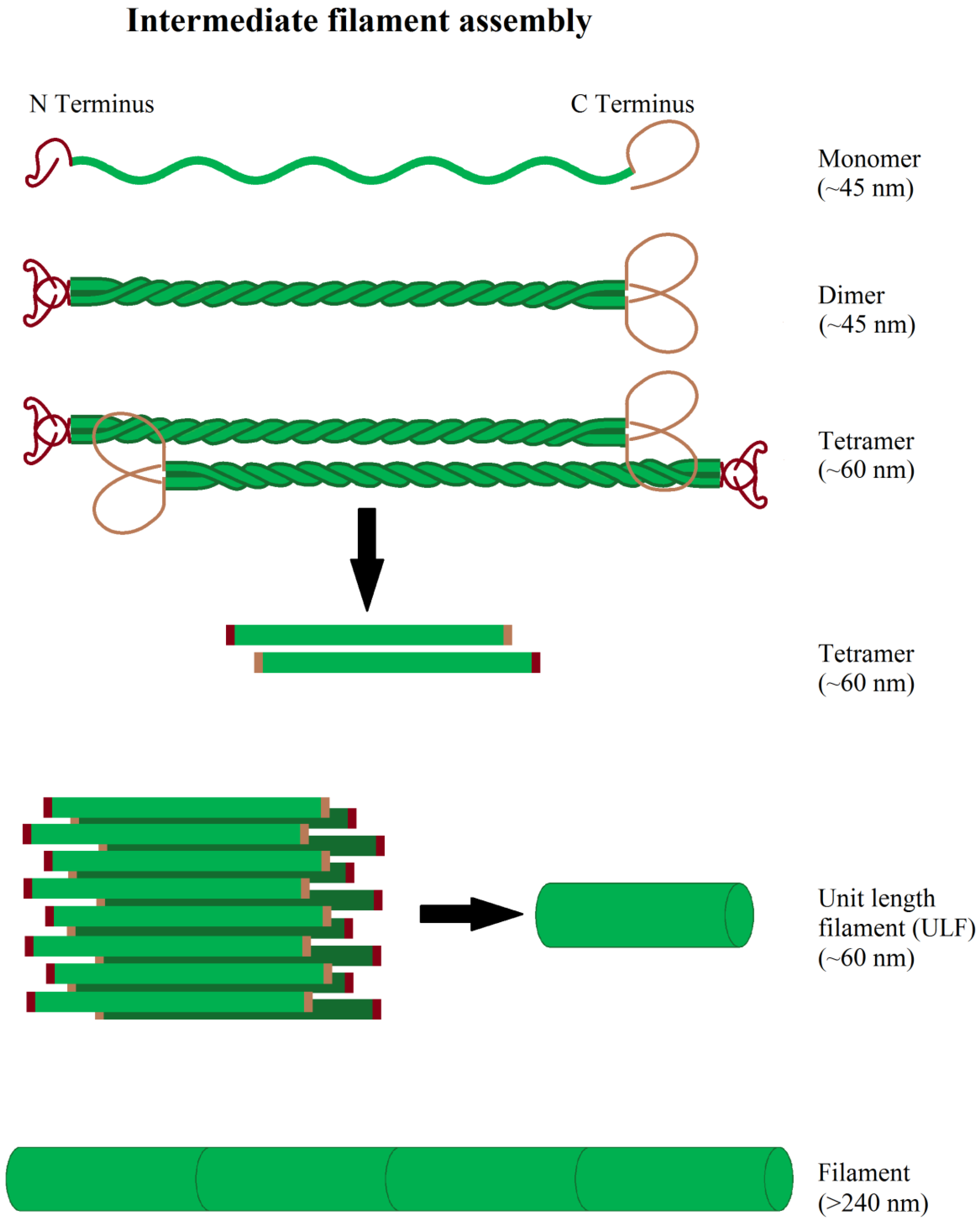{kind=link}
| Actin Associated Proteins | Function |
|---|---|
| Arp2/3 | Polymerization factor |
| Ena/VASP | Polymerization factor, anti-capping function |
| FMNL2 | Polymerization factor |
| mDia1 | Polymerization factor |
| mDia2 | Polymerization factor |
| Profilin | Inhibits actin polymerization |
| ADF/Cofilin | Actin severing |
| Arpin | Inhibits Arp2/3 |
| Myosin II | Cell/actin contractility, cross linker |
| RLC | Activates myosin II |
| MLCK | Activates myosin II |
| MHCK | Inhibits myosin II activity |
| PKC | Inhibits myosin II activity |
| CKII | Inhibits myosin II activity |
| Scruin | Cross linker |
| Fascin | Cross linker |
| α-actinin | Cross linker |
| Filamin | Cross linker |
| Fimbrin | Cross linker |
| Paladin | Cross linker |
| Ezrin | Membrane-cortex linker |
| Radixin | Membrane-cortex linker |
| Moesin | Membrane-cortex linker |
| Cdc42 | Signaling molecule, activates mDia2, WAVE, N-WASP |
| Rac1 | Signaling molecule, activates WASP/WAVE, arpin |
| RhoA | Signaling molecule, activates ROCK, mDia1, LIMK |
| ROCK | Signaling molecule, activates myosin II |
| WASP/WAVE | Signaling molecule, activates Arp2/3 |
| N-WASP | Signaling molecule, activates Arp2/3 |
| LIMK | Signaling molecule, inhibits ADF/cofilin |
| Microtubule Associated Proteins | Function |
|---|---|
| Stathmin | Depolymerization |
| XMPA215 | Polymerization factor |
| EB | Polymerization, Stabilization, Recruitment of proteins |
| DCX | Polymerization factor, Stabilization |
| CLASP | Stabilization |
| APC | Stabilization |
| mDia1 | Stabilization |
| mDia2 | Stabilization |
| Tau | Stabilization |
| MAP2 | Stabilization |
| Spastin | Microtubule severing |
| Katanin | Microtubule severing |
| Kinesin | Cargo transport |
| Dynein | Cargo transport |
| MACF1 | Actin-Microtubule interactions |
| Cdc42 | Signaling molecule, activates PAK |
| Rac1 | Signaling molecule, activates PAK |
| RhoA | Signaling molecule, mDia1 |
| PAK | Signaling molecule, inhibits stathmin |
| Intermediate Filament Associated Proteins | Function |
|---|---|
| LINC | Nucleus—intermediate filament linkage |
| Plakins | Linkage to adhesion sites |
| Plectin | Intermediate filament—integrin linkage |
| Kinesin * | Filament transport |
| Dynein * | Filament transport |
| Myosin * | Filament transport |
| Function | Expression/Activity | Sources | |
|---|---|---|---|
| Actin associated proteins | |||
| Arp2/3 | polymerization | high | [521] |
| mDia2 (formin family) | polymerization | high | [572] |
| Profilin | polymerization | low | [573] |
| Moesin/Ezrin | membrane to actin cortex linkage | high | [574,575,576] |
| Cortactin | actin cross-linker | high | [524] |
| Filamin | actin cross-linker | high | [548] |
| α-actinin | actin cross-linker | high | [577] |
| Fascin | actin cross-linker | high | [578,579] |
| Microtubule associated proteins | |||
| MAP2 | stabilization | high | [580,581] |
| Sclip (Stathmin family) | destabilization | high | [582] |
| Spastin | destabilization | high | [557] |
| MACF1 | microtubule-actin linkage | high | [571] |
| Dynein | cargo transport | unchanged | [566] |
| Kinesin-5, KiF2C, KiF14 | cargo transport | high | [568,569,570] |
| Β-III, β-IV, γ tubulin | microtubule formation and anchorage | high | [548,549,550] |
| Intermediate filaments | |||
| Vimentin/Nestin | cytoskeletal meshwork | high | [538,539,545,546,583,584,585] |
| GFAP | cytoskeletal meshwork | high | [536] |
| α-Internexin | cytoskeletal meshwork | high | [532] |
| Signaling molecules | |||
| RhoA | contractility | high/low | [499,586] |
| RhoB | contractility | low | [586] |
| RhoG | contractility, protrusion formation | high | [587] |
| Rac1 | protrusion formation | high | [499] |
| FAK | protrusion formation, adhesion turnover | high | [504,588,589] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hohmann, T.; Dehghani, F. The Cytoskeleton—A Complex Interacting Meshwork. Cells 2019, 8, 362. https://doi.org/10.3390/cells8040362
Hohmann T, Dehghani F. The Cytoskeleton—A Complex Interacting Meshwork. Cells. 2019; 8(4):362. https://doi.org/10.3390/cells8040362
Chicago/Turabian StyleHohmann, Tim, and Faramarz Dehghani. 2019. "The Cytoskeleton—A Complex Interacting Meshwork" Cells 8, no. 4: 362. https://doi.org/10.3390/cells8040362
APA StyleHohmann, T., & Dehghani, F. (2019). The Cytoskeleton—A Complex Interacting Meshwork. Cells, 8(4), 362. https://doi.org/10.3390/cells8040362