Recent Advances in the Clinical Targeting of Hedgehog/GLI Signaling in Cancer



Abstract
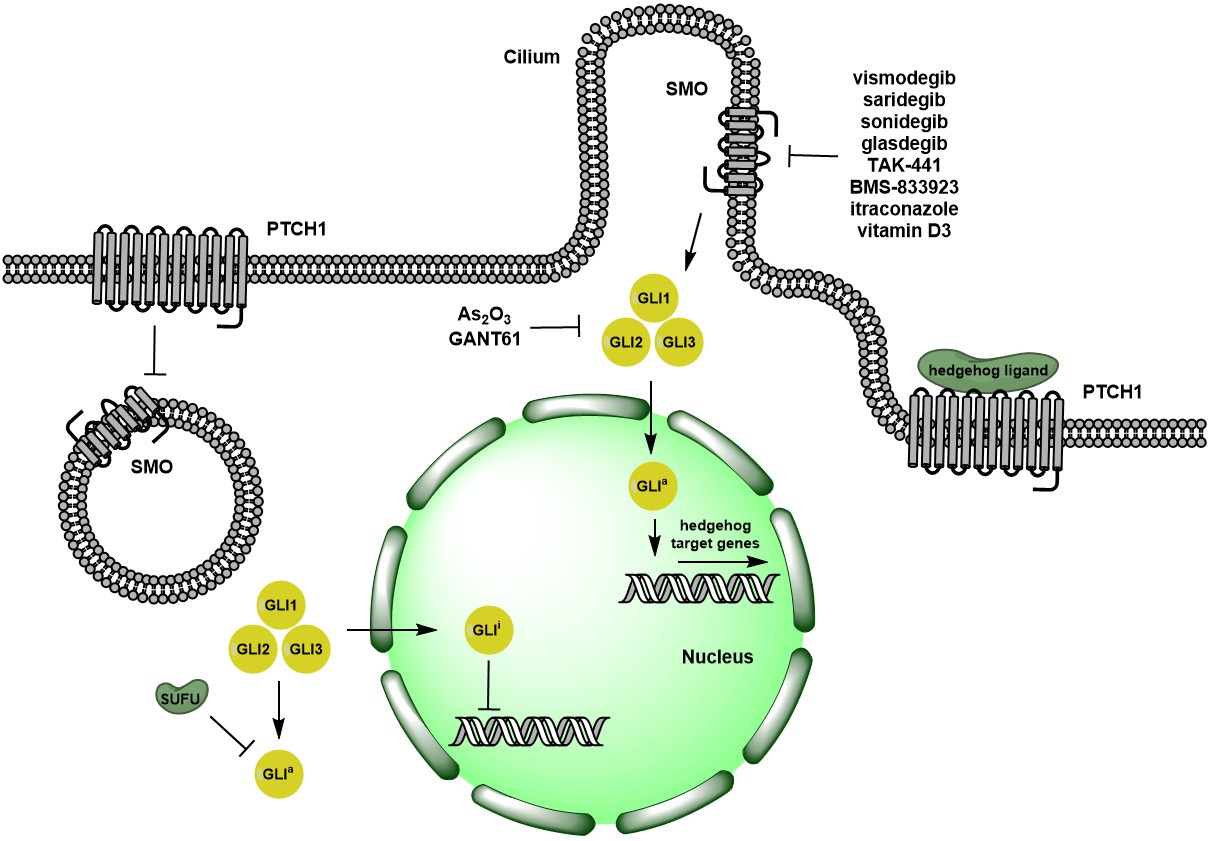
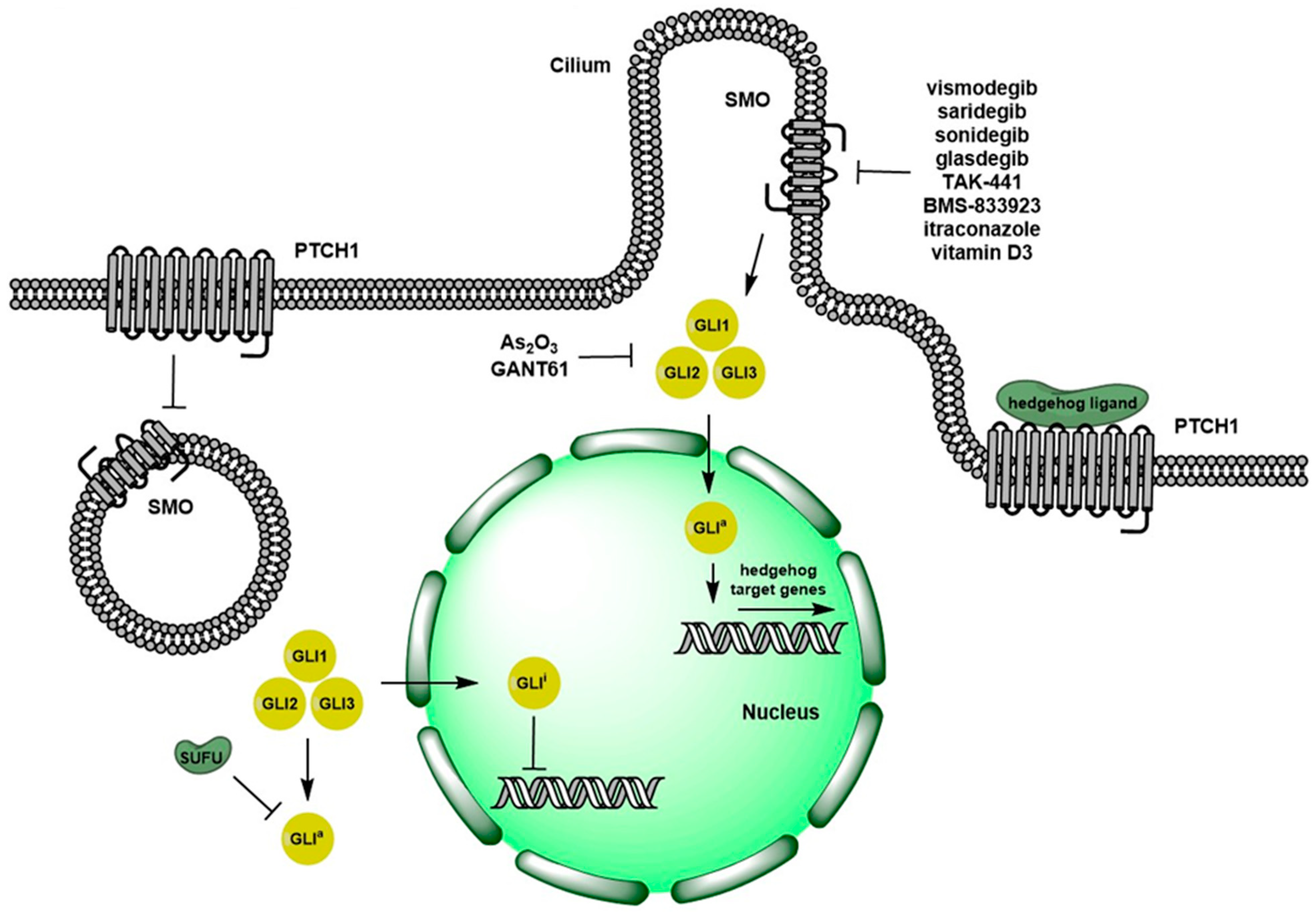
: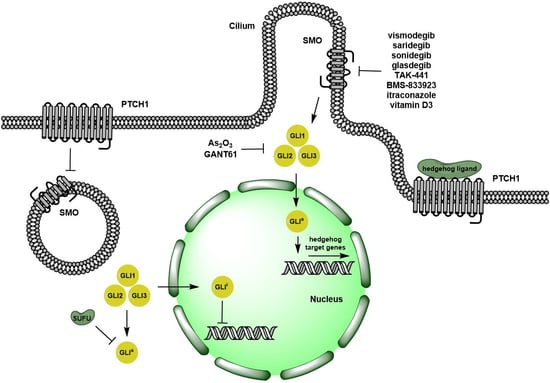
1. Overview of Hedgehog Signaling Pathway
2. Pre-Clinical Evidence on Targeting Hedgehog Signaling Pathways
2.1. Ligand-Dependent Hh Signaling Inhibition
2.2. Ligand-Independent Hh Signaling Inhibition
3. Clinical Trials Using Inhibitors of the Hedgehog Signaling Pathway
3.1. Advanced Solid Tumors
3.2. Small Cell Lung Cancer
3.3. Basal Cell Carcinoma
3.4. Medulloblastoma
3.5. Pancreatic Adenocarcinoma
3.6. Other Solid Tumors
3.7. Hematologic Malignancies
4. Conclusions and Future Directions
Funding
Acknowledgments
Conflicts of Interest
References
- Di Pasca Magliano, M.; Hebrok, M. Hedgehog signalling in cancer formation and maintenance. Nat. Rev. Cancer 2003, 3, 903–911. [Google Scholar] [CrossRef]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef]
- Alcedo, J.; Ayzenzon, M.; Von Ohlen, T.; Noll, M.; Hooper, J.E. The Drosophila smoothened gene encodes a seven-pass membrane protein, a putative receptor for the Hedgehog signal. Cell 1996, 86, 221–232. [Google Scholar] [CrossRef]
- Rubin, L.L.; de Sauvage, F.J. Targeting the Hedgehog pathway in cancer. Nat. Rev. Drug Discov. 2006, 5, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Teglund, S.; Toftgård, R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim. Et Biophys. Acta 2010, 1805, 181–208. [Google Scholar] [CrossRef]
- Zahreddine, H.A.; Culjkovic-Kraljacic, B.; Assouline, S.; Gendron, P.; Romeo, A.A.; Morris, S.J.; Cormack, G.; Jaquith, J.B.; Cerchietti, L.; Cocolakis, E.; et al. The sonic hedgehog factor GLI1 imparts drug resistance through inducible glucuronidation. Nature 2014, 511, 90–93. [Google Scholar] [CrossRef]
- McCann, C.K.; Growdon, W.B.; Kulkarni-Datar, K.; Curley, M.D.; Friel, A.M.; Proctor, J.L.; Sheikh, H.; Deyneko, I.; Ferguson, J.A.; Vathipadiekal, V.; et al. Inhibition of Hedgehog signaling antagonizes serous ovarian cancer growth in a primary xenograft model. PLoS ONE 2011, 6, e28077. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Tiet, T.D.; Hopyan, S.; Nadesan, P.; Gokgoz, N.; Poon, R.; Lin, A.C.; Yan, T.; Andrulis, I.L.; Alman, B.A.; Wunder, J.S. Constitutive hedgehog signaling in chondrosarcoma up-regulates tumor cell proliferation. Am. J. Pathol. 2006, 168, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Thayer, S.P.; Di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Fernández-del Castillo, C.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef] [Green Version]
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; Montes De Oca, R.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N.; et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003, 425, 846–851. [Google Scholar] [CrossRef]
- Pandol, S.; Edderkaoui, M.; Gukovsky, I.; Lugea, A.; Gukovskaya, A. Desmoplasia of pancreatic ductal adenocarcinoma. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2009, 7, S44–S47. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef]
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Singh, P.K.; Caffery, T.; Ouellette, M.M.; Hollingsworth, M.A. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 5995–6004. [Google Scholar] [CrossRef] [Green Version]
- Walter, K.; Omura, N.; Hong, S.-M.; Griffith, M.; Vincent, A.; Borges, M.; Goggins, M. Overexpression of smoothened activates the sonic hedgehog signaling pathway in pancreatic cancer-associated fibroblasts. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 1781–1789. [Google Scholar] [CrossRef]
- Brüggemann, L.W.; Queiroz, K.C.S.; Zamani, K.; van Straaten, A.; Spek, C.A.; Bijlsma, M.F. Assessing the efficacy of the hedgehog pathway inhibitor vitamin D3 in a murine xenograft model for pancreatic cancer. Cancer Biol. Ther. 2010, 10, 79–88. [Google Scholar] [CrossRef]
- Feldmann, G.; Fendrich, V.; McGovern, K.; Bedja, D.; Bisht, S.; Alvarez, H.; Koorstra, J.-B.M.; Habbe, N.; Karikari, C.; Mullendore, M.; et al. An orally bioavailable small-molecule inhibitor of Hedgehog signaling inhibits tumor initiation and metastasis in pancreatic cancer. Mol. Cancer Ther. 2008, 7, 2725–2735. [Google Scholar] [CrossRef] [Green Version]
- Munchhof, M.J.; Li, Q.; Shavnya, A.; Borzillo, G.V.; Boyden, T.L.; Jones, C.S.; LaGreca, S.D.; Martinez-Alsina, L.; Patel, N.; Pelletier, K.; et al. Discovery of PF-04449913, a Potent and Orally Bioavailable Inhibitor of Smoothened. Acs Med. Chem. Lett. 2012, 3, 106–111. [Google Scholar] [CrossRef]
- Fukushima, N.; Minami, Y.; Kakiuchi, S.; Kuwatsuka, Y.; Hayakawa, F.; Jamieson, C.; Kiyoi, H.; Naoe, T. Small-molecule Hedgehog inhibitor attenuates the leukemia-initiation potential of acute myeloid leukemia cells. Cancer Sci. 2016, 107, 1422–1429. [Google Scholar] [CrossRef]
- Jamieson, C.; Cortes, J.E.; Oehler, V.; Baccarani, M.; Kantarjian, H.M.; Papayannidis, C.; Rice, K.N.; Zhang, X.; Shaik, N.; Courtney, R.; et al. Phase 1 Dose-Escalation Study of PF-04449913, An Oral Hedgehog (Hh) Inhibitor, in Patients with Select Hematologic Malignancies. Blood 2011, 118, 424. [Google Scholar]
- Tremblay, M.R.; Lescarbeau, A.; Grogan, M.J.; Tan, E.; Lin, G.; Austad, B.C.; Yu, L.-C.; Behnke, M.L.; Nair, S.J.; Hagel, M.; et al. Discovery of a potent and orally active hedgehog pathway antagonist (IPI-926). J. Med. Chem. 2009, 52, 4400–4418. [Google Scholar] [CrossRef]
- Tremblay, M.; McGovern, K. Cyclopamine and Its Derivatives for Cancer Therapeutics. In Hedgehog Signaling Activation in Human Cancer and Its Clinical Implications; Xie, J., Ed.; Springer New York: New York, NY, USA, 2011; pp. 187–212. ISBN 978-1-4419-8435-7. [Google Scholar]
- Ohashi, T.; Oguro, Y.; Tanaka, T.; Shiokawa, Z.; Tanaka, Y.; Shibata, S.; Sato, Y.; Yamakawa, H.; Hattori, H.; Yamamoto, Y.; et al. Discovery of the investigational drug TAK-441, a pyrrolo3,2-cpyridine derivative, as a highly potent and orally active hedgehog signaling inhibitor: Modification of the core skeleton for improved solubility. Bioorganic Med. Chem. 2012, 20, 5507–5517. [Google Scholar] [CrossRef]
- Kogame, A.; Tagawa, Y.; Shibata, S.; Tojo, H.; Miyamoto, M.; Tohyama, K.; Kondo, T.; Prakash, S.; Shyu, W.C.; Asahi, S. Pharmacokinetic and pharmacodynamic modeling of hedgehog inhibitor TAK-441 for the inhibition of Gli1 messenger RNA expression and antitumor efficacy in xenografted tumor model mice. Drug Metab. Dispos. Biol. Fate Chem. 2013, 41, 727–734. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Heath, E.I.; Smith, D.C.; Rathkopf, D.; Blackford, A.L.; Danila, D.C.; King, S.; Frost, A.; Ajiboye, A.S.; Zhao, M.; et al. Repurposing itraconazole as a treatment for advanced prostate cancer: A noncomparative randomized phase II trial in men with metastatic castration-resistant prostate cancer. Oncology 2013, 18, 163–173. [Google Scholar] [CrossRef]
- Kim, J.; Tang, J.Y.; Gong, R.; Kim, J.; Lee, J.J.; Clemons, K.V.; Chong, C.R.; Chang, K.S.; Fereshteh, M.; Gardner, D.; et al. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell 2010, 17, 388–399. [Google Scholar] [CrossRef]
- Rudin, C.M.; Brahmer, J.R.; Juergens, R.A.; Hann, C.L.; Ettinger, D.S.; Sebree, R.; Smith, R.; Aftab, B.T.; Huang, P.; Liu, J.O. Phase 2 study of pemetrexed and itraconazole as second-line therapy for metastatic nonsquamous non-small-cell lung cancer. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2013, 8, 619–623. [Google Scholar] [CrossRef]
- Kim, J.; Lee, J.J.; Kim, J.; Gardner, D.; Beachy, P.A. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc. Natl. Acad. Sci. United States Am. 2010, 107, 13432–13437. [Google Scholar] [CrossRef] [Green Version]
- Beauchamp, E.M.; Ringer, L.; Bulut, G.; Sajwan, K.P.; Hall, M.D.; Lee, Y.-C.; Peaceman, D.; Ozdemirli, M.; Rodriguez, O.; MacDonald, T.J.; et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J. Clin. Investig. 2011, 121, 148–160. [Google Scholar] [CrossRef]
- Kim, J.; Aftab, B.T.; Tang, J.Y.; Kim, D.; Lee, A.H.; Rezaee, M.; Kim, J.; Chen, B.; King, E.M.; Borodovsky, A.; et al. Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell 2013, 23, 23–34. [Google Scholar] [CrossRef]
- Fu, J.; Rodova, M.; Roy, S.K.; Sharma, J.; Singh, K.P.; Srivastava, R.K.; Shankar, S. GANT-61 inhibits pancreatic cancer stem cell growth in vitro and in NOD/SCID/IL2R gamma null mice xenograft. Cancer Lett. 2013, 330, 22–32. [Google Scholar] [CrossRef]
- Li, J.; Cai, J.; Zhao, S.; Yao, K.; Sun, Y.; Li, Y.; Chen, L.; Li, R.; Zhai, X.; Zhang, J.; et al. GANT61, a GLI inhibitor, sensitizes glioma cells to the temozolomide treatment. J. Exp. Clin. Cancer Res. 2016, 35, 184. [Google Scholar] [CrossRef] [Green Version]
- Koike, Y.; Ohta, Y.; Saitoh, W.; Yamashita, T.; Kanomata, N.; Moriya, T.; Kurebayashi, J. Anti-cell growth and anti-cancer stem cell activities of the non-canonical hedgehog inhibitor GANT61 in triple-negative breast cancer cells. Breast Cancer (TokyoJpn. ) 2017, 24, 683–693. [Google Scholar] [CrossRef]
- Tong, W.; Qiu, L.; Qi, M.; Liu, J.; Hu, K.; Lin, W.; Huang, Y.; Fu, J. GANT-61 and GDC-0449 induce apoptosis of prostate cancer stem cells through a GLI-dependent mechanism. J. Cell. Biochem. 2018, 119, 3641–3652. [Google Scholar] [CrossRef]
- Rodon, J.; Tawbi, H.A.; Thomas, A.L.; Stoller, R.G.; Turtschi, C.P.; Baselga, J.; Sarantopoulos, J.; Mahalingam, D.; Shou, Y.; Moles, M.A.; et al. A phase I, multicenter, open-label, first-in-human, dose-escalation study of the oral smoothened inhibitor Sonidegib (LDE225) in patients with advanced solid tumors. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 2014, 20, 1900–1909. [Google Scholar] [CrossRef]
- Minami, H.; Ando, Y.; Ma, B.B.Y.; Hsiang Lee, J.; Momota, H.; Fujiwara, Y.; Li, L.; Fukino, K.; Ito, K.; Tajima, T.; et al. Phase I, multicenter, open-label, dose-escalation study of sonidegib in Asian patients with advanced solid tumors. Cancer Sci. 2016, 107, 1477–1483. [Google Scholar] [CrossRef]
- Jimeno, A.; Weiss, G.J.; Miller, W.H.; Gettinger, S.; Eigl, B.J.C.; Chang, A.L.S.; Dunbar, J.; Devens, S.; Faia, K.; Skliris, G.; et al. Phase I study of the Hedgehog pathway inhibitor IPI-926 in adult patients with solid tumors. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 2013, 19, 2766–2774. [Google Scholar] [CrossRef]
- LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; Chang, I.; Darbonne, W.C.; et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 2011, 17, 2502–2511. [Google Scholar] [CrossRef]
- Wagner, A.J.; Messersmith, W.A.; Shaik, M.N.; Li, S.; Zheng, X.; McLachlan, K.R.; Cesari, R.; Courtney, R.; Levin, W.J.; El-Khoueiry, A.B. A phase I study of PF-04449913, an oral hedgehog inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 1044–1051. [Google Scholar] [CrossRef]
- Goldman, J.; Eckhardt, S.G.; Borad, M.J.; Curtis, K.K.; Hidalgo, M.; Calvo, E.; Ryan, D.P.; Wirth, L.J.; Parikh, A.; Partyka, J.; et al. Phase I dose-escalation trial of the oral investigational Hedgehog signaling pathway inhibitor TAK-441 in patients with advanced solid tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 1002–1009. [Google Scholar] [CrossRef]
- Pietanza, M.C.; Litvak, A.M.; Varghese, A.M.; Krug, L.M.; Fleisher, M.; Teitcher, J.B.; Holodny, A.I.; Sima, C.S.; Woo, K.M.; Ng, K.K.; et al. A phase I trial of the Hedgehog inhibitor, sonidegib (LDE225), in combination with etoposide and cisplatin for the initial treatment of extensive stage small cell lung cancer. Lung Cancer (Amst. Neth.) 2016, 99, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Belani, C.P.; Dahlberg, S.E.; Rudin, C.M.; Fleisher, M.; Chen, H.X.; Takebe, N.; Velasco, M.R.; Tester, W.J.; Sturtz, K.; Hann, C.L.; et al. Vismodegib or cixutumumab in combination with standard chemotherapy for patients with extensive-stage small cell lung cancer: A trial of the ECOG-ACRIN Cancer Research Group (E1508). Cancer 2016, 122, 2371–2378. [Google Scholar] [CrossRef]
- von Hoff, D.D.; LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Yauch, R.L.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. New Engl. J. Med. 2009, 361, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. New Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef] [PubMed]
- Sekulic, A.; Migden, M.R.; Basset-Seguin, N.; Garbe, C.; Gesierich, A.; Lao, C.D.; Miller, C.; Mortier, L.; Murrell, D.F.; Hamid, O.; et al. Long-term safety and efficacy of vismodegib in patients with advanced basal cell carcinoma: final update of the pivotal ERIVANCE BCC study. BMC Cancer 2017, 17, 332. [Google Scholar] [CrossRef] [PubMed]
- Basset-Seguin, N.; Hauschild, A.; Grob, J.-J.; Kunstfeld, R.; Dréno, B.; Mortier, L.; Ascierto, P.A.; Licitra, L.; Dutriaux, C.; Thomas, L.; et al. Vismodegib in patients with advanced basal cell carcinoma (STEVIE): A pre-planned interim analysis of an international, open-label trial. Lancet. Oncol. 2015, 16, 729–736. [Google Scholar] [CrossRef]
- Basset-Séguin, N.; Hauschild, A.; Kunstfeld, R.; Grob, J.; Dréno, B.; Mortier, L.; Ascierto, P.A.; Licitra, L.; Dutriaux, C.; Thomas, L.; et al. Vismodegib in patients with advanced basal cell carcinoma: Primary analysis of STEVIE, an international, open-label trial. Eur. J. Cancer 2017, 86, 334–348. [Google Scholar] [CrossRef]
- Ally, M.S.; Aasi, S.; Wysong, A.; Teng, C.; Anderson, E.; Bailey-Healy, I.; Oro, A.; Kim, J.; Chang, A.L.; Tang, J.Y. An investigator-initiated open-label clinical trial of vismodegib as a neoadjuvant to surgery for high-risk basal cell carcinoma. J. Am. Acad. Dermatol. 2014, 71, 904–911.e1. [Google Scholar] [CrossRef]
- Sofen, H.; Gross, K.G.; Goldberg, L.H.; Sharata, H.; Hamilton, T.K.; Egbert, B.; Lyons, B.; Hou, J.; Caro, I. A phase II, multicenter, open-label, 3-cohort trial evaluating the efficacy and safety of vismodegib in operable basal cell carcinoma. J. Am. Acad. Dermatol. 2015, 73, 99–105.e1. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.Y.; Mackay-Wiggan, J.M.; Aszterbaum, M.; Yauch, R.L.; Lindgren, J.; Chang, K.; Coppola, C.; Chanana, A.M.; Marji, J.; Bickers, D.R.; et al. Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. New Engl. J. Med. 2012, 366, 2180–2188. [Google Scholar] [CrossRef]
- Tang, J.Y.; Ally, M.S.; Chanana, A.M.; Mackay-Wiggan, J.M.; Aszterbaum, M.; Lindgren, J.A.; Ulerio, G.; Rezaee, M.R.; Gildengorin, G.; Marji, J.; et al. Inhibition of the hedgehog pathway in patients with basal-cell nevus syndrome: final results from the multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. Oncol. 2016, 17, 1720–1731. [Google Scholar] [CrossRef]
- Dréno, B.; Kunstfeld, R.; Hauschild, A.; Fosko, S.; Zloty, D.; Labeille, B.; Grob, J.-J.; Puig, S.; Gilberg, F.; Bergström, D.; et al. Two intermittent vismodegib dosing regimens in patients with multiple basal-cell carcinomas (MIKIE): A randomised, regimen-controlled, double-blind, phase 2 trial. Lancet. Oncol. 2017, 18, 404–412. [Google Scholar] [CrossRef]
- Migden, M.R.; Guminski, A.; Gutzmer, R.; Dirix, L.; Lewis, K.D.; Combemale, P.; Herd, R.M.; Kudchadkar, R.; Trefzer, U.; Gogov, S.; et al. Treatment with two different doses of sonidegib in patients with locally advanced or metastatic basal cell carcinoma (BOLT): A multicentre, randomised, double-blind phase 2 trial. Lancet Oncol. 2015, 16, 716–728. [Google Scholar] [CrossRef]
- Kim, D.J.; Kim, J.; Spaunhurst, K.; Montoya, J.; Khodosh, R.; Chandra, K.; Fu, T.; Gilliam, A.; Molgo, M.; Beachy, P.A.; et al. Open-label, exploratory phase II trial of oral itraconazole for the treatment of basal cell carcinoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 32, 745–751. [Google Scholar] [CrossRef]
- Gajjar, A.; Stewart, C.F.; Ellison, D.W.; Kaste, S.; Kun, L.E.; Packer, R.J.; Goldman, S.; Chintagumpala, M.; Wallace, D.; Takebe, N.; et al. Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: A pediatric brain tumor consortium study. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 6305–6312. [Google Scholar] [CrossRef]
- Robinson, G.W.; Orr, B.A.; Wu, G.; Gururangan, S.; Lin, T.; Qaddoumi, I.; Packer, R.J.; Goldman, S.; Prados, M.D.; Desjardins, A.; et al. Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog-Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 2646–2654. [Google Scholar] [CrossRef]
- Kieran, M.W.; Chisholm, J.; Casanova, M.; Brandes, A.A.; Aerts, I.; Bouffet, E.; Bailey, S.; Leary, S.; MacDonald, T.J.; Mechinaud, F.; et al. Phase I study of oral sonidegib (LDE225) in pediatric brain and solid tumors and a phase II study in children and adults with relapsed medulloblastoma. Neuro-Oncol. 2017, 19, 1542–1552. [Google Scholar] [CrossRef]
- Kim, E.J.; Sahai, V.; Abel, E.V.; Griffith, K.A.; Greenson, J.K.; Takebe, N.; Khan, G.N.; Blau, J.L.; Craig, R.; Balis, U.G.; et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 5937–5945. [Google Scholar] [CrossRef]
- Catenacci, D.V.T.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef]
- Ko, A.H.; LoConte, N.; Tempero, M.A.; Walker, E.J.; Kate Kelley, R.; Lewis, S.; Chang, W.-C.; Kantoff, E.; Vannier, M.W.; Catenacci, D.V.; et al. A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas 2016, 45, 370–375. [Google Scholar] [CrossRef] [Green Version]
- Maughan, B.L.; Suzman, D.L.; Luber, B.; Wang, H.; Glavaris, S.; Hughes, R.; Sullivan, R.; Harb, R.; Boudadi, K.; Paller, C.; et al. Pharmacodynamic study of the oral hedgehog pathway inhibitor, vismodegib, in patients with metastatic castration-resistant prostate cancer. Cancer Chemother. Pharmacol. 2016, 78, 1297–1304. [Google Scholar] [CrossRef] [Green Version]
- Kaye, S.B.; Fehrenbacher, L.; Holloway, R.; Amit, A.; Karlan, B.; Slomovitz, B.; Sabbatini, P.; Fu, L.; Yauch, R.L.; Chang, I.; et al. A phase II, randomized, placebo-controlled study of vismodegib as maintenance therapy in patients with ovarian cancer in second or third complete remission. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 2012, 18, 6509–6518. [Google Scholar] [CrossRef]
- Berlin, J.; Bendell, J.C.; Hart, L.L.; Firdaus, I.; Gore, I.; Hermann, R.C.; Mulcahy, M.F.; Zalupski, M.M.; Mackey, H.M.; Yauch, R.L.; et al. A randomized phase II trial of vismodegib versus placebo with FOLFOX or FOLFIRI and bevacizumab in patients with previously untreated metastatic colorectal cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 258–267. [Google Scholar] [CrossRef]
- Cohen, D.J.; Christos, P.J.; Kindler, H.L.; Catenacci, D.V.T.; Bekaii-Saab, T.B.; Tahiri, S.; Janjigian, Y.Y.; Gibson, M.K.; Chan, E.; Rajdev, L.; et al. Vismodegib (V), a hedgehog (HH) pathway inhibitor, combined with FOLFOX for first-line therapy of patients (pts) with advanced gastric and gastroesophageal junction (GEJ) carcinoma: A New York Cancer Consortium led phase II randomized study. J. Clin. Oncol. 2013, 31, 4011. [Google Scholar] [CrossRef]
- Italiano, A.; Le Cesne, A.; Bellera, C.; Piperno-Neumann, S.; Duffaud, F.; Penel, N.; Cassier, P.; Domont, J.; Takebe, N.; Kind, M.; et al. GDC-0449 in patients with advanced chondrosarcomas: A French Sarcoma Group/US and French National Cancer Institute Single-Arm Phase II Collaborative Study. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. 2013, 24, 2922–2926. [Google Scholar] [CrossRef]
- Gounder, M.M.; Dickson, M.A.; Wu, N.; Ivy, S.P.; Carvajal, R.D.; D’Angelo, S.P.; Keohan, M.L.; Qin, L.-X.; Erinjeri, J.P.; Takebe, N.; et al. A first-in-human, phase Ib combination study to assess safety, pharmacokinetics (PK), and pharmacodynamics (PD) of a hedgehog inhibitor, GDC-0449, with a Notch inhibitor, RO4929097, in patients with advanced sarcoma. J. Clin. Oncol. 2012, 30, 10004. [Google Scholar] [CrossRef]
- Sloan, A.E.; Nock, C.J.; Kerstetter, A.; Supko, J.; Ye, X.; Barnholtz-Sloan, J.; Miller, R.; Rich, J.; Takebe, N.; Prados, M.; et al. Targeting glioma stem cells (GSC): A biomarker and phase II study of GDC-0449 in patients with recurrent glioblastoma multiforme (GBM). Neuro-Oncol. 2012, 14, vi101–vi105. [Google Scholar] [CrossRef]
- Minami, Y.; Minami, H.; Miyamoto, T.; Yoshimoto, G.; Kobayashi, Y.; Munakata, W.; Onishi, Y.; Kobayashi, M.; Ikuta, M.; Chan, G.; et al. Phase I study of glasdegib (PF-04449913), an oral smoothened inhibitor, in Japanese patients with select hematologic malignancies. Cancer Sci. 2017, 108, 1628–1633. [Google Scholar] [CrossRef]
- Martinelli, G.; Oehler, V.G.; Papayannidis, C.; Courtney, R.; Shaik, M.N.; Zhang, X.; O’Connell, A.; McLachlan, K.R.; Zheng, X.; Radich, J.; et al. Treatment with PF-04449913, an oral smoothened antagonist, in patients with myeloid malignancies: A phase 1 safety and pharmacokinetics study. Lancet Haematol. 2015, 2, e339–e346. [Google Scholar] [CrossRef]
- Lancet, J.E.; Komrokji, R.S.; Sweet, K.L.; Duong, V.H.; McGraw, K.L.; Zhang, L.; Nardelli, L.A.; Ma, Z.; Reich, R.R.; Padron, E.; et al. Phase 2 Trial of Smoothened (SMO) Inhibitor PF-04449913 (PF-04) in Refractory Myelodysplastic Syndromes (MDS). Blood 2016, 128, 3174. [Google Scholar]
- Cortes, J.E.; Heidel, F.H.; Heuser, M.; Fiedler, W.; Smith, B.D.; Robak, T.; Montesinos Fernandez, P.; Ma, W.W.; Shaik, M.N.; Zeremski, M.; et al. A Phase 2 Randomized Study of Low Dose Ara-C with or without Glasdegib (PF-04449913) in Untreated Patients with Acute Myeloid Leukemia or High-Risk Myelodysplastic Syndrome. Blood 2016, 128, 99. [Google Scholar]
- Sasaki, K.; Gotlib, J.R.; Mesa, R.A.; Newberry, K.J.; Ravandi, F.; Cortes, J.E.; Kelly, P.; Kutok, J.L.; Kantarjian, H.M.; Verstovsek, S. Phase II evaluation of IPI-926, an oral Hedgehog inhibitor, in patients with myelofibrosis. Leuk. Lymphoma 2015, 56, 2092–2097. [Google Scholar] [CrossRef]
- Houot, R.; Soussain, C.; Tilly, H.; Haioun, C.; Thieblemont, C.; Casasnovas, O.; Bouabdallah, K.; Morschhauser, F.; Le Gouill, S.; Salles, G.A.; et al. Inhibition of Hedgehog signaling for the treatment of lymphoma and CLL: a phase II study from the LYSA. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. 2016, 27, 1349–1350. [Google Scholar] [CrossRef]
- Savona, M.R.; Pollyea, D.A.; Stock, W.; Oehler, V.G.; Schroeder, M.A.; Lancet, J.; McCloskey, J.; Kantarjian, H.M.; Ma, W.W.; Shaik, M.N.; et al. Phase Ib Study of Glasdegib, a Hedgehog Pathway Inhibitor, in Combination with Standard Chemotherapy in Patients with AML or High-Risk MDS. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 2294–2303. [Google Scholar] [CrossRef] [Green Version]
- Iarrobino, A.; Messina, J.L.; Kudchadkar, R.; Sondak, V.K. Emergence of a squamous cell carcinoma phenotype following treatment of metastatic basal cell carcinoma with vismodegib. J. Am. Acad. Dermatol. 2013, 69, e33–e34. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Tsui, V.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M.; et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell 2015, 27, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Danial, C.; Sarin, K.Y.; Oro, A.E.; Chang, A.L.S. An Investigator-Initiated Open-Label Trial of Sonidegib in Advanced Basal Cell Carcinoma Patients Resistant to Vismodegib. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 1325–1329. [Google Scholar] [CrossRef]
- Buonamici, S.; Williams, J.; Morrissey, M.; Wang, A.; Guo, R.; Vattay, A.; Hsiao, K.; Yuan, J.; Green, J.; Ospina, B.; et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci. Transl. Med. 2010, 2, 51ra70. [Google Scholar] [CrossRef]
- Yauch, R.L.; Dijkgraaf, G.J.P.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef]
- Atwood, S.X.; Sarin, K.Y.; Whitson, R.J.; Li, J.R.; Kim, G.; Rezaee, M.; Ally, M.S.; Kim, J.; Yao, C.; Chang, A.L.S.; et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell 2015, 27, 342–353. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Zhang, Y.; Sun, B.; McMahon, A.P.; Wang, Y. Hedgehog Signaling: From Basic Biology to Cancer Therapy. Cell Chem. Biol. 2017, 24, 252–280. [Google Scholar] [CrossRef]
- Atwood, S.X.; Li, M.; Lee, A.; Tang, J.Y.; Oro, A.E. GLI activation by atypical protein kinase C ι/λ regulates the growth of basal cell carcinomas. Nature 2013, 494, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Gholamin, S.; Schubert, S.; Willardson, M.I.; Lee, A.; Bandopadhayay, P.; Bergthold, G.; Masoud, S.; Nguyen, B.; Vue, N.; et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat. Med. 2014, 20, 732–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.H.; Hempel, J.E.; Hao, J.; Frist, A.Y.; Williams, M.M.; Fleming, J.T.; Sulikowski, G.A.; Cooper, M.K.; Chiang, C.; Hong, C.C. An in vivo chemical genetic screen identifies phosphodiesterase 4 as a pharmacological target for hedgehog signaling inhibition. Cell Rep. 2015, 11, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Chu, Q.S.; Mahipal, A.; Schuler, M.; de Braud, F.G.M.; Dirix, L.; Rampersad, A.; Zhou, J.; Wu, Y.; Kalambakas, S.; Wen, P.Y. 445ODOSE-ESCALATION STUDY OF SONIDEGIB (LDE225) PLUS BUPARLISIB (BKM120) IN PATIENTS (PTS) WITH ADVANCED SOLID TUMORS. Ann. Oncol. 2014, 25, iv147–iv148. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Disease Type | Clinical Trial Phase (# Patients) | Dosing and Schedule | Adverse Events (G3-5 ≥ 10%) | Activities |
|---|---|---|---|---|
| Extensive stage SCLC | II (152) [42] | Cisplatin/etoposide q3w, with or without vismodegib 150 mg daily x 4 cycles | G3-5 neutropenia (53%), febrile neutropenia (12%) | ORR 56%, PFS 4.4 months, OS 9.8 months, similar to cisplatin, etoposide arm |
| I (15) [41] | Cisplatin/etoposide q3w, sonidegib 400 mg and 800 mg daily (MTD: 800 mg) | G3/4 anemia (33%), neutropenia (53%), CK elevation (13%), fatigue (13%), nausea (13%). DLT: nausea, febrile neutropenia | PR: 79% | |
| Advanced solid tumors | I (103) [35] | Sonidegib 100 to 3000 mg daily and 250 to 750 mg twice daily (MTD: 800 mg daily and 250 mg twice daily) | G3/4 nausea (25%), dysgeusia (29%), anorexia (29%), muscle spasms (32%), fatigue or asthenia (27%). DLT: G3/4 CK elevation (18%) | CR/PR: 37% for BCC, 33% for medulloblastoma; SD: 23% |
| I (45) [36] | Sonidegib 400 to 800 mg daily (RD: 400 mg daily) | G3/4 elevated LFT (15%). DLT: G3/4 CK elevation (24%), rhabdomyolysis (10%) | SD: 33% | |
| I (94) [37] | IPI-926 20 to 210 mg daily (RD: 160 mg daily) | G3/4 anemia (18%), elevated LFT (66%), fatigue (37%). DLT: G3 LFT elevation, fatigue, anorexia | ORR 29% in BCC cohort | |
| I (23) [39] | Glasdegib 80 to 640 mg daily (MTD: 320 mg daily) | DLT: G2 fatigue, hypotension and G3 nausea, vomiting, dehydration at 640 mg daily | SD: 35% | |
| I (34) [40] | TAK-441 50 to 1600 mg daily (MTD: 1600 mg daily) | G3/4: hyponatremia (12%), DLT: muscle spasms and fatigue | PR: 3%, SD: 21% | |
| Advanced or metastatic BCC or Basal-cell nevus syndrome or Resectable BCC | II (230) [53] | Sonidegib 200 mg vs. 800 mg daily | G3/4 elevated CK (13% in 800 mg arm) | ORR: 36% in 200 mg arm, 34% in 800 mg arm |
| II (41) [50,51] | Vismodegib 150 mg daily vs. placebo for 18 months | G3/4 weight loss (15%) | New surgically eligible BCC: 2 (vismodegib) vs. 29 (placebo) cases per year | |
| II (229) [52] | vismodegib 150 mg daily x 12 wks, then placebo x 24 wks, then 150 mg daily x 12 wks (arm A) vs. vismodegib 150 mg daily x 24 wks, then placebo x 24 wks, then 150 mg daily x 8 wks (arm B) | G3/4 muscle spasm (4% in arm A,11% in arm B) | Number of lesion reduction: 63% in arm A, 54% in arm B. | |
| II (1215) open-label safety trial [46,47] | Vismodegib 150 mg daily | ORR: 68% in locally advanced BCC, 37% in metastatic BCC | ||
| II (15), neoadjuvant [48] | Vismodegib 150 mg daily x 3-6 months before surgery | Surgical defect area reduction: 27% | ||
| II (24) before surgery [49] | Vismodegib 150 mg daily before surgery for cohort 1: 12 wks, cohort 2: 12 wks, then 24 wks observation, cohort 3: 8 wks on, 4 wks off, 8 wks on | Most frequent adverse events: muscle spasms (76%), alopecia (58%), and dysgeusia (50%). | Complete histologic clearance: 42% for cohort 1, 16% for cohort 2, 44% for cohort 3. | |
| II (29) open-label [54] | Itraconazole oral 200 mg twice daily x 1-month vs. 100 mg twice daily x 2.3 months | G4 congestive heart failure | cell proliferation reduction: 45%, Hh activity reduction: 65%, tumor size reduction: 24% | |
| Recurrent or refractory medulloblastoma | I/II (55) [57] | Adult: Sonidegib 800 mg daily, pediatric: 680 mg/m2 (RD) | In adult: G3/4 elevated CK (31%), elevated LFT (12%) | ORR: 50% in patients with activated Hh pathway |
| I (33) [55] | Vismodegib 85 to 170 mg/m2, revised to 150 and 300 mg daily (RD) | DLT: G3 γ-glutamyl transferase elevation, thrombocytopenia, G4 hypokalemia | One patient with SHH- subgroup had response | |
| II (43) [56] | Vismodegib 150 mg daily | G3/4 lymphopenia (30%), seizure (12%) | No response in non-SHH-subgroup. 15% in adult patients with SHH-subgroup, 41% with prolonged disease stabilization | |
| Advanced or metastatic pancreatic adenocarcinoma | I (15) [60] | FOLFIRINOX and IPI-926 130 to 160 mg daily (MTD: 130 mg daily) | G3/4 infection (13%), thrombocytopenia (13%), DLT: G3 elevated LFTs (20%) | ORR: 67% |
| Ib/II (113) [59] | Gemcitabine with or without vismodegib 150 mg daily | G3-5 neutropenia (28%), fatigue (13%), thrombocytopenia (11%) | Similar ORR, PFS, and OS | |
| I (25) [58] | Vismodegib 150 mg daily x 3 wks, then vismodegib + gemcitabine | G3 anemia (12%), LFT elevation (12%) | GLI1 inhibition: 96%, PTCH1 inhibition: 83%, ORR 22%, disease control rate: 65%. | |
| Metastatic castration-resistant prostate cancer | I (9) [61] | Vismodegib 150 mg daily x 4 wks | G3/4 anemia (11%), dehydration (11%), dyspnea (11%), pain (22%), pneumonia (11%), vomiting (11%) | GLI1 inhibition: 57% in tumor, 75% in normal skin. No response. Median PFS 1.9 months, OS 7.0 months |
| II (46) [25] | Itraconazole 200 mg vs. 600 mg daily | G3 (600 mg arm) hypokalemia (10%) | PSA PFS at 24 weeks (200 vs. 600 mg): 12% vs. 48%. | |
| Metastatic colorectal cancer | II (199) [63] | Vismodegib 150 mg daily or placebo with FOLFOX or FOLFIRI and bevacizumab | G3-5 neutropenia (22%), diarrhea (12%), nausea (10%), fatigue (18%), weight loss (10%), dehydration (12%) | median PFS HR 1.25 (p = 0.3), ORR 46% vs. 51% for vismodegib vs. placebo |
| Advanced chondrosarcoma | II (45) [65] | Vismodegib 150 mg daily | 6-month clinical benefit rate 25.6%. median PFS 3.5 months | |
| Advanced gastric or GEJ adenocarcinoma | II (124), [64] | FOLFOX with or without vismodegib 150 mg daily | G3-5 neutropenia (83%), neuropathy (32%), fatigue (25%), thrombosis (23%), anemia (17%), GI bleeding (13%), hypokalemia (17%), nausea (13%). | ORR 58%, median PFS 7.3 months, OS 11.5 months. |
| Lung adenocarcinoma | II (23), 2nd-line setting [27] | Pemetrexed with or without itraconazole 200 mg daily (stopped early due to 1st-line pemetrexed | G3/4 (itraconazole arm) lymphopenia (20%) | PFS at 3 months (itraconazole vs. no itraconazole): 67% vs. 29% |
| Hematologic malignancies or myelofibrosis | I (13), Japanese patients [68] | Glasdegib 25 to 100 mg daily (RD: 100 mg daily) | G3-4 thrombocytopenia (23%), hypokalemia (15%), DLT: none | AML: CR 8%, SD 31%; MDS: CR 8%, SD 16%. |
| I (47) [69] | Glasdegib 5 to 600 mg daily (MTD: 400 mg daily, RD: 200 mg daily or lower) | G3-4 anorexia (11%) DLT: G3 hypoxia, pleural effusion, peripheral edema | CML: partial cytogenetic response 20%; MDS/CMML: SD 57%; myelofibrosis: improvement 29%; AML: ORR 32%, SD 25% | |
| II (14) [72] | IPI-926 160 mg daily | G3-4 bilirubin elevation (21%) | <50% spleen size reduction: 86%; 64% had no response | |
| NHL and CLL | II (31) [73] | Vismodegib 150 mg daily | G3-5 29% | Indolent lymphoma: (17%) |
| AML and high risk MDS | II (35) [70] | Glasdegib 100 mg daily x 4 months, 200 mg daily allowed for SD | G3-4 infection (11%) | ORR: 6%; SD: 54%; median OS: 10.2 months |
| Ib (52) [74] | Glasdegib 100 or 200 mg daily with low-dose cytarabine (arm A) or decitabine (arm B) or cytarabine/daunorubicin (arm C). RD: 100 mg daily | G3-4 febrile neutropenia (A: 39%, C: 54%), fatigue (A: 22%), neutropenia (A: 22%, B: 57%), anemia (B: 29%), thrombocytopenia (A: 30%, B: 43%), pyrexia (C: 18%). No DLT in arms A, B, grade 4 neuropathy in arm C. | Arm A: CR 8.7% Arm B: CR 29% Arm C: CR 54% | |
| II (132), ineligible for intensive chemotherapy [71] | Glasdegib 100 mg daily and low-dose cytarabine versus low-dose cytarabine alone | Glasdegib arm: more frequent febrile neutropenia. | Glasdegib + cytarabine vs. cytarabine: CR 15% vs. 2%; median OS: 8.3 vs. 4.9 months |
| Agent | Tumor Types | Phase of Development | Clinicaltrials.Gov Identifier |
|---|---|---|---|
| BMS-833923 | Advanced or metastatic cancer | I | NCT00670189 |
| Extensive stage small cell lung cancer | I: carboplatin, etoposide and BMS-833923 | NCT00927875 | |
| Metastatic gastric, gastroesophageal, or esophageal adenocarcinomas | I: BMS-833923, cisplatin and capecitabine | NCT00909402 | |
| Itraconazole | Esophageal cancer | I | NCT02749513 |
| Prostate cancer | II | NCT01787331 | |
| Skin basal cell carcinoma | I | NCT02735356 | |
| Non-small cell lung cancer | II: itraconazole and chemotherapy | NCT03664115 | |
| Non-small cell lung cancer | I: neoadjuvant setting | NCT02357836 | |
| Basal cell carcinoma | II: SUBA-Itraconazole | NCT02354261 | |
| Various tumors | I: volasertib and itraconazole | NCT01772563 | |
| Glioblastoma | I: itraconazole and temozolomide | NCT02770378 | |
| Saridegib | Recurrent head and neck cancer | I: saridegib and cetuximab | NCT01255800 |
| Metastatic solid tumor | I | NCT00761696 | |
| Metastatic pancreatic cancer | I/II: saridegib and gemcitabine | NCT01130142 | |
| Advanced chondrosarcoma | II: saridegib or placebo | NCT01310816 | |
| Sonidegib | Advanced or metastatic hepatocellular carcinoma | I | NCT02151864 |
| Basal cell carcinoma | II: neoadjuvant sonidegib followed by surgery or imiquimod | NCT03534947 | |
| Extensive stage small cell lung cancer | I: sonidegib, etoposide and cisplatin | NCT01579929 | |
| Resectable pancreatic adenocarcinoma | I/II: sonidegib, gemcitabine, nab-paclitaxel in neoadjuvant setting | NCT01431794 | |
| Localized prostate cancer | I | NCT02111187 | |
| Multiple myeloma | II: sonidegib and lenalidomide | NCT02086552 | |
| Esophageal cancer | I: sonidegib and everolimus | NCT02138929 | |
| Advanced pancreatic cancer | I: sonidegib, fluorouracil, leucovorin, oxaliplatin, irinotecan | NCT01485744 | |
| Pancreatic cancer | I/II: sonidegib, gemcitabine, and nab-paclitaxel | NCT02358161 | |
| Advanced solid tumor | I | NCT01208831 | |
| Advanced solid tumor | I | NCT00880308 | |
| Solid tumors | I: sonidegib and paclitaxel | NCT01954355 | |
| Advanced solid tumors | I: sonidegib and BKM120 | NCT01576666 | |
| Myeloid malignancies | I: sonidegib with azacytidine or decitabine | NCT02129101 | |
| Advanced or metastatic basal cell carcinoma | II: sonidegib and buparlisib | NCT02303041 | |
| LEQ-506 | Advanced solid tumors | I | NCT01106508 |
| Taladegib | Advanced cancers | I | NCT01919398 |
| Esophageal cancer | I/II: Taladegib, paclitaxel, carboplatin, and radiation | NCT02530437 | |
| Advanced solid tumors | I | NCT02784795 | |
| Glasdegib | Hematologic malignancies | I | NCT00953758 |
| Solid tumors | I | NCT01286467 | |
| Acute myeloid leukemia | II | NCT01841333 | |
| Hematologic malignancies | I: with standard chemotherapy agents | NCT02038777 | |
| Acute myeloid leukemia | III: chemotherapy or azacytidine with or without glasdegib | NCT03416179 | |
| Glioblastoma | I/II: glasdegib and temozolomide | NCT03466450 | |
| TAK-441 | Advanced nonhematologic malignancies | I | NCT01204073 |
| Vismodegib | Metastatic pancreatic adenocarcinoma | II: vismodegib, gemcitabine and nab-paclitaxel | NCT01088815 |
| Solid and hematologic malignancies | II: Canadian profiling and targeted agent utilization trial | NCT03297606 | |
| Keratocystic odontogenic tumors | II | NCT02366312 | |
| Acute myeloid leukemia | II: ribavirin, vismodegib with or without decitabine | NCT02073838 | |
| Pancreatic adenocarcinoma | I: vismodegib and gemcitabine in neoadjuvant setting | NCT01713218 | |
| Basal cell nevus syndrome, Gorlin syndrome | II | NCT00957229 | |
| Breast cancer | II: neoadjuvant paclitaxel, epirubicin, cyclophosphamide with or without vismodegib | NCT02694224 | |
| Glioblastoma | I/II: Neuro Master Match | NCT03158389 | |
| Recurrent medulloblastoma | I | NCT00822458 | |
| Metastatic pancreatic cancer or solid tumors | I: vismodegib, erlotinib, and gemcitabine | NCT00878163 | |
| Advanced chondrosarcoma | Phase 1 | NCT01267955 | |
| Advanced basal cell skin cancer | I/II: pembrolizumab with or without vismodegib | NCT02690948 | |
| Advanced solid tumors | II: My Pathway | NCT02091141 | |
| Advanced head/neck basal cell carcinoma | II: vismodegib and radiation | NCT01835626 | |
| Advanced gastric cancer | II | NCT03052478 | |
| Multiple myeloma | I | NCT01330173 | |
| Solid tumors, lymphomas or multiple myeloma | II: MATCH | NCT02465060 | |
| Orbital and periocular basal cell carcinoma | IV | NCT02436408 | |
| Medulloblastoma | II: with radiation and chemotherapy | NCT01878617 | |
| Vitamin D3 | Basal cell carcinoma | I: with photodynamic therapy | NCT03483441 |
| Pancreatic cancer | III: high dose versus standard dose | NCT03472833 | |
| Acute myeloid leukemia | II: deferasirox, vitamin D3, and azacitidine | NCT02341495 | |
| Chronic lymphocytic leukemia, non-Hodgkin lymphoma | I | NCT02553447 | |
| Indolent lymphoma | III: rituximab with or without vitamin D3 | NCT03078855 | |
| Arsenic trioxide | High-grade glioma | I: with temozolomide and radiation therapy | NCT00720564 |
| Glioma | I: with radiation therapy | NCT00095771 | |
| Glioma | I: stereotactic radiotherapy | NCT00185861 | |
| Neuroblastoma and other childhood solid tumors | II | NCT00024258 | |
| Basal cell carcinoma | I/II | NCT01791894 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, H.; Paradise, B.D.; Ma, W.W.; Fernandez-Zapico, M.E. Recent Advances in the Clinical Targeting of Hedgehog/GLI Signaling in Cancer. Cells 2019, 8, 394. https://doi.org/10.3390/cells8050394
Xie H, Paradise BD, Ma WW, Fernandez-Zapico ME. Recent Advances in the Clinical Targeting of Hedgehog/GLI Signaling in Cancer. Cells. 2019; 8(5):394. https://doi.org/10.3390/cells8050394
Chicago/Turabian StyleXie, Hao, Brooke D. Paradise, Wen Wee Ma, and Martin E. Fernandez-Zapico. 2019. "Recent Advances in the Clinical Targeting of Hedgehog/GLI Signaling in Cancer" Cells 8, no. 5: 394. https://doi.org/10.3390/cells8050394
APA StyleXie, H., Paradise, B. D., Ma, W. W., & Fernandez-Zapico, M. E. (2019). Recent Advances in the Clinical Targeting of Hedgehog/GLI Signaling in Cancer. Cells, 8(5), 394. https://doi.org/10.3390/cells8050394