Genetic and Real-World Clinical Data, Combined with Empirical Validation, Nominate Jak-Stat Signaling as a Target for Alzheimer’s Disease Therapeutic Development

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
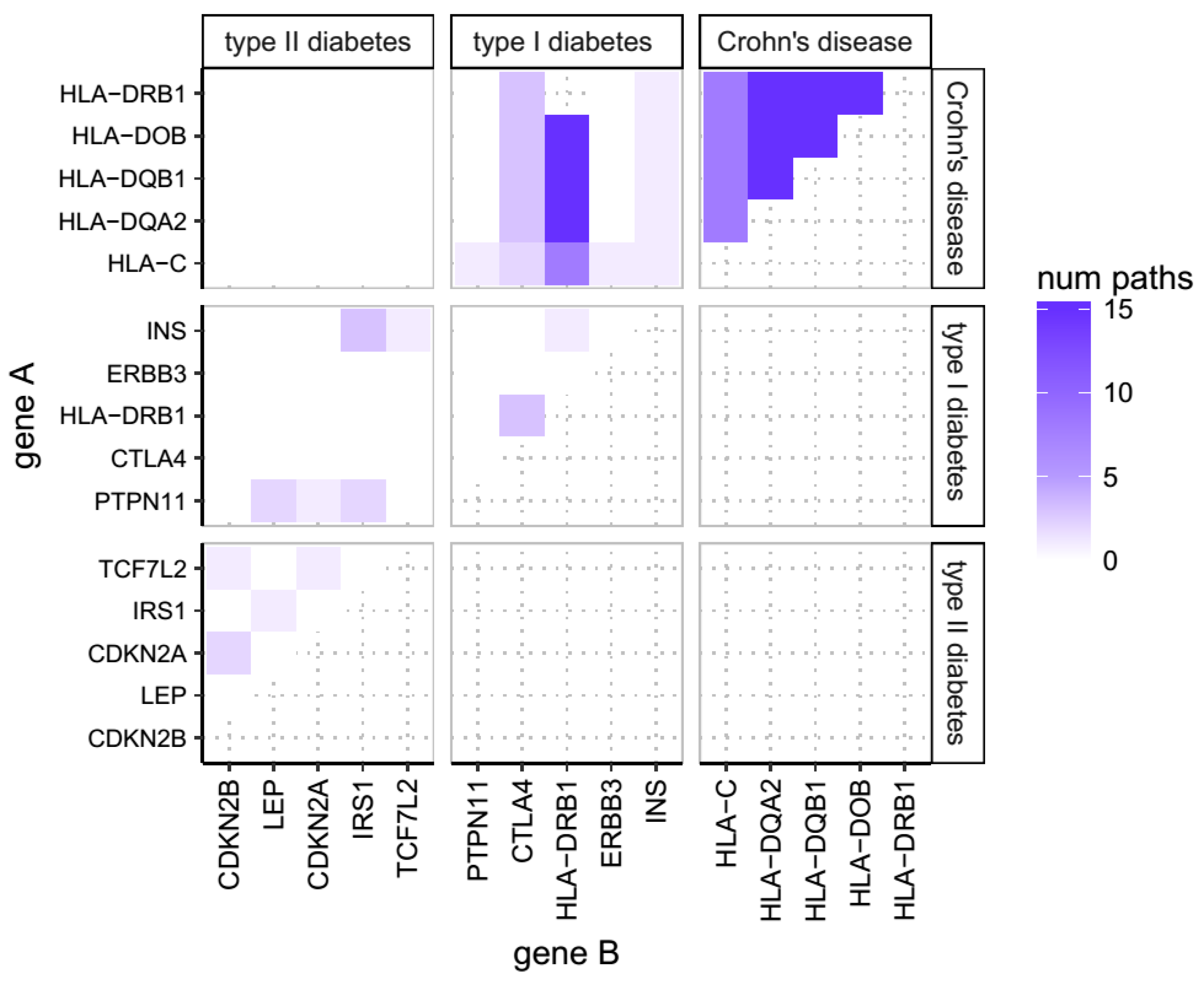
2.1. Overlap of Susceptibility Genes Across Human Disease
2.2. Contribution of Biological Pathways to Diseases
2.3. Shared Contribution of Biological Pathways to Disease Comorbidity with AD
2.4. Pathway Gene Expression Dysregulation in AD
2.5. Proof of Concept in an In Vitro Rat Model of Aβ Exposure
2.6. Proof of Concept in an In Vivo Rat Model of Aβ Exposure
2.7. Ethical Considerations
3. Results
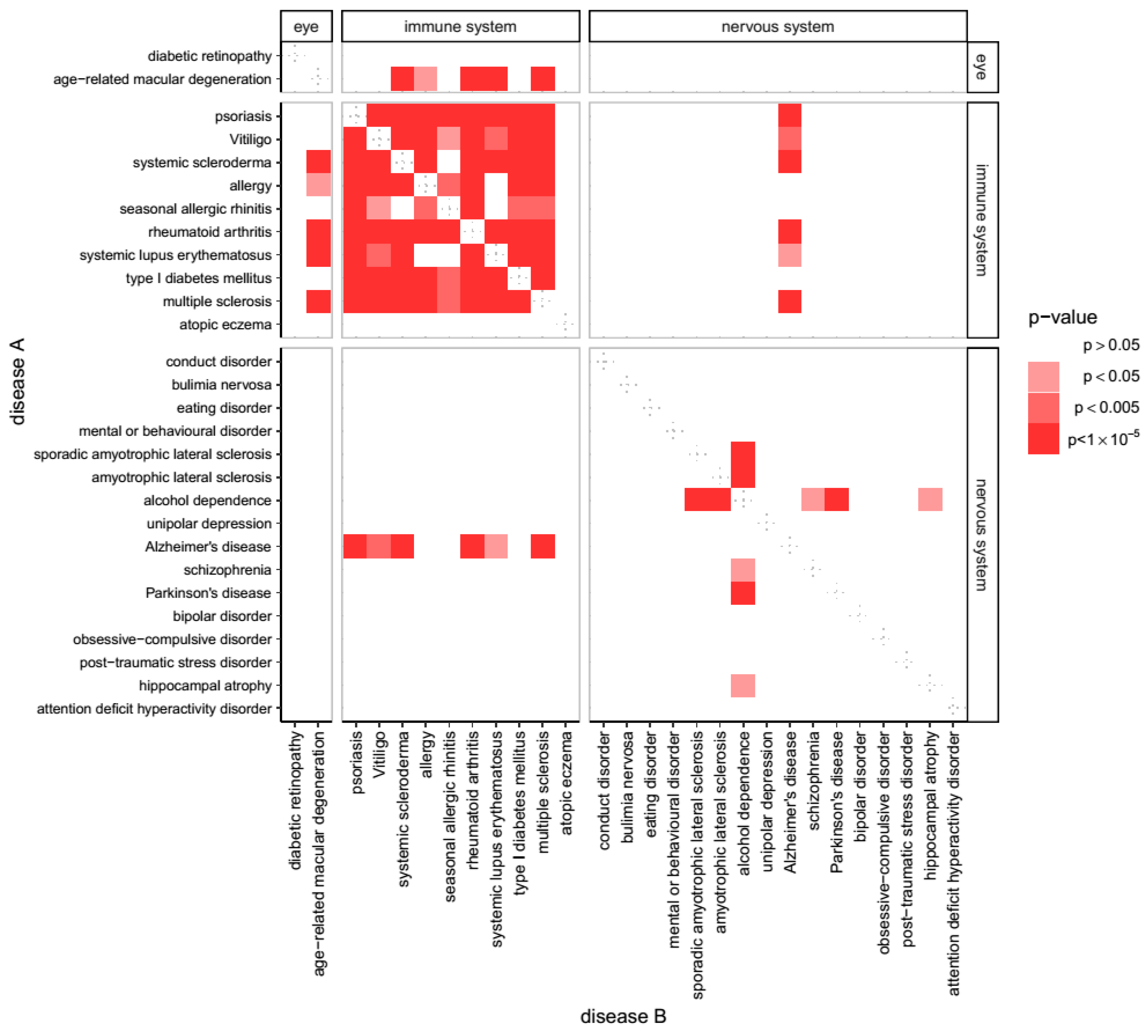
3.1. Genes Associated with AD Show Shared Susceptibility to Diseases of Immunity
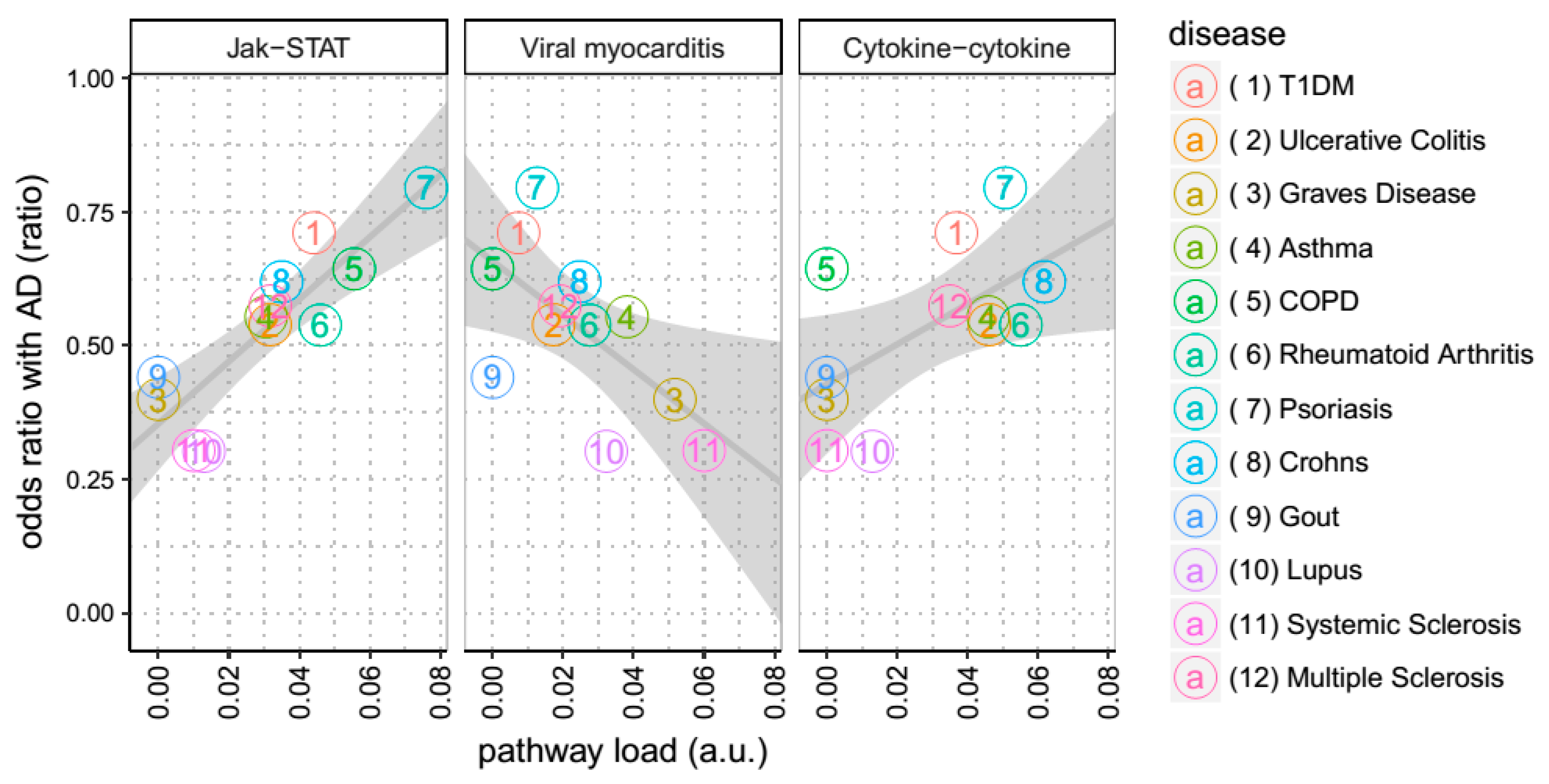
3.2. Among all KEGG Pathways, Associations with JAK-STAT Signaling are Shared between Diseases Co-Morbid with AD
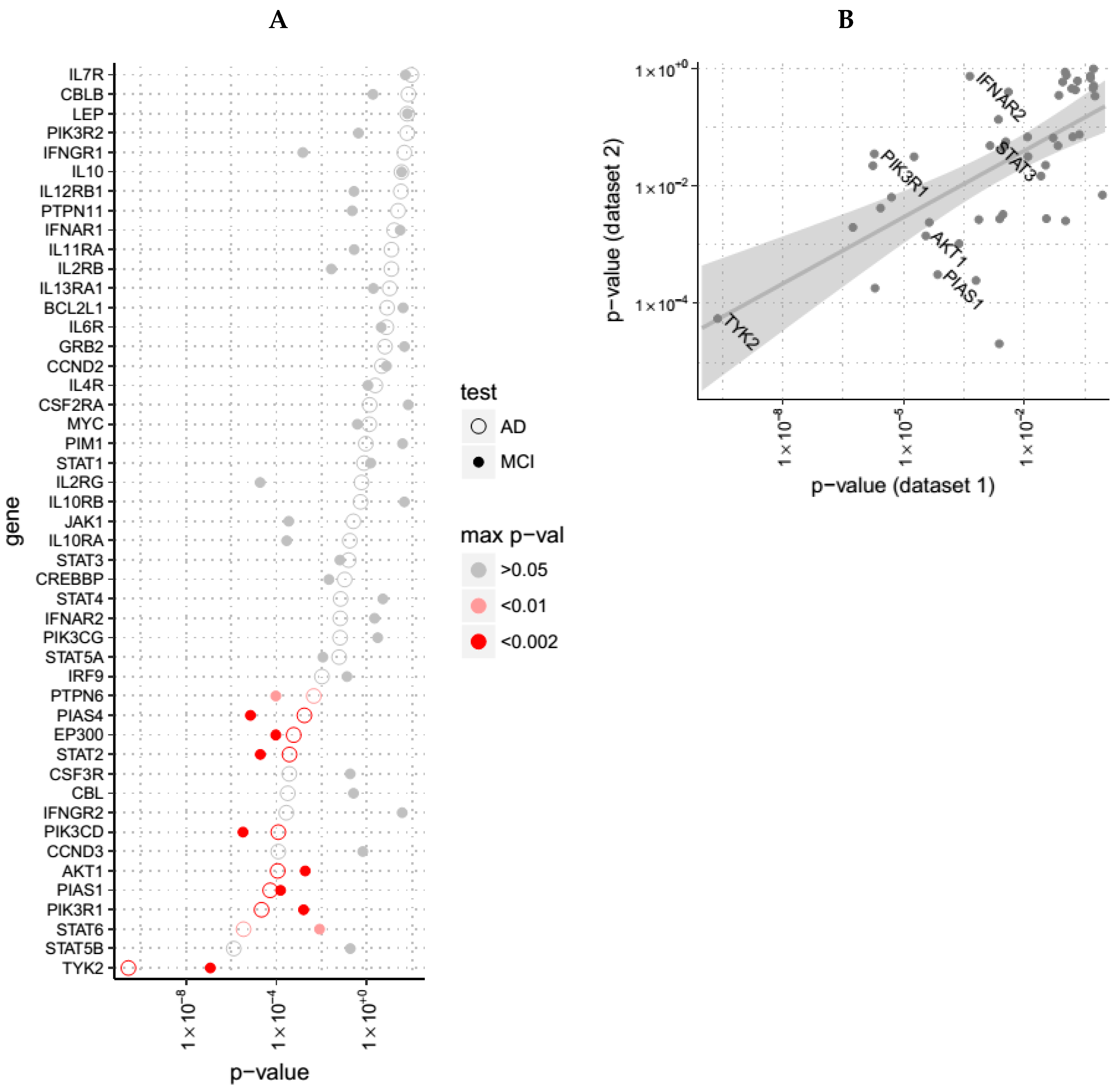
3.3. Evidence for Altered JAK-STAT Pathway Gene Expression in AD Blood, Brain and in an In Vitro Model with Established Relevance to AD
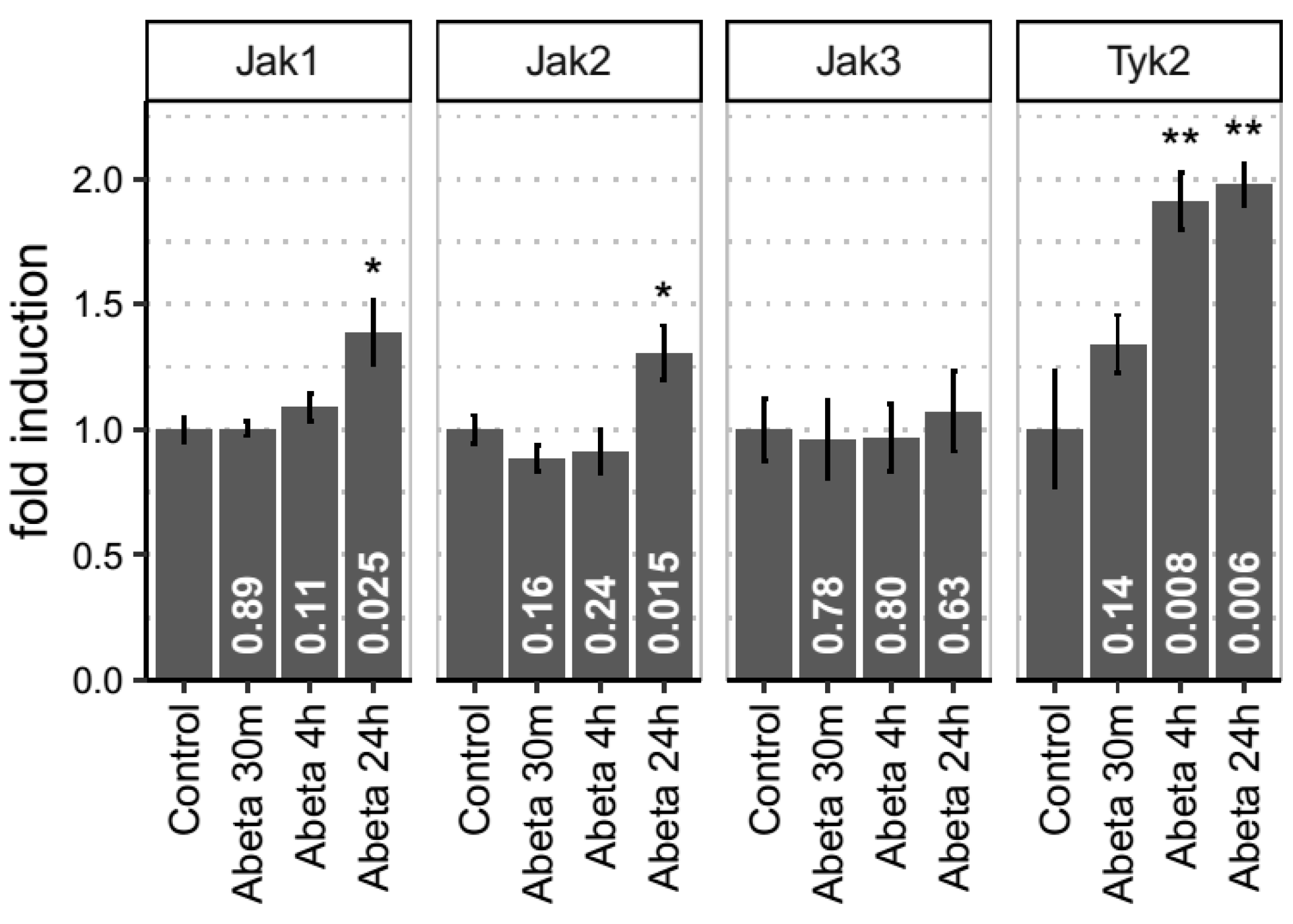
3.4. Empirical Evidence for JAK-STAT Dysregulation in Both In Vitro and In Vivo Models of Aβ-Induced Neurotoxicity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Ramanan, V.K.; Saykin, A.J. Pathways to neurodegeneration: Mechanistic insights from GWAS in Alzheimer’s disease, Parkinson’s disease, and related disorders. Am. J. Neurodegener. Dis. 2013, 2, 145–175. [Google Scholar]
- Naj, A.C.; Schellenberg, G.D. Alzheimer’s Disease Genetics Consortium (ADGC). Genomic variants, genes, and pathways of Alzheimer’s disease: An overview. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2017, 174, 5–26. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.; Holmans, P.A.; Hamshere, M.L.; Harold, D.; Moskvina, V.; Ivanov, D.; Pocklington, A.; Abraham, R.; Hollingworth, P.; Sims, R.; et al. Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer’s disease. PLoS ONE 2010, 5, e13950. [Google Scholar] [CrossRef]
- Rohne, P.; Prochnow, H.; Koch-Brandt, C. The CLU-files: Disentanglement of a mystery. Biomol. Concepts 2016, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Welter, D.; MacArthur, J.; Morales, J.; Burdett, T.; Hall, P.; Junkins, H.; Klemm, A.; Flicek, P.; Manolio, T.; Hindorff, L.; et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014, 42, D1001–D1006. [Google Scholar] [CrossRef] [PubMed]
- Malone, J.; Holloway, E.; Adamusiak, T.; Kapushesky, M.; Zheng, J.; Kolesnikov, N.; Zhukova, A.; Brazma, A.; Parkinson, H. Modeling sample variables with an Experimental Factor Ontology. Bioinformatics 2010, 26, 1112–1118. [Google Scholar] [CrossRef]
- DeFrances, C.J.; Cullen, K.A.; Kozak, L.J. National Hospital Discharge Survey: 2005 annual summary with detailed diagnosis and procedure data. Vital Health Stat. 13. 2007, 165, 1–209. [Google Scholar]
- United States Department of Health and Human Services. National Center for Health Statistics National Hospital Discharge Survey, 1979–2006: Multi-Year Public Use File 2009; Inter-university Consortium for Political and Social Research: Ann Arbor, MI, USA, 2009. [Google Scholar]
- Simmons, A.; Westman, E.; Muehlboeck, S.; Mecocci, P.; Vellas, B.; Tsolaki, M.; Kłoszewska, I.; Wahlund, L.-O.; Soininen, H.; Lovestone, S.; et al. MRI measures of Alzheimer’s disease and the AddNeuroMed study. Ann. N.Y. Acad. Sci. 2009, 1180, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Lovestone, S.; Francis, P.; Kloszewska, I.; Mecocci, P.; Simmons, A.; Soininen, H.; Spenger, C.; Tsolaki, M.; Vellas, B.; Wahlund, L.-O.; et al. AddNeuroMed-The European Collaboration for the Discovery of Novel Biomarkers for Alzheimer’s Disease. Ann. N.Y. Acad. Sci. 2009, 1180, 36–46. [Google Scholar] [CrossRef]
- Zhang, B.; Gaiteri, C.; Bodea, L.-G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [PubMed]
- Killick, R.; Ribe, E.M.; Al-Shawi, R.; Malik, B.; Hooper, C.; Fernandes, C.; Dobson, R.; Nolan, P.M.; Lourdusamy, A.; Furney, S.; et al. Clusterin regulates β-amyloid toxicity via Dickkopf-1-driven induction of the wnt–PCP–JNK pathway. Mol. Psychiatry 2014, 19, 88–98. [Google Scholar] [CrossRef]
- Thambisetty, M.; Simmons, A.; Hye, A.; Campbell, J.; Westman, E.; Zhang, Y.; Wahlund, L.-O.; Kinsey, A.; Causevic, M.; Killick, R.; et al. Plasma Biomarkers of Brain Atrophy in Alzheimer’s Disease. PLoS ONE 2011, 6, e28527. [Google Scholar] [CrossRef] [PubMed]
- Tizon, B.; Ribe, E.M.; Mi, W.; Troy, C.M.; Levy, E. Cystatin C Protects Neuronal Cells from Amyloid-β-induced Toxicity. J. Alzheimers Dis. 2010, 19, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Barghorn, S.; Nimmrich, V.; Striebinger, A.; Krantz, C.; Keller, P.; Janson, B.; Bahr, M.; Schmidt, M.; Bitner, R.S.; Harlan, J.; et al. Globular amyloid beta-peptide1-42 oligomer—A homogenous and stable neuropathological protein in Alzheimer’s disease. J. Neurochem. 2005, 95, 834–847. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Ribe, E.; Jean, Y.; Goldstein, R.; Manzl, C.; Stefanis, L.; Villunger, A.; Troy, C. Neuronal caspase 2 activity and function requires RAIDD, but not PIDD. Biochem. J. 2012, 444, 591–599. [Google Scholar] [CrossRef]
- Jean, Y.; Ribe, E.; Pero, M.; Moskalenko, M.; Iqbal, Z.; Marks, L.; Greene, L.; Troy, C. Caspase-2 is essential for c-Jun transcriptional activation and Bim induction in neuron death. Biochem. J. 2013, 455, 15–25. [Google Scholar] [CrossRef]
- Amar, F.; Sherman, M.A.; Rush, T.; Larson, M.; Boyle, G.; Chang, L.; Götz, J.; Buisson, A.; Lesné, S.E. The amyloid-β oligomer Aβ*56 induces specific alterations in neuronal signaling that lead to tau phosphorylation and aggregation. Sci. Signal. 2017, 10, eaal2021. [Google Scholar] [CrossRef]
- De Felice, F.G.; Wu, D.; Lambert, M.P.; Fernandez, S.J.; Velasco, P.T.; Lacor, P.N.; Bigio, E.H.; Jerecic, J.; Acton, P.J.; Shughrue, P.J.; et al. Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by Aβ oligomers. Neurobiol. Aging 2008, 29, 1334–1347. [Google Scholar] [CrossRef]
- Zempel, H.; Thies, E.; Mandelkow, E.; Mandelkow, E.-M. Aβ Oligomers Cause Localized Ca2+ Elevation, Missorting of Endogenous Tau into Dendrites, Tau Phosphorylation, and Destruction of Microtubules and Spines. J. Neurosci. 2010, 30, 11938–11950. [Google Scholar] [CrossRef]
- Jin, M.; Shepardson, N.; Yang, T.; Chen, G.; Walsh, D.; Selkoe, D.J. Soluble amyloid -protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 5819–5824. [Google Scholar] [CrossRef]
- Lacor, P.N.; Buniel, M.C.; Furlow, P.W.; Sanz Clemente, A.; Velasco, P.T.; Wood, M.; Viola, K.L.; Klein, W.L. Aβ Oligomer-Induced Aberrations in Synapse Composition, Shape, and Density Provide a Molecular Basis for Loss of Connectivity in Alzheimer’s Disease. J. Neurosci. 2007, 27, 796–807. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Salminen, A.; Haapasalo, A.; Soininen, H.; Hiltunen, M. Age-related macular degeneration (AMD): Alzheimer’s disease in the eye? J. Alzheimers Dis. 2011, 24, 615–631. [Google Scholar] [CrossRef]
- Frost, S.; Guymer, R.; Aung, K.Z.; Macaulay, S.L.; Sohrabi, H.R.; Bourgeat, P.; Salvado, O.; Rowe, C.C.; Ames, D.; Masters, C.L.; et al. Alzheimer’s Disease and the Early Signs of Age-Related Macular Degeneration. Curr. Alzheimer Res. 2016, 13, 1259–1266. [Google Scholar] [CrossRef]
- Isas, J.M.; Luibl, V.; Johnson, L.V.; Kayed, R.; Wetzel, R.; Glabe, C.G.; Langen, R.; Chen, J. Soluble and Mature Amyloid Fibrils in Drusen Deposits. Investig. Opthalmology Vis. Sci. 2010, 51, 1304. [Google Scholar] [CrossRef]
- Dentchev, T.; Milam, A.H.; Lee, V.M.-Y.; Trojanowski, J.Q.; Dunaief, J.L. Amyloid-beta is found in drusen from some age-related macular degeneration retinas, but not in drusen from normal retinas. Mol. Vis. 2003, 9, 184–190. [Google Scholar]
- Hye, A.; Lynham, S.; Thambisetty, M.; Causevic, M.; Campbell, J.; Byers, H.L.; Hooper, C.; Rijsdijk, F.; Tabrizi, S.J.; Banner, S.; et al. Proteome-based plasma biomarkers for Alzheimer’s disease. Brain 2006, 129, 3042–3050. [Google Scholar] [CrossRef]
- Wang, D.; Di, X.; Fu, L.; Li, Y.; Han, X.; Wu, H.; Cai, L.; Meng, X.; Jiang, C.; Kong, W.; et al. Analysis of serum β-amyloid peptides, α2-macroglobulin, complement factor H, and clusterin levels in APP/PS1 transgenic mice during progression of Alzheimer’s disease. NeuroReport 2016, 27, 1114–1119. [Google Scholar] [CrossRef]
- Horie-Inoue, K.; Inoue, S. Genomic aspects of age-related macular degeneration. Biochem. Biophys. Res. Commun. 2014, 452, 263–275. [Google Scholar] [CrossRef]
- Bellou, V.; Belbasis, L.; Tzoulaki, I.; Middleton, L.T.; Ioannidis, J.P.A.; Evangelou, E. Systematic evaluation of the associations between environmental risk factors and dementia: An umbrella review of systematic reviews and meta-analyses. Alzheimers Dement. 2017, 13, 406–418. [Google Scholar] [CrossRef]
- McManus, R.M.; Heneka, M.T. Role of neuroinflammation in neurodegeneration: New insights. Alzheimers Res. Ther. 2017, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Schulzer, M.; McGeer, E.G. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: A review of 17 epidemiologic studies. Neurology 1996, 47, 425–432. [Google Scholar] [CrossRef]
- Myllykangas-LuosujäRvi, R.; IsomäKi, H. Alzheimer’s Disease and Rheumatoid Arthritis. Rheumatology 1994, 33, 501–502. [Google Scholar] [CrossRef]
- Anthony, J.C.; Breitner, J.C.S.; Zandi, P.P.; Meyer, M.R.; Jurasova, I.; Norton, M.C.; Stone, S.V. Reduced prevalence of AD in users of NSAIDs and H2 receptor antagonists: The Cache County Study. Neurology 2000, 54, 2066–2071. [Google Scholar] [CrossRef]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.T.; Clarke, J.R.; Bomfim, T.R.; De Felice, F.G. Inflammation, defective insulin signaling, and neuronal dysfunction in Alzheimer’s disease. Alzheimers Dement. J. Alzheimers Assoc. 2014, 10, S76–S83. [Google Scholar] [CrossRef] [PubMed]
- Leung, R.; Proitsi, P.; Simmons, A.; Lunnon, K.; Güntert, A.; Kronenberg, D.; Pritchard, M.; Tsolaki, M.; Mecocci, P.; Kloszewska, I.; et al. Inflammatory Proteins in Plasma Are Associated with Severity of Alzheimer’s Disease. PLoS ONE 2013, 8, e64971. [Google Scholar] [CrossRef]
- Clark, L.F.; Kodadek, T. The Immune System and Neuroinflammation as Potential Sources of Blood-Based Biomarkers for Alzheimer’s Disease, Parkinson’s Disease, and Huntington’s Disease. ACS Chem. Neurosci. 2016, 7, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Liu, W.; Wang, X. TREM2 variants and risk of Alzheimer’s disease: A meta-analysis. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2015, 36, 1881–1888. [Google Scholar] [CrossRef] [PubMed]
- Aiyaz, M.; Lupton, M.K.; Proitsi, P.; Powell, J.F.; Lovestone, S. Complement activation as a biomarker for Alzheimer’s disease. Immunobiology 2012, 217, 204–215. [Google Scholar] [CrossRef]
- Wang, K.; Gaitsch, H.; Poon, H.; Cox, N.J.; Rzhetsky, A. Classification of common human diseases derived from shared genetic and environmental determinants. Nat. Genet. 2017, 49, 1319. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Liang, X.; Shaikh, A.S.; Zang, J.; Xu, W.; Zhang, Y. JAK/STAT signal transduction: Promising attractive targets for immune, inflammatory and hematopoietic diseases. Curr. Drug Targets 2016, 19, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Popiolek-Barczyk, K.; Mika, J. Targeting the Microglial Signaling Pathways: New Insights in the Modulation of Neuropathic Pain. Curr. Med. Chem. 2016, 23, 2908–2928. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Janus kinase (JAK) inhibitors in the treatment of inflammatory and neoplastic diseases. Pharmacol. Res. 2016, 111, 784–803. [Google Scholar] [CrossRef] [PubMed]
- Gurzov, E.N.; Stanley, W.J.; Pappas, E.G.; Thomas, H.E.; Gough, D.J. The JAK/STAT pathway in obesity and diabetes. FEBS J. 2016, 283, 3002–3015. [Google Scholar] [CrossRef]
- Wan, J.; Fu, A.K.Y.; Ip, F.C.F.; Ng, H.-K.; Hugon, J.; Page, G.; Wang, J.H.; Lai, K.-O.; Wu, Z.; Ip, N.Y. Tyk2/STAT3 signaling mediates beta-amyloid-induced neuronal cell death: Implications in Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 6873–6881. [Google Scholar] [CrossRef] [PubMed]
- Ben Haim, L.; Ceyzeriat, K.; Carrillo-de Sauvage, M.A.; Aubry, F.; Auregan, G.; Guillermier, M.; Ruiz, M.; Petit, F.; Houitte, D.; Faivre, E.; et al. The JAK/STAT3 Pathway Is a Common Inducer of Astrocyte Reactivity in Alzheimer’s and Huntington’s Diseases. J. Neurosci. 2015, 35, 2817–2829. [Google Scholar] [CrossRef]
- Nicolas, C.S.; Peineau, S.; Amici, M.; Csaba, Z.; Fafouri, A.; Javalet, C.; Collett, V.J.; Hildebrandt, L.; Seaton, G.; Choi, S.-L.; et al. The Jak/STAT pathway is involved in synaptic plasticity. Neuron 2012, 73, 374–390. [Google Scholar] [CrossRef]
- McGregor, G.; Irving, A.J.; Harvey, J. Canonical JAK-STAT signaling is pivotal for long-term depression at adult hippocampal temporoammonic-CA1 synapses. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 3449–3466. [Google Scholar] [CrossRef]
- Peineau, S.; Taghibiglou, C.; Bradley, C.; Wong, T.P.; Liu, L.; Lu, J.; Lo, E.; Wu, D.; Saule, E.; Bouschet, T.; et al. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 2007, 53, 703–717. [Google Scholar] [CrossRef]
- Hooper, C.; Markevich, V.; Plattner, F.; Killick, R.; Schofield, E.; Engel, T.; Hernandez, F.; Anderton, B.; Rosenblum, K.; Bliss, T.; et al. Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur. J. Neurosci. 2007, 25, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Yamada, M.; Aiso, S. Targeting the JAK2/STAT3 axis in Alzheimer’s disease. Expert Opin. Ther. Targets 2009, 13, 1155–1167. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset 1 (Blood, ANM) | Dataset 2 (Blood, DCR) | Dataset 3 (Brain, AE) | |
|---|---|---|---|
| Species | |||
| Homo Sapiens | 344 | 228 | 229 |
| Gender (n) | |||
| Female | 209 | 148 | 108 |
| Male | 135 | 80 | 121 |
| Age (years) | |||
| mean ± SD | 75.9 ± 6.8 | 78.1 ± 7.0 | 73.8 ± 12 |
| Centre (n) | |||
| 1 | 44 | 44 | 229 |
| 2 | 34 | 11 | - |
| 3 | 84 | 62 | - |
| 4 | 43 | 43 | - |
| 5 | 45 | 25 | - |
| 6 | 94 | 43 | - |
| Diagnosis (n) | |||
| AD | 105 | 90 | 129 |
| control | 114 | 73 | 100 |
| MCI | 125 | 65 | 0 |
| Gene | Dataset 1 (Blood, ANM) | Dataset 2 (Blood, DCR) | Dataset 3 (Brain, AE) | Dataset 4 (In-Vitro) |
|---|---|---|---|---|
| TYK2 | 5·10−10 | 0.0005 | 0.0002 | - |
| PIK3R1 | 1·10−6 | 0.03 | 0.3 | 0.5 |
| IFNAR2 | 0.0004 | 0.7 | 0.04 | 0.007 |
| AKT1 | 0.0003 | 0.001 | 0.5 | 0.001 |
| PIAS1 | 0.0007 | 0.0003 | 8·10−5 | 0.016 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nevado-Holgado, A.J.; Ribe, E.; Thei, L.; Furlong, L.; Mayer, M.-A.; Quan, J.; Richardson, J.C.; Cavanagh, J.; NIMA Consortium; Lovestone, S. Genetic and Real-World Clinical Data, Combined with Empirical Validation, Nominate Jak-Stat Signaling as a Target for Alzheimer’s Disease Therapeutic Development. Cells 2019, 8, 425. https://doi.org/10.3390/cells8050425
Nevado-Holgado AJ, Ribe E, Thei L, Furlong L, Mayer M-A, Quan J, Richardson JC, Cavanagh J, NIMA Consortium, Lovestone S. Genetic and Real-World Clinical Data, Combined with Empirical Validation, Nominate Jak-Stat Signaling as a Target for Alzheimer’s Disease Therapeutic Development. Cells. 2019; 8(5):425. https://doi.org/10.3390/cells8050425
Chicago/Turabian StyleNevado-Holgado, Alejo J., Elena Ribe, Laura Thei, Laura Furlong, Miguel-Angel Mayer, Jie Quan, Jill C. Richardson, Jonathan Cavanagh, NIMA Consortium, and Simon Lovestone. 2019. "Genetic and Real-World Clinical Data, Combined with Empirical Validation, Nominate Jak-Stat Signaling as a Target for Alzheimer’s Disease Therapeutic Development" Cells 8, no. 5: 425. https://doi.org/10.3390/cells8050425
APA StyleNevado-Holgado, A. J., Ribe, E., Thei, L., Furlong, L., Mayer, M. -A., Quan, J., Richardson, J. C., Cavanagh, J., NIMA Consortium, & Lovestone, S. (2019). Genetic and Real-World Clinical Data, Combined with Empirical Validation, Nominate Jak-Stat Signaling as a Target for Alzheimer’s Disease Therapeutic Development. Cells, 8(5), 425. https://doi.org/10.3390/cells8050425