Long Noncoding RNA HCP5, a Hybrid HLA Class I Endogenous Retroviral Gene: Structure, Expression, and Disease Associations


Abstract
:1. Introduction
2. HCP5 Genomic Organization, Gene Structure, and the HERV16 Antisense Transcript
2.1. Human MHC Class I Genomic Structure as Duplication Blocks and HCP5 Location within an HERV16 Duplicated Sequence
2.2. HCP5 RNA, a Complementary Sequence of ERV16 (an ERV Class III (ERVL))
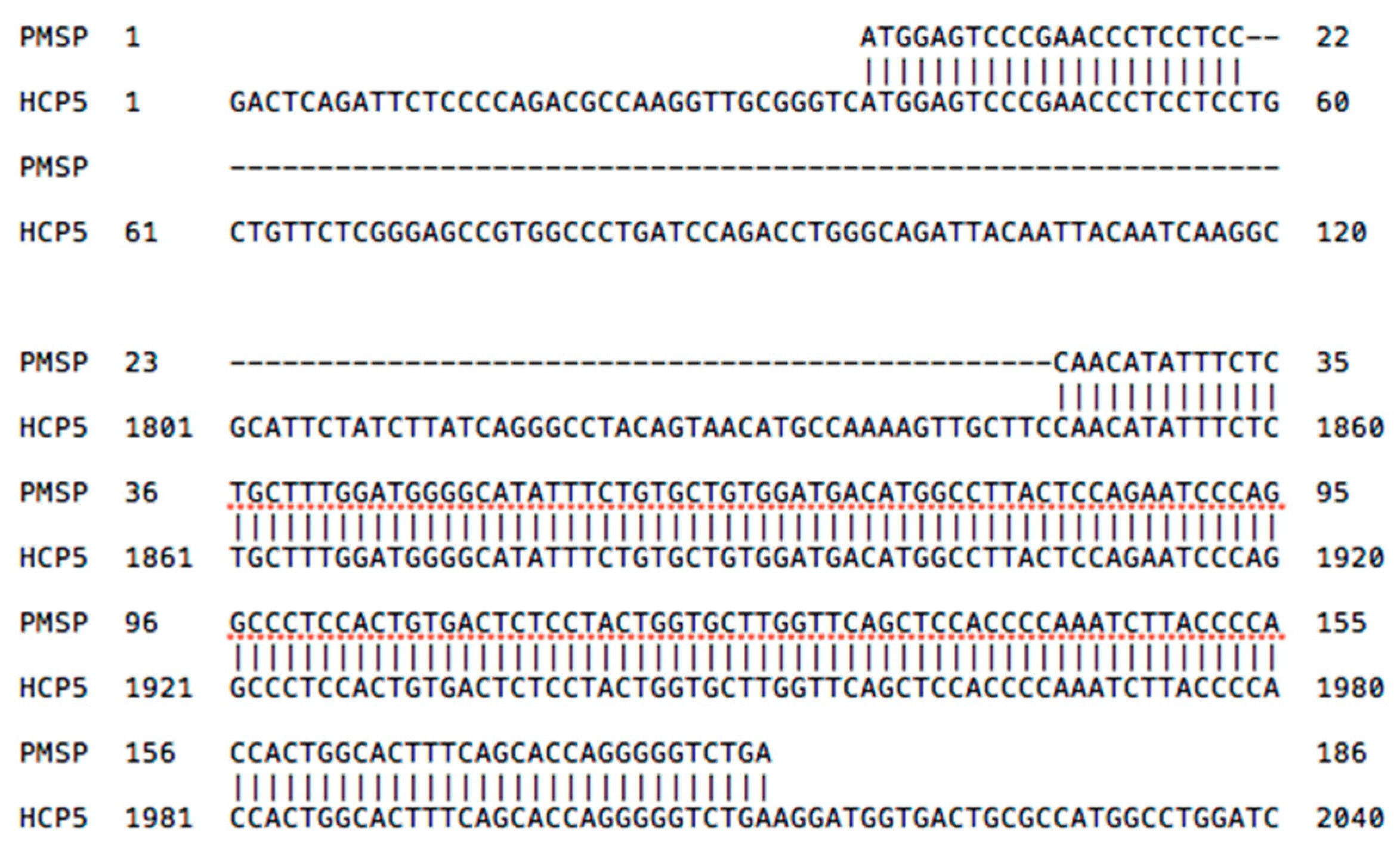
2.3. HCP5 and the Human Papillomavirus (HPV) Minor Structural Protein Interacting Protein (PMSP) Gene
2.4. HLA Class I Leader Sequence and Cytomegalovirus (CMV) Signal Peptides
2.5. HLA-Class I Gene Promoters and Enhancers within HCP5
2.6. HCP5-CCCTC-Binding Factor (CTCF) Connection and Chromatin Structural Regulation
2.7. Structural Deletion of HCP5 and MICA Genes
3. HCP5 Single Nucleotide Variant (SNV) Associations with Disease
3.1. HIV and AIDS
3.2. Herpes Zoster
3.3. Autoimmune Disease and Drug Hypersensitivity
3.4. Transplantation
3.5. Cancer
4. HCP5 Methylomics
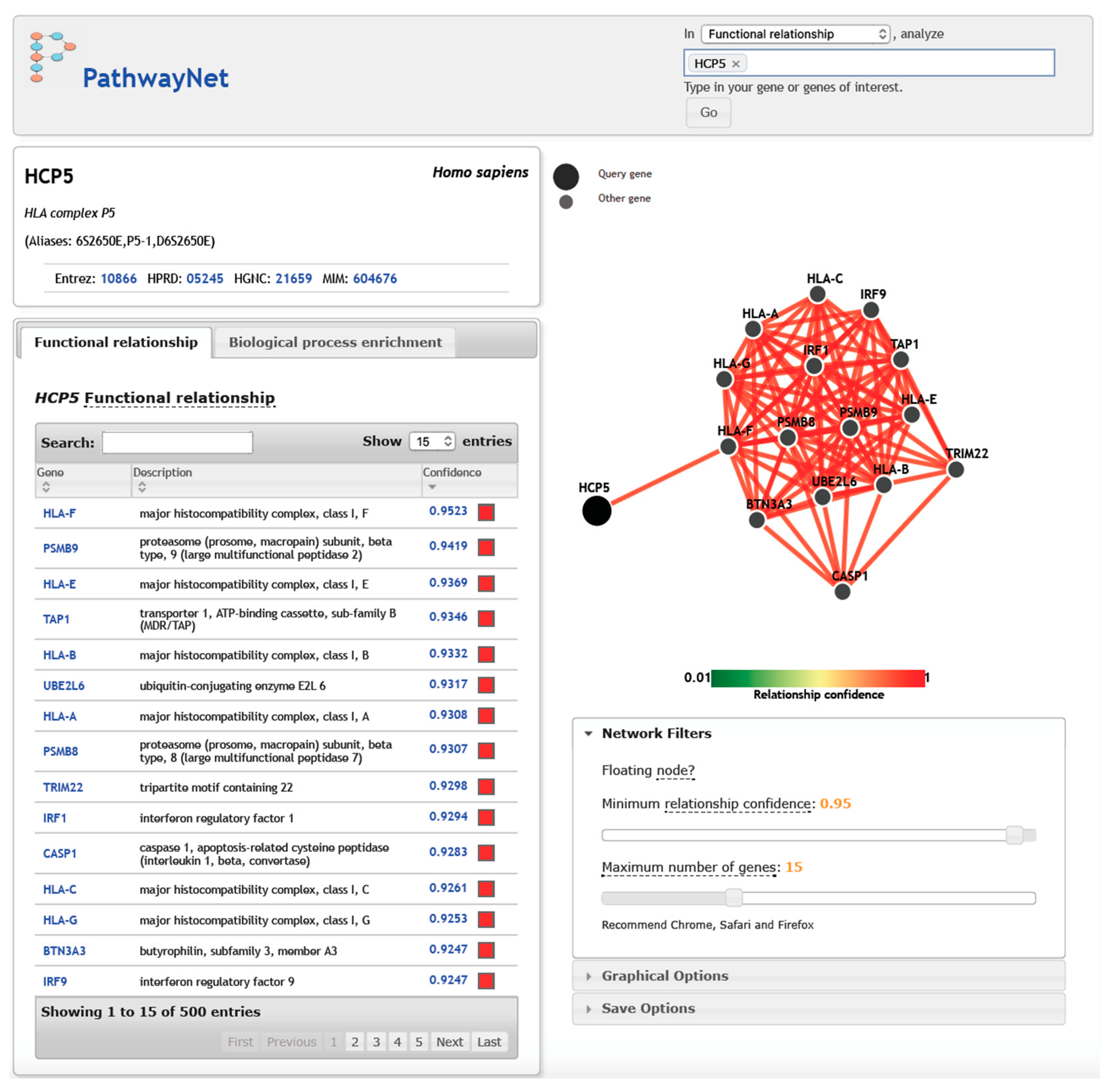
5. HCP5, Gene Targets, and Transcription Factors in Interaction Networks
6. HCP5 Gene Expression and Gene Interactions
6.1. HCP5 Expression in HIV-Infected Cells
6.2. HCP5 Expression in Cancer
7. HCP5 lncRNA Interactions with Regulatory miRNA in Cancer
8. Perspective
9. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Shiina, T.; Inoko, H.; Kulski, J.K. An update of the HLA genomic region, locus information and disease associations: 2004. Tissue Antigens 2004, 64, 631–649. [Google Scholar] [CrossRef]
- Shiina, T.; Hosomichi, K.; Inoko, H.; Kulski, J.K. The HLA genomic loci map: Expression, interaction, diversity and disease. J. Hum. Genet. 2009, 54, 15–39. [Google Scholar] [CrossRef]
- Alter, I.; Gragert, L.; Fingerson, S.; Maiers, M.; Louzoun, Y. HLA class I haplotype diversity is consistent with selection for frequent existing haplotypes. PLoS Comput. Biol. 2017, 13, e1005693. [Google Scholar] [CrossRef]
- Meyer, D.; Single, R.M.; Mack, S.J.; Erlich, H.A.; Thomson, G. Signatures of Demographic History and Natural Selection in the Human Major Histocompatibility Complex Loci. Genetics 2006, 173, 2121–2142. [Google Scholar] [CrossRef] [Green Version]
- Shiina, T.; Ota, M.; Shimizu, S.; Katsuyama, Y.; Hashimoto, N.; Takasu, M.; Anzai, T.; Kulski, J.K.; Kikkawa, E.; Naruse, T.; et al. Rapid Evolution of Major Histocompatibility Complex Class I Genes in Primates Generates New Disease Alleles in Humans via Hitchhiking Diversity. Genetics 2006, 173, 1555–1570. [Google Scholar] [CrossRef] [Green Version]
- Trowsdale, J.; Knight, J.C. Major Histocompatibility Complex Genomics and Human Disease. Annu. Rev. Genom. Hum. Genet. 2013, 14, 301–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosaad, Y.M. Clinical Role of Human Leukocyte Antigen in Health and Disease. Scand. J. Immunol. 2015, 82, 283–306. [Google Scholar] [CrossRef]
- Matzaraki, V.; Kumar, V.; Wijmenga, C.; Zhernakova, A. The MHC locus and genetic susceptibility to autoimmune and infectious diseases. Genome Biol. 2017, 18, 76–92. [Google Scholar] [CrossRef] [PubMed]
- Cabarrocas, J.; Bauer, J.; Piaggio, E.; Liblau, R.; Lassmann, H. Effective and selective immune surveillance of the brain by MHC class I-restricted cytotoxic T lymphocytes. Eur. J. Immunol. 2003, 33, 1174–1182. [Google Scholar] [CrossRef] [Green Version]
- Lazarczyk, M.J.; Kemmler, J.E.; Eyford, B.A.; Short, J.A.; Varghese, M.; Sowa, A.; Dickstein, D.R.; Yuk, F.J.; Puri, R.; Biron, K.E.; et al. Major Histocompatibility Complex class I proteins are critical for maintaining neuronal structural complexity in the aging brain. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Nelson, P.A.; Sage, J.R.; Wood, S.C.; Davenport, C.M.; Anagnostaras, S.G.; Boulanger, L.M. MHC class I immune proteins are critical for hippocampus-dependent memory and gate NMDAR-dependent hippocampal long-term depression. Learn. Mem. 2013, 20, 505–517. [Google Scholar] [CrossRef] [Green Version]
- Ohtsuka, M.; Inoko, H.; Kulski, J.K.; Yoshimura, S. Major histocompatibility complex (Mhc) class Ib gene duplications, organization and expression patterns in mouse strain C57BL/6. BMC Genom. 2008, 9, 178. [Google Scholar] [CrossRef] [PubMed]
- Prugnolle, F.; Manica, A.; Charpentier, M.; Guégan, J.F.; Guernier, V.; Balloux, F. Pathogen-Driven Selection and Worldwide HLA Class I Diversity. Curr. Biol. 2005, 15, 1022–1027. [Google Scholar] [CrossRef] [Green Version]
- Garrido, F.; Perea, F.; Bernal, M.; Sánchez-Palencia, A.; Aptsiauri, N.; Ruiz-Cabello, F. The Escape of Cancer from T Cell-Mediated Immune Surveillance: HLA Class I Loss and Tumor Tissue Architecture. Vaccines 2017, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Morrison, B.J.; Steel, J.C.; Morris, J.C. Reduction of MHC-I expression limits T-lymphocyte-mediated killing of Cancer-initiating cells. BMC Cancer 2018, 18, 469. [Google Scholar] [CrossRef] [PubMed]
- Borchert, G.M.; Holton, N.W.; Williams, J.D.; Hernan, W.L.; Bishop, I.P.; Dembosky, J.A.; Elste, J.E.; Gregoire, N.S.; Kim, J.-A.; Koehler, W.W.; et al. Comprehensive analysis of microRNA genomic loci identifies pervasive repetitive-element origins. Mob. Genet. Elem. 2011, 1, 8–17. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Kraus, W.L. From Discovery to Function: The Expanding Roles of Long NonCoding RNAs in Physiology and Disease. Endocr. Rev. 2015, 36, 25–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, T.R.; Mattick, J.S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 2013, 20, 300–307. [Google Scholar] [CrossRef]
- Mattick, J.S. The State of Long Non-Coding RNA Biology. Non-Coding RNA 2018, 4, 17. [Google Scholar] [CrossRef]
- Liu, S.J.; Horlbeck, M.A.; Cho, S.W.; Birk, H.S.; Malatesta, M.; He, D.; Attenello, F.J.; Villalta, J.E.; Cho, M.Y.; Chen, Y.; et al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science 2017, 355, eaah7111. [Google Scholar] [CrossRef]
- Sigdel, K.R.; Cheng, A.; Wang, Y.; Duan, L.; Zhang, Y. The Emerging Functions of Long Noncoding RNA in Immune Cells: Autoimmune Diseases. J. Immunol. Res. 2015, 2015, 1–9. [Google Scholar] [CrossRef]
- Geng, H.; Tan, X.-D. Functional diversity of long non-coding RNAs in immune regulation. Genes Dis. 2016, 3, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Spurlock, C.F.; Crooke, P.S.; Aune, T.M. Biogenesis and Transcriptional Regulation of Long Noncoding RNAs in the Human Immune System. J. Immunol. 2016, 197, 4509–4517. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.G.; Satpathy, A.T.; Chang, H.Y. Gene regulation in the immune system by long noncoding RNAs. Nat. Immunol. 2017, 18, 962–972. [Google Scholar] [CrossRef] [PubMed]
- Clark, P.M.; Chitnis, N.; Shieh, M.; Kamoun, M.; Johnson, F.B.; Monos, D. Novel and Haplotype Specific MicroRNAs Encoded by the Major Histocompatibility Complex. Sci. Rep. 2018, 8, 3832. [Google Scholar] [CrossRef] [PubMed]
- Gensterblum-Miller, E.; Wu, W.; Sawalha, A.H. Novel Transcriptional Activity and Extensive Allelic Imbalance in the Human MHC Region. J. Immunol. 2018, 200, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Vernet, C.; Ribouchon, M.-T.; Chimini, G.; Jouanolle, A.-M.; Sidibe, I.; Pontarotti, P. A novel coding sequence belonging to a new multicopy gene family mapping within the human MHC class I region. Immunogenetics 1993, 38, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Avoustin, P.; Ribouchon, M.T.; Vernet, C.; N’Guyen, B.; Crouau-Roy, B.; Pontarotti, P. Non-homologous recombination within the Major Histocompatibility Complex creates a transcribed hybrid sequence. Mamm. Genome 1994, 5, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Shiina, T.; Tamiya, G.; Oka, A.; Yamagata, T.; Yamagata, N.; Kikkawa, E.; Goto, K.; Mizuki, N.; Watanabe, K.; Fukuzumi, Y.; et al. Nucleotide Sequencing Analysis of the 146-Kilobase Segment around the IkBL and MICA Genes at the Centromeric End of the HLA Class I Region. Genomics 1998, 47, 372–382. [Google Scholar] [CrossRef]
- Kulski, J.K.; Dawkins, R.L. The P5 multicopy gene family in the MHC is related in sequence to human endogenous retroviruses HERV-L and HERV-16. Immunogenetics 1999, 49, 404–412. [Google Scholar] [CrossRef]
- Babaian, A.; Mager, D.L. Endogenous retroviral promoter exaptation in human cancer. Mob. DNA 2016, 7, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, J.; Sugimoto, R.; Nakaoka, H.; Yamada, S.; Kimura, T.; Hayano, T.; Inoue, I. Systematic identification and characterization of regulatory elements derived from human endogenous retroviruses. PLos Genet. 2017, 13, e1006883. [Google Scholar] [CrossRef]
- Fellay, J.; Shianna, K.V.; Ge, D.; Colombo, S.; Ledergerber, B.; Weale, M.; Zhang, K.; Gumbs, C.; Castagna, A.; Cossarizza, A.; et al. A Whole-Genome Association Study of Major Determinants for Host Control of HIV-1. Science 2007, 317, 944–947. [Google Scholar] [CrossRef] [Green Version]
- Fellay, J.; Ge, D.; Shianna, K.V.; Colombo, S.; Ledergerber, B.; Cirulli, E.T.; Urban, T.J.; Zhang, K.; Gumbs, C.E.; Smith, J.P.; et al. Common Genetic Variation and the Control of HIV-1 in Humans. PLoS Genet. 2009, 5, e1000791. [Google Scholar] [CrossRef]
- Yoon, W.; Ma, B.-J.; Fellay, J.; Huang, W.; Xia, S.-M.; Zhang, R.; Shianna, K.V.; Liao, H.-X.; Haynes, B.F.; Goldstein, D.B. A polymorphism in the HCP5 gene associated with HLA-B*5701 does not restrict HIV-1 in vitro. AIDS 2010, 24, 155–157. [Google Scholar] [CrossRef] [Green Version]
- Dawkins, R.; Leelayuwat, C.; Gaudieri, S.; Tay, G.; Hui, J.; Cattley, S.; Martinez, P.; Kulski, J. Genomics of the major histocompatibility complex: Haplotypes, duplication, retroviruses and disease. Immunol. Rev. 1999, 167, 275–304. [Google Scholar] [CrossRef] [PubMed]
- Kulski, J.K.; Gaudieri, S.; Martin, A.; Dawkins, R.L. Coevolution of PERB11 (MIC) and HLA Class I Genes with HERV-16 and Retroelements by Extended Genomic Duplication. J. Mol. Evol. 1999, 49, 84–97. [Google Scholar] [CrossRef]
- Kulski, J.K.; Gaudieri, S.; Inoko, H.; Dawkins, R.L. Comparison Between Two Human Endogenous Retrovirus (HERV)-Rich Regions Within the Major Histocompatibility Complex. J. Mol. Evol. 1999, 48, 675–683. [Google Scholar] [CrossRef]
- Kim, J.-J.; Yun, S.W.; Yu, J.J.; Yoon, K.L.; Lee, K.-Y.; Kil, H.-R.; Kim, G.B.; Han, M.-K.; Song, M.S.; Lee, H.D.; et al. A genome-wide association analysis identifies NMNAT2 and HCP5 as susceptibility loci for Kawasaki disease. J. Hum. Genet. 2017, 62, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Kapitonov, V.; Jurka, J. The age of Alu subfamilies. J. Mol. Evol. 1996, 42, 59–65. [Google Scholar] [CrossRef]
- Görnemann, J.; Hofmann, T.G.; Will, H.; Müller, M. Interaction of Human Papillomavirus Type 16 L2 with Cellular Proteins: Identification of Novel Nuclear Body-Associated Proteins. Virology 2002, 303, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Komatsu-Wakui, M.; Tokunaga, K.; Ishikawa, Y.; Kashiwase, K.; Moriyama, S.; Tsuchiya, N.; Ando, H.; Shiina, T.; Geraghty, D.E.; Inoko, H.; et al. MIC-A polymorphism in Japanese and a MIC-A-MIC-B null haplotype. Immunogenetics 1999, 49, 620–628. [Google Scholar] [CrossRef]
- Tian, C.; Hinds, D.A.; Hromatka, B.S.; Kiefer, A.K.; Eriksson, N.; Tung, J.Y. Genome-wide association and HLA region fine-mapping studies identify susceptibility loci for multiple common infections. BioRxiv 2016, 8, 599. [Google Scholar] [CrossRef] [PubMed]
- Karim, R.; Meyers, C.; Backendorf, C.; Ludigs, K.; Offringa, R.; van Ommen, G.-J.B.; Melief, C.J.M.; van der Burg, S.H.; Boer, J.M. Human Papillomavirus Deregulates the Response of a Cellular Network Comprising of Chemotactic and Proinflammatory Genes. PLoS ONE 2011, 6, e17848. [Google Scholar] [CrossRef] [PubMed]
- Prod’homme, V.; Tomasec, P.; Cunningham, C.; Lemberg, M.K.; Stanton, R.J.; McSharry, B.P.; Wang, E.C.Y.; Cuff, S.; Martoglio, B.; Davison, A.J.; et al. Human Cytomegalovirus UL40 Signal Peptide Regulates Cell Surface Expression of the NK Cell Ligands HLA-E and gpUL18. J. Immunol. 2012, 188, 2794–2804. [Google Scholar] [CrossRef] [PubMed]
- Halenius, A.; Gerke, C.; Hengel, H. Classical and non-classical MHC I molecule manipulation by human cytomegalovirus: So many targets—But how many arrows in the quiver? Cell. Mol. Immunol. 2015, 12, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Ramsuran, V.; Hernández-Sanchez, P.G.; O’hUigin, C.; Sharma, G.; Spence, N.; Augusto, D.G.; Gao, X.; García-Sepúlveda, C.A.; Kaur, G.; Mehra, N.K.; et al. Sequence and Phylogenetic Analysis of the Untranslated Promoter Regions for HLA Class I Genes. J. Immunol. 2017, 198, 2320–2329. [Google Scholar] [CrossRef]
- Barbash, Z.S.; Weissman, J.D.; Campbell, J.A.; Mu, J.; Singer, D.S. Major Histocompatibility Complex Class I Core Promoter Elements Are Not Essential for Transcription in vivo. Mol. Cell. Biol. 2013, 33, 4395–4407. [Google Scholar] [CrossRef] [Green Version]
- van den Elsen, P.J. Expression Regulation of Major Histocompatibility Complex Class I and Class II Encoding Genes. Front. Immunol. 2011, 2, 48. [Google Scholar] [CrossRef]
- Jongsma, M.L.M.; Guarda, G.; Spaapen, R.M. The regulatory network behind MHC class I expression. Mol. Immunol. 2017. [Google Scholar] [CrossRef]
- Gobin, S.J.P.; Peijnenburg, A.; Keijsers, V.; van den Elsen, P.J. Site alpha Is Crucial for Two Routes of IFNgamma-Induced MHC Class I Transactivation: The ISRE-Mediated Route and a Novel Pathway Involving CIITA. Immunity 1997, 6, 601–61111. [Google Scholar] [CrossRef]
- Zhou, F. Molecular Mechanisms of IFN-γ to Up-Regulate MHC Class I Antigen Processing and Presentation. Int. Rev. Immunol. 2009, 28, 239–260. [Google Scholar] [CrossRef]
- Indraccolo, S.; Pfeffer, U.; Minuzzo, S.; Esposito, G.; Roni, V.; Mandruzzato, S.; Ferrari, N.; Anfosso, L.; Dell’Eva, R.; Noonan, D.M.; et al. Identification of genes selectively regulated by IFNs in endothelial cells. J. Immunol. 2007, 178, 1122–1135. [Google Scholar] [CrossRef]
- Johnson-Huang, L.M.; Suárez-Fariñas, M.; Pierson, K.C.; Fuentes-Duculan, J.; Cueto, I.; Lentini, T.; Sullivan-Whalen, M.; Gilleaudeau, P.; Krueger, J.G.; Haider, A.S.; et al. A Single Intradermal Injection of IFN-γ Induces an Inflammatory State in Both Non-Lesional Psoriatic and Healthy Skin. J. Invest. Dermatol. 2012, 132, 1177–1187. [Google Scholar] [CrossRef] [Green Version]
- Rock, R.B.; Hu, S.; Deshpande, A.; Munir, S.; May, B.J.; Baker, C.A.; Peterson, P.K.; Kapur, V. Transcriptional response of human microglial cells to interferon-γ. Genes Immun. 2005, 6, 712–719. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Wang, R.; Fang, L.; Ge, X.; Chen, L.; Zhou, M.; Zhou, Y.; Xiong, W.; Hu, Y.; Tang, X.; et al. HCP5 is a SMAD3-responsive long non-coding RNA that promotes lung adenocarcinoma metastasis via miR-203/SNAI axis. Theranostics 2019, 9, 2460–2474. [Google Scholar] [CrossRef]
- Zhao, W.; Li, L. SP1-induced upregulation of long non-coding RNA HCP5 promotes the development of osteosarcoma. Pathol. Res. Pract. 2019, 215, 439–445. [Google Scholar] [CrossRef]
- Tilburgs, T.; Meissner, T.B.; Ferreira, L.M.R.; Mulder, A.; Musunuru, K.; Ye, J.; Strominger, J.L. NLRP2 is a suppressor of NF-ƙB signaling and HLA-C expression in human trophoblasts. Biol. Reprod. 2017, 96, 831–842. [Google Scholar] [CrossRef]
- Gobin, S.J.P.; Keijsers, V.; van Zutphen, M.; van den Elsen, P.J. The Role of Enhancer A in the Locus-Specific Transactivation of Classical and Nonclassical HLA Class I Genes by Nuclear Factor κB. J. Immunol. 1998, 161, 2276–2283. [Google Scholar]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the Human Tissue-specific Expression by Genome-wide Integration of Transcriptomics and Antibody-based Proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef]
- Downs, I.; Vijayan, S.; Sidiq, T.; Kobayashi, K.S. CITA/NLRC5: A critical transcriptional regulator of MHC class I gene expression: CITA/NLRC5. BioFactors 2016, 42, 349–357. [Google Scholar] [CrossRef]
- Kobayashi, K.S.; van den Elsen, P.J. NLRC5: A key regulator of MHC class I-dependent immune responses. Nat. Rev. Immunol. 2012, 12, 813–820. [Google Scholar] [CrossRef]
- Vijayan, S.; Sidiq, T.; Yousuf, S.; van den Elsen, P.J.; Kobayashi, K.S. Class I transactivator, NLRC5: A central player in the MHC class I pathway and cancer immune surveillance. Immunogenetics 2019, 71, 273–282. [Google Scholar] [CrossRef]
- Meeks, K.A.C.; Henneman, P.; Venema, A.; Burr, T.; Galbete, C.; Danquah, I.; Schulze, M.B.; Mockenhaupt, F.P.; Owusu-Dabo, E.; Rotimi, C.N.; et al. An epigenome-wide association study in whole blood of measures of adiposity among Ghanaians: The RODAM study. Clin. Epigenetics 2017, 9, 103. [Google Scholar] [CrossRef]
- Wiesner, T.; Lee, W.; Obenauf, A.C.; Ran, L.; Murali, R.; Zhang, Q.F.; Wong, E.W.P.; Hu, W.; Scott, S.N.; Shah, R.H.; et al. Alternative transcription initiation leads to expression of a novel ALK isoform in cancer. Nature 2015, 526, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Majumder, P.; Boss, J.M. CTCF Controls Expression and Chromatin Architecture of the Human Major Histocompatibility Complex Class II Locus. Mol. Cell. Biol. 2010, 30, 4211–4223. [Google Scholar] [CrossRef]
- Ottaviani, D.; Lever, E.; Mao, S.; Christova, R.; Ogunkolade, B.W.; Jones, T.A.; Szary, J.; Aarum, J.; Mumin, M.A.; Pieri, C.A.; et al. CTCF binds to sites in the major histocompatibility complex that are rapidly reconfigured in response to interferon-gamma. Nucleic Acids Res. 2012, 40, 5262–5270. [Google Scholar] [CrossRef] [Green Version]
- Chaumeil, J.; Skok, J.A. The role of CTCF in regulating V(D)J recombination. Curr. Opin. Immunol. 2012, 24, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.E.; Corces, V.G. CTCF: Master Weaver of the Genome. Cell 2009, 137, 1194–1211. [Google Scholar] [CrossRef] [Green Version]
- Guelen, L.; Pagie, L.; Brasset, E.; Meuleman, W.; Faza, M.B.; Talhout, W.; Eussen, B.H.; de Klein, A.; Wessels, L.; de Laat, W.; et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 2008, 453, 948–951. [Google Scholar] [CrossRef]
- Bailey, C.; Metierre, C.; Feng, Y.; Baidya, K.; Filippova, G.; Loukinov, D.; Lobanenkov, V.; Semaan, C.; Rasko, J. CTCF Expression is Essential for Somatic Cell Viability and Protection Against Cancer. Int. J. Mol. Sci. 2018, 19, 3832. [Google Scholar] [CrossRef]
- Cross, S.H.; Clark, V.H.; Simmen, M.W.; Bickmore, W.A.; Maroon, H.; Langford, C.F.; Carter, N.P.; Bird, A.P. CpG island libraries from human chromosomes 18 and 22: Landmarks for novel genes. Mamm. Genome 2000, 11, 373–383. [Google Scholar] [CrossRef]
- Ota, M.; Bahram, S.; Katsuyama, Y.; Saito, S.; Nose, Y.; Sada, M.; Ando, H.; Inoko, H. On the MICA deleted-MICB null, HLA-B*4801 haplotype. Tissue Antigens 2000, 56, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Middleton, D.; Menchaca, L.; Rood, H.; Komerofsky, R. New allele frequency database: http://www.allelefrequencies.net. Tissue Antigens 2003, 61, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Aida, K.; Russomando, G.; Kikuchi, M.; Candia, N.; Franco, L.; Almiron, M.; Ubalee, R.; Hirayama, K. High frequency of MIC null haplotype (HLA-B48-MICA-del-MICB*0107 N) in the Angaite Amerindian community in Paraguay. Immunogenetics 2002, 54, 439–441. [Google Scholar] [CrossRef]
- Shaikh, T.H.; Gai, X.; Perin, J.C.; Glessner, J.T.; Xie, H.; Murphy, K.; O’Hara, R.; Casalunovo, T.; Conlin, L.K.; D’Arcy, M.; et al. High-resolution mapping and analysis of copy number variations in the human genome: A data resource for clinical and research applications. Genome Res. 2009, 19, 1682–1690. [Google Scholar] [CrossRef] [Green Version]
- Itsara, A.; Cooper, G.M.; Baker, C.; Girirajan, S.; Li, J.; Absher, D.; Krauss, R.M.; Myers, R.M.; Ridker, P.M.; Chasman, D.I.; et al. Population Analysis of Large Copy Number Variants and Hotspots of Human Genetic Disease. Am. J. Hum. Genet. 2009, 84, 148–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melis, R.; Lewis, T.; Millson, A.; Lyon, E.; McMillin, G.A.; Slev, P.R.; Swensen, J. Copy Number Variation and Incomplete Linkage Disequilibrium Interfere with the HCP5 Genotyping Assay for Abacavir Hypersensitivity. Genet. Test. Mol. Biomark. 2012, 16, 1111–1114. [Google Scholar] [CrossRef]
- Dunn, D.; Ota, M.; Inoko, H.; Kulski, J.K. Association of MHC dimorphic Alu insertions with HLA class I and MIC genes in Japanese HLA-B48 haplotypes. Tissue Antigens 2003, 62, 259–262. [Google Scholar] [CrossRef]
- Anzai, T.; Shiina, T.; Kimura, N.; Yanagiya, K.; Kohara, S.; Shigenari, A.; Yamagata, T.; Kulski, J.K.; Naruse, T.K.; Fujimori, Y.; et al. Comparative sequencing of human and chimpanzee MHC class I regions unveils insertions/deletions as the major path to genomic divergence. Proc. Natl. Acad. Sci. 2003, 100, 7708–7713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulski, J.K.; Shiina, T.; Anzai, T.; Kohara, S.; Inoko, H. Comparative genomic analysis of the MHC: The evolution of class I duplication blocks, diversity and complexity from shark to man. Immunol. Rev. 2002, 190, 95–122. [Google Scholar] [CrossRef] [PubMed]
- Wilming, L.G.; Hart, E.A.; Coggill, P.C.; Horton, R.; Gilbert, J.G.R.; Clee, C.; Jones, M.; Lloyd, C.; Palmer, S.; Sims, S.; et al. Sequencing and comparative analysis of the gorilla MHC genomic sequence. Database 2013, 2013, bat011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudieri, S.; Giles, K.M.; Kulski, J.K.; Dawkins, R.L. Duplication and polymorphism in the MHC: Alu generated diversity and polymorphism within the PERB11 gene family. Hereditas 1997, 127, 37–46. [Google Scholar] [CrossRef]
- Hughes, A.L.; Yeager, M.; Ten Elshof, A.E.; Chorney, M.J. A new taxonomy of mammalian MHC class I molecules. Immunol. Today 1999, 20, 22–26. [Google Scholar] [CrossRef]
- Bahram, S. MIC genes: From genetics to biology. Adv. Immunol. 2000, 76, 1–60. [Google Scholar] [PubMed]
- Vejbaesya, S.; Luangtrakool, P.; Luangtrakool, K.; Kalayanarooj, S.; Vaughn, D.W.; Endy, T.P.; Mammen, M.P.; Green, S.; Libraty, D.H.; Ennis, F.A.; et al. TNF and LTA Gene, Allele, and Extended HLA Haplotype Associations with Severe Dengue Virus Infection in Ethnic Thais. J. Infect. Dis. 2009, 199, 1442–1448. [Google Scholar] [CrossRef]
- Low, J.S.Y.; Chin, Y.M.; Mushiroda, T.; Kubo, M.; Govindasamy, G.K.; Pua, K.C.; Yap, Y.Y.; Yap, L.F.; Subramaniam, S.K.; Ong, C.A.; et al. A Genome Wide Study of Copy Number Variation Associated with Nasopharyngeal Carcinoma in Malaysian Chinese Identifies CNVs at 11q14.3 and 6p21.3 as Candidate Loci. PLoS ONE 2016, 11, e0145774. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Tian, W.; Zhu, F.; Li, L.; Cai, J.; Wang, F.; Liu, K.; Jin, H.; Wang, J. MICA Gene Deletion in 3411 DNA Samples from Five Distinct Populations in Mainland China and Lack of Association with Nasopharyngeal Carcinoma (NPC) in a Southern Chinese Han population. Ann. Hum. Genet. 2016, 80, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Caillat-Zucman, S. New insights into the understanding of MHC associations with immune-mediated disorders: MHC-associated immune diseases. HLA 2017, 89, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef]
- Medici, M.; Porcu, E.; Pistis, G.; Teumer, A.; Brown, S.J.; Jensen, R.A.; Rawal, R.; Roef, G.L.; Plantinga, T.S.; Vermeulen, S.H.; et al. Identification of Novel Genetic Loci Associated with Thyroid Peroxidase Antibodies and Clinical Thyroid Disease. PLoS Genet. 2014, 10, e1004123. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Liu, H.; Liu, Z.; Owzar, K.; Han, Y.; Su, L.; Wei, Y.; Hung, R.J.; McLaughlin, J.; Brhane, Y.; et al. A Novel Genetic Variant in Long Non-coding RNA Gene NEXN-AS1 is Associated with Risk of Lung Cancer. Sci. Rep. 2016, 6, 34234. [Google Scholar] [CrossRef] [Green Version]
- The International HIV Controllers Study. The Major Genetic Determinants of HIV-1 Control Affect HLA Class I Peptide Presentation. Science 2010, 330, 1551–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daly, A.K.; Donaldson, P.T.; Bhatnagar, P.; Shen, Y.; Pe’er, I.; Floratos, A.; Daly, M.J.; Goldstein, D.B.; John, S.; Nelson, M.R.; et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat. Genet. 2009, 41, 816–819. [Google Scholar] [CrossRef]
- Limou, S.; Le Clerc, S.; Coulonges, C.; Carpentier, W.; Dina, C.; Delaneau, O.; Labib, T.; Taing, L.; Sladek, R.; ANRS Genomic Group; et al. Genomewide Association Study of an AIDS-Nonprogression Cohort Emphasizes the Role Played by HLA Genes (ANRS Genomewide Association Study 02). J. Infect. Dis. 2009, 199, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Helms, C.; Liao, W.; Zaba, L.C.; Duan, S.; Gardner, J.; Wise, C.; Miner, A.; Malloy, M.J.; Pullinger, C.R.; et al. A Genome-Wide Association Study of Psoriasis and Psoriatic Arthritis Identifies New Disease Loci. PLoS Genet. 2008, 4, e1000041. [Google Scholar] [CrossRef]
- Rodriguez-Novoa, S.; Cuenca, L.; Morello, J.; Cordoba, M.; Blanco, F.; Jimenez-Nacher, I.; Soriano, V. Use of the HCP5 single nucleotide polymorphism to predict hypersensitivity reactions to abacavir: Correlation with HLA-B*5701. J. Antimicrob. Chemother. 2010, 65, 1567–1569. [Google Scholar] [CrossRef]
- Suhre, K.; Arnold, M.; Bhagwat, A.M.; Cotton, R.J.; Engelke, R.; Raffler, J.; Sarwath, H.; Thareja, G.; Wahl, A.; DeLisle, R.K.; et al. Connecting genetic risk to disease end points through the human blood plasma proteome. Nat. Commun. 2017, 8, 14357. [Google Scholar] [CrossRef] [Green Version]
- Crosslin, D.R.; Carrell, D.S.; Burt, A.; Kim, D.S.; Underwood, J.G.; Hanna, D.S.; Comstock, B.A.; Baldwin, E.; de Andrade, M.; Kullo, I.J.; et al. Genetic variation in the HLA region is associated with susceptibility to herpes zoster. Genes Immun. 2015, 16, 1–7. [Google Scholar] [CrossRef]
- Astle, W.J.; Elding, H.; Jiang, T.; Allen, D.; Ruklisa, D.; Mann, A.L.; Mead, D.; Bouman, H.; Riveros-Mckay, F.; Kostadima, M.A.; et al. The Allelic Landscape of Human Blood Cell Trait Variation and Links to Common Complex Disease. Cell 2016, 167, 1415–1429. [Google Scholar] [CrossRef] [PubMed]
- McKay, J.D.; Hung, R.J.; Han, Y.; Zong, X.; Carreras-Torres, R.; Christiani, D.C.; Caporaso, N.E.; Johansson, M.; Xiao, X.; Li, Y.; et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat. Genet. 2017, 49, 1126–1132. [Google Scholar] [CrossRef] [Green Version]
- Feenstra, B.; Pasternak, B.; Geller, F.; Carstensen, L.; Wang, T.; Huang, F.; Eitson, J.L.; Hollegaard, M.V.; Svanström, H.; Vestergaard, M.; et al. Common variants associated with general and MMR vaccine–related febrile seizures. Nat. Genet. 2014, 46, 1274–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, F.W.; Chen, W.; O’Hanlon, T.P.; Cooper, R.G.; Vencovsky, J.; Rider, L.G.; Danko, K.; Wedderburn, L.R.; Lundberg, I.E.; Pachman, L.M.; et al. Genome-wide association study identifies HLA 8.1 ancestral haplotype alleles as major genetic risk factors for myositis phenotypes. Genes Immun. 2015, 16, 470–480. [Google Scholar] [CrossRef] [Green Version]
- Rothwell, S.; Cooper, R.G.; Lundberg, I.E.; Miller, F.W.; Gregersen, P.K.; Bowes, J.; Vencovsky, J.; Danko, K.; Limaye, V.; Selva-O’Callaghan, A.; et al. Dense genotyping of immune-related loci in idiopathic inflammatory myopathies confirms HLA alleles as the strongest genetic risk factor and suggests different genetic background for major clinical subgroups. Ann. Rheum. Dis. 2016, 75, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.-P.; Chang, S.-W.; Jaing, T.-H.; Wang, W.-T.; Hus, F.-P.; Tseng, C.-P. Single nucleotide polymorphisms within HLA region are associated with disease relapse for patients with unrelated cord blood transplantation. PeerJ 2018, 6, e5228. [Google Scholar] [CrossRef]
- van Manen, D.; Kootstra, N.A.; Boeser-Nunnink, B.; Handulle, M.A.; van’t Wout, A.B.; Schuitemaker, H. Association of HLA-C and HCP5 gene regions with the clinical course of HIV-1 infection. AIDS 2009, 23, 19–28. [Google Scholar] [CrossRef] [Green Version]
- van Manen, D.; van ‘t Wout, A.B.; Schuitemaker, H. Genome-wide association studies on HIV susceptibility, pathogenesis and pharmacogenomics. Retrovirology 2012, 9, 70. [Google Scholar] [CrossRef]
- Shrestha, S.; Aissani, B.; Song, W.; Wilson, C.M.; Kaslow, R.A.; Tang, J. Host genetics and HIV-1 viral load set-point in African–Americans. AIDS 2009, 23, 673–677. [Google Scholar] [CrossRef]
- Thørner, L.W.; Erikstrup, C.; Harritshøj, L.H.; Larsen, M.H.; Kronborg, G.; Pedersen, C.; Larsen, C.S.; Pedersen, G.; Gerstoft, J.; Obel, N.; et al. Impact of polymorphisms in the HCP5 and HLA-C, and ZNRD1 genes on HIV viral load. Infect. Genet. Evol. 2016, 41, 185–190. [Google Scholar] [CrossRef]
- Catano, G.; Kulkarni, H.; He, W.; Marconi, V.C.; Agan, B.K.; Landrum, M.; Anderson, S.; Delmar, J.; Telles, V.; Song, L.; et al. HIV-1 Disease-Influencing Effects Associated with ZNRD1, HCP5 and HLA-C Alleles Are Attributable Mainly to Either HLA-A10 or HLA-B*57 Alleles. PLoS ONE 2008, 3, e3636. [Google Scholar] [CrossRef]
- Colombo, S.; Rauch, A.; Rotger, M.; Fellay, J.; Martinez, R.; Fux, C.; Thurnheer, C.; Günthard, H.F.; Goldstein, D.B.; Furrer, H.; et al. The HCP5 Single-Nucleotide Polymorphism: A Simple Screening Tool for Prediction of Hypersensitivity Reaction to Abacavir. J. Infect. Dis. 2008, 198, 864–867. [Google Scholar] [CrossRef] [PubMed]
- Sousa-Pinto, B.; Correia, C.; Gomes, L.; Gil-Mata, S.; Araújo, L.; Correia, O.; Delgado, L. HLA and Delayed Drug-Induced Hypersensitivity. Int. Arch. Allergy Immunol. 2016, 170, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Illing, P.T.; Vivian, J.P.; Dudek, N.L.; Kostenko, L.; Chen, Z.; Bharadwaj, M.; Miles, J.J.; Kjer-Nielsen, L.; Gras, S.; Williamson, N.A.; et al. Immune self-reactivity triggered by drug-modified HLA-peptide repertoire. Nature 2012, 486, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Ostrov, D.A.; Grant, B.J.; Pompeu, Y.A.; Sidney, J.; Harndahl, M.; Southwood, S.; Oseroff, C.; Lu, S.; Jakoncic, J.; de Oliveira, C.A.F.; et al. Drug hypersensitivity caused by alteration of the MHC-presented self-peptide repertoire. Proc. Natl. Acad. Sci. 2012, 109, 9959–9964. [Google Scholar] [CrossRef] [Green Version]
- Denny, J.C.; Bastarache, L.; Ritchie, M.D.; Carroll, R.J.; Zink, R.; Mosley, J.D.; Field, J.R.; Pulley, J.M.; Ramirez, A.H.; Bowton, E.; et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat. Biotechnol. 2013, 31, 1102–1111. [Google Scholar] [CrossRef] [Green Version]
- Kisiel, B.; Kisiel, K.; Szymański, K.; Mackiewicz, W.; Biało-Wójcicka, E.; Uczniak, S.; Fogtman, A.; Iwanicka-Nowicka, R.; Koblowska, M.; Kossowska, H.; et al. The association between 38 previously reported polymorphisms and psoriasis in a Polish population: High predicative accuracy of a genetic risk score combining 16 loci. PLoS ONE 2017, 12, e0179348. [Google Scholar] [CrossRef] [PubMed]
- Borgiani, P.; Di Fusco, D.; Erba, F.; Marazzi, M.C.; Mancinelli, S.; Novelli, G.; Palombi, L.; Ciccacci, C. HCP5 genetic variant (RS3099844) contributes to Nevirapine-induced Stevens Johnsons Syndrome/Toxic Epidermal Necrolysis susceptibility in a population from Mozambique. Eur. J. Clin. Pharmacol. 2014, 70, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, F.; Perricone, C.; Borgiani, P.; Ciccacci, C.; Rufini, S.; Cipriano, E.; Alessandri, C.; Spinelli, F.R.; Sili Scavalli, A.; Novelli, G.; et al. Genetic Factors in Systemic Lupus Erythematosus: Contribution to Disease Phenotype. J. Immunol. Res. 2015, 2015, 1–11. [Google Scholar] [CrossRef]
- Colafrancesco, S.; Ciccacci, C.; Priori, R.; Latini, A.; Picarelli, G.; Arienzo, F.; Novelli, G.; Valesini, G.; Perricone, C.; Borgiani, P. STAT4, TRAF3IP2, IL10, and HCP5 Polymorphisms in Sjögren’s Syndrome: Association with Disease Susceptibility and Clinical Aspects. J. Immunol. Res. 2019, 2019, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Reynolds, L.M.; Taylor, J.R.; Ding, J.; Lohman, K.; Johnson, C.; Siscovick, D.; Burke, G.; Post, W.; Shea, S.; Jacobs, D.R.; et al. Age-related variations in the methylome associated with gene expression in human monocytes and T cells. Nat. Commun. 2014, 5, 5366. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.A.; Nititham, J.; Elboudwarej, E.; Quach, H.L.; Taylor, K.E.; Barcellos, L.F.; Criswell, L.A. Genome-Wide Assessment of Differential DNA Methylation Associated with Autoantibody Production in Systemic Lupus Erythematosus. PLoS ONE 2015, 10, e0129813. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.M.; Jaeger, P.A.; Kreisberg, J.F.; Licon, K.; Jepsen, K.L.; Khosroheidari, M.; Morsey, B.M.; Swindells, S.; Shen, H.; Ng, C.T.; et al. Methylome-wide Analysis of Chronic HIV Infection Reveals Five-Year Increase in Biological Age and Epigenetic Targeting of HLA. Mol. Cell 2016, 62, 157–168. [Google Scholar] [CrossRef]
- Zimmermann, M.T.; Oberg, A.L.; Grill, D.E.; Ovsyannikova, I.G.; Haralambieva, I.H.; Kennedy, R.B.; Poland, G.A. System-Wide Associations between DNA-Methylation, Gene Expression, and Humoral Immune Response to Influenza Vaccination. PLoS ONE 2016, 11, e0152034. [Google Scholar] [CrossRef] [PubMed]
- Kukushkina, V.; Modhukur, V.; Suhorutšenko, M.; Peters, M.; Mägi, R.; Rahmioglu, N.; Velthut-Meikas, A.; Altmäe, S.; Esteban, F.J.; Vilo, J.; et al. DNA methylation changes in endometrium and correlation with gene expression during the transition from pre-receptive to receptive phase. Sci. Rep. 2017, 7, 3916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.; Sui, W.; Tan, Q.; Chen, J.; Zhang, Y.; Ou, M.; Xue, W.; Li, F.; Cao, C.; Sun, Y.; et al. Integrated analyses of a major histocompatibility complex, methylation and transcribed ultra-conserved regions in systemic lupus erythematosus. Int. J. Mol. Med. 2016, 37, 139–148. [Google Scholar] [CrossRef]
- Cheng, L.; Sun, B.; Xiong, Y.; Hu, L.; Gao, L.; Lv, Q.; Zhou, M.; Li, J.; Chen, X.; Zhang, W.; et al. The minor alleles HCP5 rs3099844 A and PSORS1C1 rs3131003 G are associated with allopurinol-induced severe cutaneous adverse reactions in Han Chinese: A multicentre retrospective case-control clinical study. Br. J. Dermatol. 2018, 178, e191–e193. [Google Scholar] [CrossRef]
- Cvitic, S.; Longtine, M.S.; Hackl, H.; Wagner, K.; Nelson, M.D.; Desoye, G.; Hiden, U. The Human Placental Sexome Differs between Trophoblast Epithelium and Villous Vessel Endothelium. PLoS ONE 2013, 8, 12. [Google Scholar] [CrossRef]
- Rosenfeld, C.S. Sex-Specific Placental Responses in Fetal Development. Endocrinology 2015, 156, 3422–3434. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Yan, Z.; Wu, C.; Zhang, Q.; Zhu, Y.; Li, K.; Xu, Y. Integrated analysis of dosage effect lncRNAs in lung adenocarcinoma based on comprehensive network. Oncotarget 2017, 8, 71430–71446. [Google Scholar] [CrossRef] [Green Version]
- Coit, P.; Kaushik, P.; Kerr, G.; Walsh, J.; Dubreuil, M.; Reimold, A.; Sawalha, A. Genome-Wide DNA Methylation Analysis in Ankylosing Spondylitis [abstract]. Arthritis Rheumatol. 2018, 70. abstract number 2061. [Google Scholar]
- Koga, M.; Kawasaki, A.; Ito, I.; Furuya, T.; Ohashi, J.; Kyogoku, C.; Ito, S.; Hayashi, T.; Matsumoto, I.; Kusaoi, M.; et al. Cumulative association of eight susceptibility genes with systemic lupus erythematosus in a Japanese female population. J. Hum. Genet. 2011, 56, 503–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houshdaran, S.; Zelenko, Z.; Irwin, J.C.; Giudice, L.C. Human endometrial DNA methylome is cycle-dependent and is associated with gene expression regulation. Mol. Endocrinol. 2014, 28, 1118–1135. [Google Scholar] [CrossRef] [PubMed]
- Saare, M.; Modhukur, V.; Suhorutshenko, M.; Rajashekar, B.; Rekker, K.; Sõritsa, D.; Karro, H.; Soplepmann, P.; Sõritsa, A.; Lindgren, C.M.; et al. The influence of menstrual cycle and endometriosis on endometrial methylome. Clin. Epigenetics 2016, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Orvis, T.; Hepperla, A.; Walter, V.; Song, S.; Simon, J.; Parker, J.; Wilkerson, M.D.; Desai, N.; Major, M.B.; Hayes, D.N.; et al. BRG1/SMARCA4 Inactivation Promotes Non-Small Cell Lung Cancer Aggressiveness by Altering Chromatin Organization. Cancer Res. 2014, 74, 6486–6498. [Google Scholar] [CrossRef]
- Warner, N.; Burberry, A.; Pliakas, M.; McDonald, C.; Núñez, G. A Genome-wide Small Interfering RNA (siRNA) Screen Reveals Nuclear Factor-κB (NF-κB)-independent Regulators of NOD2-induced Interleukin-8 (IL-8) Secretion. J. Biol. Chem. 2014, 289, 28213–28224. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Krishnan, A.; Zhu, Q.; Wong, A.K.; Lee, Y.-S.; Troyanskaya, O.G. Tissue-aware data integration approach for the inference of pathway interactions in metazoan organisms. Bioinformatics 2015, 31, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016, 2016, baw100. [Google Scholar] [CrossRef]
- Davis, A.P.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; McMorran, R.; Wiegers, J.; Wiegers, T.C.; Mattingly, C.J. The Comparative Toxicogenomics Database: Update 2019. Nucleic Acids Res. 2019, 47, D948–D954. [Google Scholar] [CrossRef]
- Rieswijk, L.; Claessen, S.M.H.; Bekers, O.; van Herwijnen, M.; Theunissen, D.H.J.; Jennen, D.G.J.; de Kok, T.M.C.M.; Kleinjans, J.C.S.; van Breda, S.G.J. Aflatoxin B1 induces persistent epigenomic effects in primary human hepatocytes associated with hepatocellular carcinoma. Toxicology 2016, 350–352, 31–39. [Google Scholar] [CrossRef]
- Lerner, P.; Guadalupe, M.; Donovan, R.; Hung, J.; Flamm, J.; Prindiville, T.; Sankaran-Walters, S.; Syvanen, M.; Wong, J.K.; George, M.D.; et al. The Gut Mucosal Viral Reservoir in HIV-Infected Patients Is Not the Major Source of Rebound Plasma Viremia following Interruption of Highly Active Antiretroviral Therapy. J. Virol. 2011, 85, 4772–4782. [Google Scholar] [CrossRef]
- Mitchell, R.S.; Beitzel, B.F.; Schroder, A.R.W.; Shinn, P.; Chen, H.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Retroviral DNA Integration: ASLV, HIV, and MLV Show Distinct Target Site Preferences. PLoS Biol. 2004, 2, e234. [Google Scholar] [CrossRef]
- Borjabad, A.; Morgello, S.; Chao, W.; Kim, S.-Y.; Brooks, A.I.; Murray, J.; Potash, M.J.; Volsky, D.J. Significant Effects of Antiretroviral Therapy on Global Gene Expression in Brain Tissues of Patients with HIV-1-Associated Neurocognitive Disorders. Plos Pathog. 2011, 7, e1002213. [Google Scholar] [CrossRef]
- Ockenhouse, C.F.; Bernstein, W.B.; Wang, Z.; Vahey, M.T. Functional Genomic Relationships in HIV-1 Disease Revealed by Gene-Expression Profiling of Primary Human Peripheral Blood Mononuclear Cells. J. Infect. Dis. 2005, 191, 2064–2074. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.N.; Kohler, J.J.; Coberley, C.R.; Sleasman, J.W.; Goodenow, M.M. HIV-1 Activates Macrophages Independent of Toll-Like Receptors. PLoS ONE 2008, 3, e3664. [Google Scholar] [CrossRef]
- Hyrcza, M.D.; Kovacs, C.; Loutfy, M.; Halpenny, R.; Heisler, L.; Yang, S.; Wilkins, O.; Ostrowski, M.; Der, S.D. Distinct Transcriptional Profiles in Ex Vivo CD4+ and CD8+ T Cells Are Established Early in Human Immunodeficiency Virus Type 1 Infection and Are Characterized by a Chronic Interferon Response as Well as Extensive Transcriptional Changes in CD8+ T Cells. J. Virol. 2007, 81, 3477–3486. [Google Scholar] [CrossRef]
- Lewinski, M.K.; Bisgrove, D.; Shinn, P.; Chen, H.; Hoffmann, C.; Hannenhalli, S.; Verdin, E.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Genome-Wide Analysis of Chromosomal Features Repressing Human Immunodeficiency Virus Transcription. J. Virol. 2005, 79, 6610–6619. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhang, R.; Zhao, X.; Su, J.; Bian, X.; Ni, J.; Yue, Y.; Cai, Y.; Jin, J. A potential diagnostic marker for ovarian cancer: Involvement of the histone acetyltransferase, human males absent on the first. Oncol. Lett. 2013, 6, 393–400. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Shenoy, A.K.; Li, X.; Jin, Y.; Jin, L.; Cai, Q.; Tang, M.; Liu, Y.; Chen, H.; Reisman, D.; et al. MOF Acetylates the Histone Demethylase LSD1 to Suppress Epithelial-to-Mesenchymal Transition. Cell Rep. 2016, 15, 2665–2678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, H.; Wang, P.; Xue, Y.; Liu, X.; Ma, J.; Cai, H.; Xi, Z.; Li, Z.; Liu, Y. Role of HCP5-miR-139-RUNX1 Feedback Loop in Regulating Malignant Behavior of Glioma Cells. Mol. Ther. 2016, 24, 1806–1822. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-J.; Kim, M.; Kim, H.-P. Epigenetic regulation of long noncoding RNA UCA1 by SATB1 in breast cancer. Bmb Rep. 2016, 49, 578–583. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Lu, B.; Sun, L.; Yan, X.; Xu, J. Identification of candidate genes or microRNAs associated with the lymph node metastasis of SCLC. Cancer Cell Int. 2018, 18, 161–171. [Google Scholar] [CrossRef]
- Yang, C.; Sun, J.; Liu, W.; Yang, Y.; Chu, Z.; Yang, T.; Gui, Y.; Wang, D. Long noncoding RNA HCP5 contributes to epithelial-mesenchymal transition in colorectal cancer through ZEB1 activation and interacting with miR-139-5p. 11. Am. J. Transl. Res. 2019, 11, 953–963. [Google Scholar]
- Yun, W.-K.; Hu, Y.-M.; Zhao, C.-B.; Yu, D.-Y.; Tang, J.-B. HCP5 promotes colon cancer development by activating AP1G1 via PI3K/AKT pathway. Eur. Rev. Med Pharmacol. Sci. 2019, 23, 2786–2793. [Google Scholar]
- Liang, L.; Xu, J.; Wang, M.; Xu, G.; Zhang, N.; Wang, G.; Zhao, Y. LncRNA HCP5 promotes follicular thyroid carcinoma progression via miRNAs sponge. Cell Death Dis. 2018, 9, 372. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Shen, H.; Fang, D.; Meng, Q.; Xin, Y. LncRNA HCP5 promotes the development of cervical cancer. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 4812–4819. [Google Scholar] [PubMed]
- Olgun, G.; Sahin, O.; Tastan, O. Discovering lncRNA mediated sponge interactions in breast cancer molecular subtypes. BMC Genomics 2018, 19, 650. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Ma, R.; Wang, J.; Wu, X.; Jin, S.; Peng, J.; Tan, R.; Zhang, T.; Li, Y.; Wang, Y. LncRNA2Function: A comprehensive resource for functional investigation of human lncRNAs based on RNA-seq data. BMC Genom. 2015, 16, S2. [Google Scholar] [CrossRef]
- Yan, X.; Hu, Z.; Feng, Y.; Hu, X.; Yuan, J.; Zhao, S.D.; Zhang, Y.; Yang, L.; Shan, W.; He, Q.; et al. Comprehensive Genomic Characterization of Long Non-coding RNAs across Human Cancers. Cancer Cell 2015, 28, 529–540. [Google Scholar] [CrossRef]
- Lanzós, A.; Carlevaro-Fita, J.; Mularoni, L.; Reverter, F.; Palumbo, E.; Guigó, R.; Johnson, R. Discovery of Cancer Driver Long Noncoding RNAs across 1112 Tumour Genomes: New Candidates and Distinguishing Features. Sci. Rep. 2017, 7, 41544. [Google Scholar] [CrossRef] [Green Version]
- De Giorgio, A.; Krell, J.; Harding, V.; Stebbing, J.; Castellano, L. Emerging Roles of Competing Endogenous RNAs in Cancer: Insights from the Regulation of PTEN. Mol. Cell. Biol. 2013, 33, 3976–3982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]
- Mo, X.; Li, T.; Xie, Y.; Zhu, L.; Xiao, B.; Liao, Q.; Guo, J. Identification and functional annotation of metabolism-associated lncRNAs and their related protein-coding genes in gastric cancer. Mol. Genet. Genom. Med. 2018, 6, 728–738. [Google Scholar] [CrossRef]
- Tian, L.; He, Y.; Zhang, H.; Wu, Z.; Li, D.; Zheng, C. Comprehensive analysis of differentially expressed profiles of lncRNAs and mRNAs reveals ceRNA networks in the transformation of diffuse large B-cell lymphoma. Oncol. Lett. 2018, 16, 882–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhu, Y.; Wang, H.; Ji, X. Targeting Long Noncoding RNA in Glioma: A Pathway Perspective. Mol. Ther. Nucleic Acids 2018, 13, 431–441. [Google Scholar] [CrossRef]
- Klase, Z.; Yedavalli, V.S.R.K.; Houzet, L.; Perkins, M.; Maldarelli, F.; Brenchley, J.; Strebel, K.; Liu, P.; Jeang, K.-T. Activation of HIV-1 from Latent Infection via Synergy of RUNX1 Inhibitor Ro5-3335 and SAHA. PLoS Pathog. 2014, 10, e1003997. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; He, Z.; Li, L.; Yang, D.; Liu, G. Expression profiles analysis of long non-coding RNAs identified novel lncRNA biomarkers with predictive value in outcome of cutaneous melanoma. Oncotarget 2017, 8, 1–10. [Google Scholar] [CrossRef]
- Nohata, N.; Abba, M.C.; Gutkind, J.S. Unraveling the oral cancer lncRNAome: Identification of novel lncRNAs associated with malignant progression and HPV infection. Oral Oncol. 2016, 59, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Gallant-Behm, C.; Espinosa, J. ΔNp63α utilizes multiple mechanisms to repress transcription in squamous cell carcinoma cells. Cell Cycle 2013, 12, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Goossens, N.; Hoshida, Y. Hepatitis C virus-induced hepatocellular carcinoma. Clin. Mol. Hepatol. 2015, 21, 105. [Google Scholar] [CrossRef]
- Sanford, T.; Porten, S.; Meng, M.V. Molecular Analysis of Upper Tract and Bladder Urothelial Carcinoma: Results from a Microarray Comparison. PLoS ONE 2015, 10, e0137141. [Google Scholar] [CrossRef] [PubMed]
- Sood, R.; Wong, W.; Jeng, M.; Zehnder, J.L. Gene expression profile of idiopathic thrombocytopenic purpura (ITP). Pediatr. Blood Cancer 2006, 47, 675–677. [Google Scholar] [CrossRef] [PubMed]
- Gaudieri, S.; Kulski, J.K.; Dawkins, R.L. MHC and Disease Associations in Nonhuman Primates. In Molecular Biology and Evolution of Blood Group and MHC Antigens in Primates; Blancher, A., Klein, J., Socha, W.W., Eds.; Springer: Berlin, Germany, 1997; pp. 464–490. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence Name | Location on Chr6 * | Length bp | Orient. | Feature |
|---|---|---|---|---|
| MERC21 | 31,461,669–31,462,057 | 389 | + | LTR fragment |
| HLA-X | 31,461,846–31,462,490 | 645 | + | Silent pseudogene |
| MERC21 | 31,462,544–31,462,625 | 82 | + | LTR fragment |
| HLA-UTR | 31,462,625–31,463,419 | 794 | + | HLA-5′UTR + exon 1 |
| HCP5-5′ncr | 31,462,464–31,463,413 | 950 | + | HLA class I promoter homologs |
| DNase region | 31,463,001–31,463,190 | 190 | + | 100% reliability score |
| HCP5 | 31,463,180–31,465,809 | 2630 | + | lncRNA gene |
| 3′LTR16B | 31,463,420–31,463,715 | 296 | − | 3′LTR (nt 150–446 in HCP5 RNA) |
| ERV3-16A3 | 31,463,716–31,464,933 | 1218 | − | Internal (nt 447–2547 in HCP5 RNA) |
| AluSp | 31,465,920–31,466,225 | 306 | − | Insertion within ERV3-16A3 |
| ERV3-16A3 | 31,466,237–31,467,707 | 1471 | − | Internal |
| THE1B | 31,467,708–31,468,050 | 343 | + | Fragmented insertion within ERV3-16A3 |
| ERV3-16A3 | 31,468,051–31,468,201 | 151 | − | Internal |
| ERV3-16A3 | 31,468,362–31,468,644 | 283 | − | Internal |
| 5′LTR16B2 | 31,468,881–31,469,043 | 163 | − | 5′LTR |
| AluSx | 31,469,044–31,469,282 | 239 | − | Insertion within LTR16B |
| 5′LTR16B2 | 31,469,283–31,469,592 | 310 | − | 5′LTR |
| L2 | 31,469,593–31,470,361 | 769 | − | L2 LINE |
| MLT1E2 | 31,470,734–31,471,317 | 584 | − | ERL-MaLR, LTR fragment |
| L2 | 31,471,325–31,472,047 | 723 | − | L2 LINE |
| HCG26 | 31,471,229–31,472,408 | 1180 | + | LncRNA |
| Cytokine | Cell Type | No: Controls/Tests | Up or Down | Geoprofile (206082_at) | Reference PMID |
|---|---|---|---|---|---|
| IL10 | Peripheral blood mononuclear cells | 4/4 | down | GDS4551 | 23449998 |
| KSHV-IRF4 | lymphoma | 2/2 | up | GDS4956 | 24335298 |
| IFN-alpha | natural killer cells | 1/3 | up | GDS4163 | 20334827 |
| IFN-gamma | keratinocytes | 5/4 | up | GDS1846 | 14760888 |
| IFN-alpha | hepatocytes (24 h) | 3/3 | up | GDS4390 | 22248663 |
| IL28B | hepatocytes (24 h) | 3/3 | up | GDS4390 | 22248663 |
| IFN-II | lung epithelium (24 h | 4/4 | up | GDS2341 | 16800785 |
| IFN-gamma | bronchial epithelium (24 h) | 5/5 | up | GDS1256 | 15985639 |
| IFNg + DXM | bronchial epithelium (24 h) | 5/5 | up | GDS1256 | 15985639 |
| IFN-gamma | macrophages | 6/6 | up | GDS4232 | 22140520 |
| IFN-gamma | microglia (24 h) | 4/4 | up | GDS1036 | 16163375 |
| TNF | endothelial | 4/4 | nd | GDS1542 | 16617158 |
| TNF | keratinocytes | 4/4 | up | GDS1289 | 15722350 |
| IL1 | macrophages | 5/5 | nd | GDS3005 | 18498781 |
| IL6 | macrophages | 5/5 | nd | GDS3005 | 18498781 |
| IL6 | macrophages | 3/3 | nd | GDS3290 | 18511485 |
| Cigarette smoke | small airway cells | 12/12 | down | GDS2486 | 19106307 |
| SNV ID | Chr Position | RE or Pseudogene | HCP5 Position | Ref | Disease or Phenotype |
|---|---|---|---|---|---|
| rs3094228 | 6:31462150 | HLA-X | - | [91] | Thyroid peroxidase antibody positivity |
| rs115846244 | 6:31462283 | HLA-X | - | [43] | Tonsillectomy |
| rs4360170 | 6:31462582 | MERC21 | - | [43] | Cold sores |
| rs3094605 | 6:31462917 | HLA promoter | 5’UTR | [92] | Lung cancer |
| rs3099840 | 6:31462944 | HLA promoter | 5’UTR | [92] | Lung cancer |
| rs3132090 | 6:31462975 | HLA promoter | 5’UTR | [92] | Lung cancer |
| rs2255221 | 6:31463914 | ERV3-16A3 | internal | [93] | HIV-1 control |
| rs2395029 | 6:31464003 | ERV3-16A3 | internal | [94] | Flucloxacillin drug liver injury |
| rs2395029 | 6:31464003 | ERV3-16A3 | internal | [33,34] | HIV-1 control |
| rs2395029 | 6:31464003 | ERV3-16A3 | internal | [95] | AIDS and HIV-1 control |
| rs2395029 | 6:31464003 | ERV3-16A3 | internal | [96] | Psoriasis and psoriatic arthritis |
| rs2395029 | 6:31464003 | ERV3-16A3 | internal | [93] | HIV-1 control |
| rs2395029 | 6:31464003 | ERV3-16A3 | internal | [97] | Abacavir-induced hypersensitivity |
| rs3130907 | 6:31464036 | ERV3-16A3 | internal | [92] | Lung cancer |
| rs2284178 | 6:31464348 | ERV3-16A3 | internal | [98] | MICB measurement |
| rs3094014 | 6:31465781 | ERV3-16A3 | 3′UTR | [92] | Lung cancer |
| rs75640364 | 6:31465789 | ERV3-16A3 | 3′UTR | [99] | Herpes zoster |
| rs3128986 | 6:31465916 | ERV3-16A3 | 3′UTR | [92] | Lung cancer |
| rs79022003 | 6:31465935 | AluSp | 3’UTR | [100] | Eosinophil counts |
| rs3131620 | 6:31466054 | AluSp | 3′UTR | [92] | Lung cancer |
| rs3094604 | 6:31466334 | ERV3-16A3 | 3’UTR | [101] | Squamous cell lung carcinoma |
| rs3094604 | 6:31466334 | ERV3-16A3 | 3′UTR | [92] | Lung cancer |
| rs3094604 | 6:31466334 | ERV3-16A3 | 3’UTR | [102] | MMR vaccine-related febrile seizures |
| rs3131619 | 6:31466554 | ERV3-16A3 | 3’UTR | [103] | Myositis |
| rs3094013 | 6:31466589 | ERV3-16A3 | 3’UTR | [104] | Idiopathic inflammatory myopathies |
| rs3131618 | 6:31466589 | ERV3-16A3 | 3’UTR | [103] | Myositis |
| rs3094013 | 6:31466844 | ERV3-16A3 | 3’UTR | [43] | Tonsillectomy |
| rs2523675 | 6:31468255 | ERV3-16A3 | 3’UTR | [105] | Relapse after cord blood transplantation |
| rs2518028 | 6:31468270 | ERV3-16A3 | 3’UTR | [105] | Relapse after cord blood transplantation |
| rs2523673 | 6:31469218 | LTR16B/AluSx | 3’UTR | [100] | Platelet count |
| Disease/Phenotype | CpG Site | Position Genecode | Feature | Delta | p-Value | Reference |
|---|---|---|---|---|---|---|
| Age-related expression | cg25843003 | 31431312 | LTR16B2 | −0.23 | 2.3 × 10−13 | [121] |
| in monocytes and T cells | cg01082299 | 31431969 | ERV3-16 | −0.001 | 6.86 × 10−6 | |
| SLE: Anti-dsDNA’ | cg25843003 | 31431312 | LTR16B2 | −0.044 | 5.2 × 10−10 | [122] |
| cg00218406 | 31431407 | LTR16B2 | −0.067 | 8.7 × 10−9 | ||
| cg01082299 | 31431969 | ERV3-16 | −0.04 | 1.2 × 10−7 | ||
| cg18808777 | 31431503 | ERV3-16 | −0.051 | 3.0 × 10−10 | ||
| HIV associated marker | cg00218406 | 31431407 | LTR16B2 | [123] | ||
| Influenza vaccination | CpGsite | 31428956 | MER21B | −0.301 | 1.31 × 10−4 | [124] |
| 31431456 | LTR16B2 | |||||
| 31431457 | LTR16B2 | −0.401 | 2.0 × 10−7 | |||
| 31433586 | 3′UTR | |||||
| Endometrium | cg25843003 | 31431312 | LTR16B2 | −0.064 | [125] | |
| Pre to receptive | cg00218406 | 31431407 | LTR16B2 | −0.058 | ||
| cg21684411 | 31431573 | ERV3-16 | −0.071 | |||
| BMI | cg00218406 | 31431407 | LTR16B2 | 0.005 | 8.03 × 10−7 | [64] |
| BMI | cg25843003 | 31431312 | LTR16B2 | 0.002 | 1.51 × 10−6 | |
| Obesity | cg00218406 | 31431407 | LTR16B2 | 0.048 | 4.34 × 10−5 | |
| Obesity | cg25843003 | 31431312 | LTR16B2 | 0.020 | 5.98 × 10−5 |
| Gene Symbol | Gene ID | Interaction Count | Gene Symbol | Gene ID | Interaction Count |
|---|---|---|---|---|---|
| PTGS2 | 5743 | 71 | ICAM1 | 3383 | 9 |
| TNF | 7124 | 55 | NOS3 | 4846 | 9 |
| IL1B | 3553 | 52 | PON1 | 5444 | 9 |
| PTGS1 | 5742 | 42 | SCARB1 | 949 | 9 |
| CASP3 | 836 | 28 | AHR | 196 | 8 |
| BCL2 | 596 | 21 | APOA1 | 335 | 8 |
| AGT | 183 | 20 | TXN1 | 22166 | 8 |
| CTNNB1 | 1499 | 20 | VEGFA | 7422 | 8 |
| CCND1 | 595 | 18 | ABCA1 | 19 | 7 |
| NFKB1 | 4790 | 18 | ALB | 213 | 7 |
| RELA | 5970 | 17 | CCL2 | 6347 | 7 |
| TNFSF10 | 8743 | 17 | IKBKB | 3551 | 7 |
| NOS2 | 4843 | 14 | ITGA2B | 3674 | 7 |
| PARP1 | 142 | 14 | MAPK1 | 5594 | 7 |
| NFKBIA | 4792 | 13 | MAPK3 | 5595 | 7 |
| BAX | 581 | 12 | MYC | 4609 | 7 |
| CDKN1A | 1026 | 12 | PPARG | 5468 | 7 |
| IL4 | 3565 | 12 | SELP | 6403 | 7 |
| IL6 | 3569 | 11 | TCF4 | 6925 | 7 |
| ITGB3 | 3690 | 11 | ABCB1 | 5243 | 6 |
| MMP9 | 4318 | 11 | AKT1 | 207 | 6 |
| CASP9 | 842 | 10 | CYSLTR1 | 10800 | 6 |
| IL1A | 3552 | 10 | JUN | 3725 | 6 |
| PPARA | 5465 | 10 | MMP2 | 4313 | 6 |
| BIRC5 | 332 | 9 | PLAUR | 5329 | 6 |
| CASP8 | 841 | 9 | RB1 | 5925 | 6 |
| Antioncogenic miRNA | HCP5 Action | Gene Symbol and Up- or Downregulated | Cancer Type | Reference |
|---|---|---|---|---|
| miR-203 | sponge | TGFb/SMAD3 | Lung | [56] |
| miR-106b-5p | network | CTSS/FGL2 | Lung | [130] |
| miR-17-5b | network | PDCD1LG2/PDL2 | Lung | [130] |
| miR-126 | GSR/ASCL1 ↑ | Lung metastasis | [152] | |
| MET/GRM8/DACH1 ↓ | Lung metastasis | [152] | ||
| miR-139 | absorption | RUNX1 ↑ | Glioma | [150] |
| miR-139-5p | ZEB1 | Colorectal | [153] | |
| APIG1 ↓ | Colon | [154] | ||
| miR-22-3p | sponge | ST6GAL2 ↑ | Thyroid | [155] |
| mi-186-5p | sponge | ST6GAL2 ↑ | Thyroid | [155] |
| miR-216a-5p | sponge | ST6GAL2 ↑ | Thyroid | [155] |
| miR-15a | adsorption | MACC1 ↑ | Cervical | [156] |
| miR-155 | sponge | Complement genes | Breast | [157] |
| miR-128 | sponge | MACC1 ↑ | Gastric | [163] |
| miR-101 | sponge | NDUFB6 ↓ | Gastric | [163] |
| miR-103a | sponge | NDUFB6 ↓ | Gastric | [163] |
| 24 different mIR | ceRNA | TAP1 ↑ | Lymphoma | [164] |
| ceRNA | PSMB9 ↑ | Lymphoma | [164] | |
| ceRNA | KLF2 ↑ | Lymphoma | [164] | |
| ceRNA | GIPC1 ↑ | Lymphoma | [164] | |
| ceRNA | ETN3A3 ↑ | Lymphoma | [164] | |
| ceRNA | ETN3A1 ↑ | Lymphoma | [164] | |
| ceRNA | CD47 ↑ | Lymphoma | [164] | |
| ceRNA | CCDC50 ↑ | Lymphoma | [164] | |
| ceRNA | LMBR1L ↑ | Lymphoma | [164] | |
| ceRNA | HERC6 ↑ | Lymphoma | [164] |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulski, J.K. Long Noncoding RNA HCP5, a Hybrid HLA Class I Endogenous Retroviral Gene: Structure, Expression, and Disease Associations. Cells 2019, 8, 480. https://doi.org/10.3390/cells8050480
Kulski JK. Long Noncoding RNA HCP5, a Hybrid HLA Class I Endogenous Retroviral Gene: Structure, Expression, and Disease Associations. Cells. 2019; 8(5):480. https://doi.org/10.3390/cells8050480
Chicago/Turabian StyleKulski, Jerzy K. 2019. "Long Noncoding RNA HCP5, a Hybrid HLA Class I Endogenous Retroviral Gene: Structure, Expression, and Disease Associations" Cells 8, no. 5: 480. https://doi.org/10.3390/cells8050480
APA StyleKulski, J. K. (2019). Long Noncoding RNA HCP5, a Hybrid HLA Class I Endogenous Retroviral Gene: Structure, Expression, and Disease Associations. Cells, 8(5), 480. https://doi.org/10.3390/cells8050480