Capturing the Biologic Onset of Inflammatory Bowel Diseases: Impact on Translational and Clinical Science


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. What We Know about Preclinical CD
3. Is Screening for IBD Feasible? In Whom? And How Should it be Done?
4. Can IBD Be “Intercepted” or “Predicted”?
5. What Is the Potential Impact of Translational Research in Preclinical IBD? The Example of the Microbiome
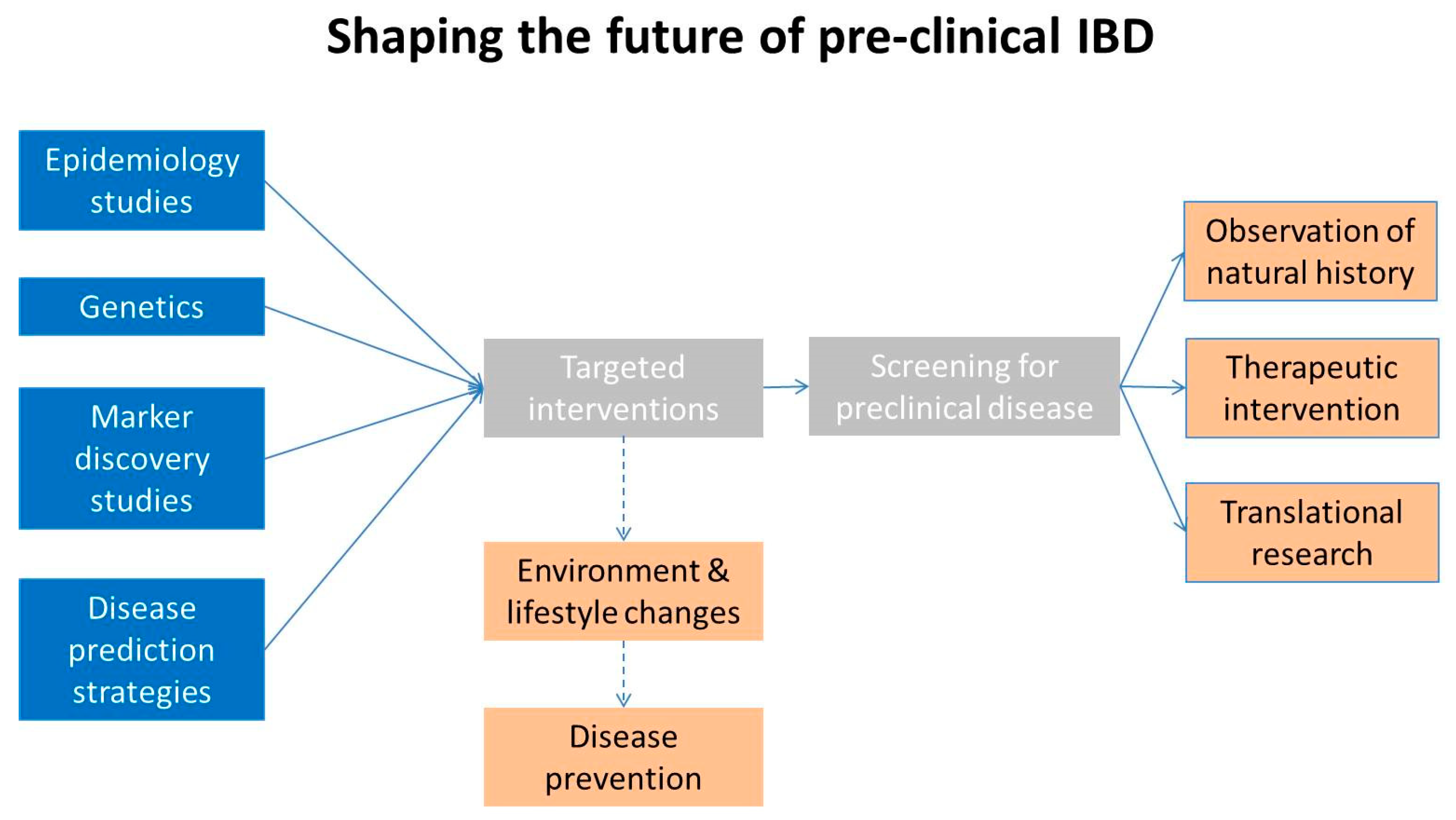
6. The Future
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Molodecky, N.A.; Soon, I.S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Barkema, H.W.; et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012, 142, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, D. The Coming of Age of Inflammatory Bowel Diseases in Asia. Inflamm. Intest. Dis. 2017, 2, 93–94. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.K. How Does the Epidemiology of Inflammatory Bowel Disease Differ between East and West? A Korean Perspective. Inflamm. Intest. Dis. 2017, 2, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.Q.; Jiang, D.; Hoffman, S.N.; Guntaka, S.; Mays, J.L.; Wang, A.; Gomes, J.; Sorrentino, D. Impact of Diagnostic Delay and Associated Factors on Clinical Outcomes in a U.S. Inflammatory Bowel Disease Cohort. Inflamm. Bowel Dis. 2017, 23, 1825–1831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benevento, G.; Avellini, C.; Terrosu, G.; Geraci, M.; Lodolo, I.; Sorrentino, D. Diagnosis and assessment of Crohn’s disease: The present and the future. Expert Rev. Gastroenterol. Hepatol. 2010, 4, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Cosnes, J.; Cattan, S.; Blain, A.; Beaugerie, L.; Carbonnel, F.; Parc, R.; Gendre, J.P. Longterm evolution of disease behavior of Crohn’s disease. Inflamm. Bowel Dis. 2002, 8, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Hovde, O.; Moum, B.A. Epidemiology and clinical course of Crohn’s disease: Results from observational studies. World J. Gastroenterol. 2012, 18, 1723–1731. [Google Scholar] [CrossRef] [PubMed]
- Fiorino, G.; Peyrin-Biroulet, L.; Naccarato, P.; Szabò, H.; Sociale, O.R.; Vetrano, S.; Fries, W.; Montanelli, A.; Repici, A.; Malesci, A.; et al. Mo1041 MRE Findings at Crohn’s Disease diagnosis: Disease activity, bowel damage and predictive value for long-term outcomes. Gastroenterology 2012, 142, S579–S580. [Google Scholar] [CrossRef]
- Prantera, C.; Levenstein, S.; Capocaccia, R.; Mariotti, S.; Luzi, C.; Cosintino, R.; Simi, M. Prediction of surgery for obstruction in Crohn’s ileitis. A study of 64 patients. Dig. Dis. Sci. 1987, 32, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Sands, B.E.; Arsenault, J.E.; Rosen, M.J.; Alsahli, M.; Bailen, L.; Banks, P.; Bensen, S.; Bousvaros, A.; Cave, D.; Cooley, J.S.; et al. Risk of early surgery for Crohn’s disease: Implications for early treatment strategies. Am. J. Gastroenterol. 2003, 98, 2712–2718. [Google Scholar] [CrossRef] [PubMed]
- Rieder, F.; Brenmoehl, J.; Leeb, S.; Scholmerich, J.; Rogler, G. Wound healing and fibrosis in intestinal disease. Gut 2007, 56, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, D. Role of Biologics and Other Therapies in Stricturing Crohn’s Disease: What Have We Learnt So Far? Digestion 2008, 77, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Louis, E.; Collard, A.; Oger, A.F.; Degroote, E.; Aboul Nasr, E.l.; Yafi, F.A.; Belaiche, J. Behaviour of Crohn’s disease according to the Vienna classification: Changing pattern over the course of the disease. Gut 2001, 49, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Beasley, S.; Cameron, D.S.; McLain, B. Diagnostic delay in Crohn’s disease. Pediatr. Surg. Int. 1992, 7, 171–173. [Google Scholar] [CrossRef]
- Vavricka, S.R.; Spigaglia, S.M.; Rogler, G.; Pittet, V.; Michetti, P.; Felley, C.; Mottet, C.; Braegger, C.P.; Rogler, D.; Straumann, A.; et al. Systemic evaluation of risk factors for diagnostic delay in inflammatory bowel disease. Inflamm. Bowel Dis. 2012, 18, 496–505. [Google Scholar] [CrossRef] [PubMed]
- D’Haens, G.R.; Geboes, K.; Peeters, M.; Baert, F.; Penninckx, F.; Rutgeerts, P. Early lesions of recurrent Crohn’s disease caused by infusion of intestinal contents in excluded ileum. Gastroenterology 1998, 114, 262–267. [Google Scholar] [CrossRef]
- Kelly, J.K.; Sutherland, L.R. The chronological sequence in the pathology of Crohn’s disease. J. Clin. Gastroenterol. 1988, 10, 28–33. [Google Scholar] [CrossRef]
- Greenstein, A.J.; Lachman, P.; Sachar, D.B.; Springhorn, J.; Heimann, T.; Janowitz, H.D.; Aufses, A.H., Jr. Perforating and nonperforating indications for repeated operations in Crohn’s disease: Evidence for two clinical forms. Gut 1988, 29, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Limketkai, B.N.; Bayless, T.M. Editorial: Can stenosis in ileal Crohn’s disease be prevented by current therapy? Am. J. Gastroenterol. 2013, 108, 1755–1756. [Google Scholar] [CrossRef]
- Jacene, H.A.; Ginsburg, P.; Kwon, J.; Nguyen, G.C.; Montgomery, E.A.; Bayless, T.M.; Wahl, R.L. Prediction of the need for surgical intervention in obstructive Crohn’s disease by 18F-FDG PET/CT. J. Nucl. Med. 2009, 50, 1751–1759. [Google Scholar] [CrossRef]
- Esch, A.; Bourrier, A.; Seksik, P.; Nion-Larmurier, I.; Sokol, H.; Beaugerie, L.; Cosnes, J. What is the prognosis of silent Crohn’s Disease? Gastroenterology 2013, 144, S-102. [Google Scholar] [CrossRef]
- Mayberry, J.F.; Ballantyne, K.C.; Hardcastle, J.D.; Mangham, C.; Pye, G. Epidemiological study of asymptomatic inflammatory bowel disease: The identification of cases during a screening programme for colorectal cancer. Gut 1989, 30, 481–483. [Google Scholar] [CrossRef] [PubMed]
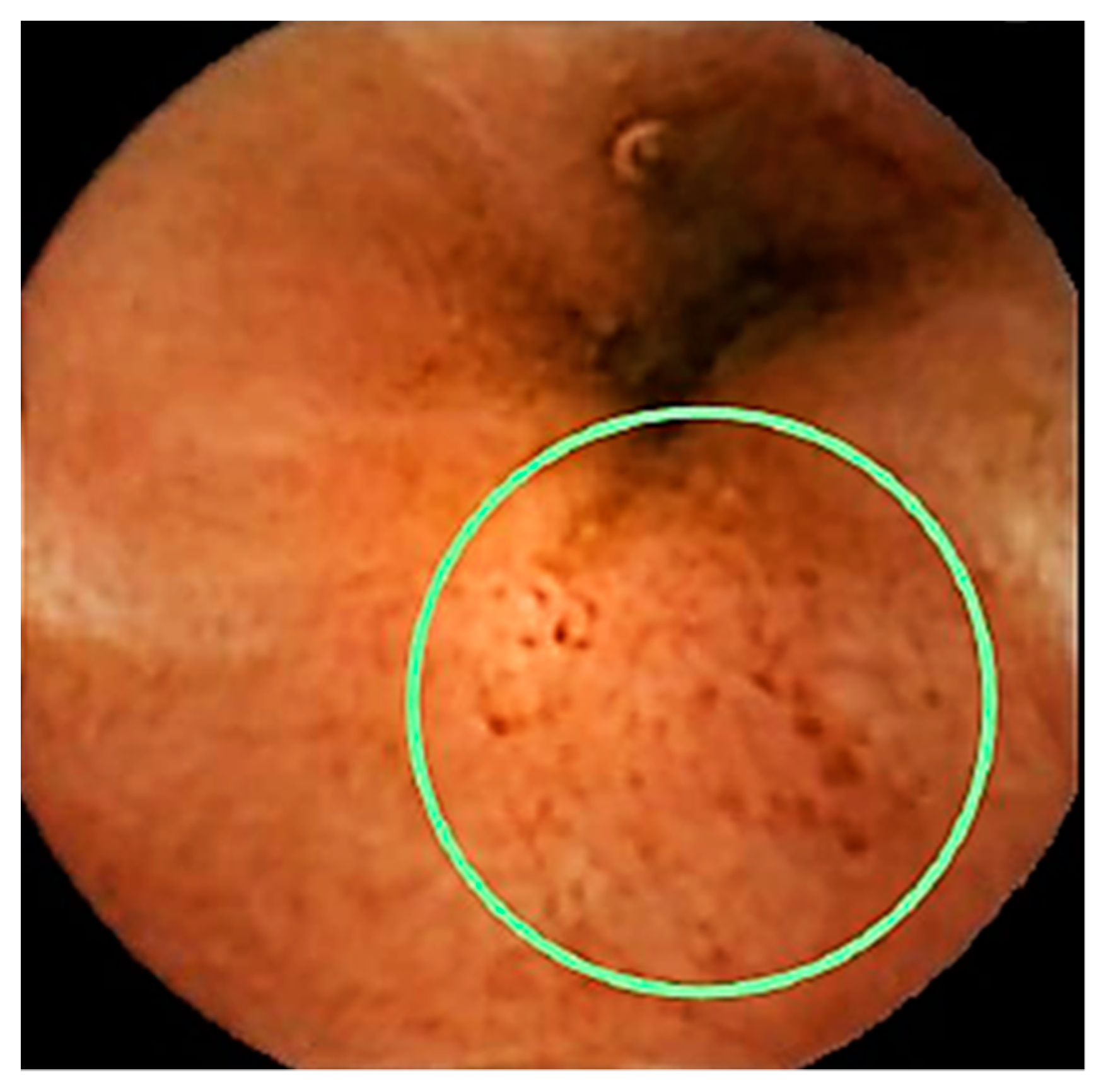
- Rodríguez-Lago, I.; Merino, O.; Azagra, I.; Maiz, A.; Zapata, E.; Higuera, R.; Montalvo, I.; Fernández-Calderón, M.; Arreba, P.; Carrascosa, J.; et al. Characteristics and Progression of Preclinical Inflammatory Bowel Disease. Clin. Gastroenterol. Hepatol. 2018, 16, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.; Sawaya, G.F.; Moyer, V.A.; Calonge, N. Reconsidering the criteria for evaluating proposed screening programs: Reflections from 4 current and former members of the U.S. Preventive services task force. Epidemiol. Rev. 2011, 33, 20–35. [Google Scholar] [CrossRef] [PubMed]
- Orholm, M.; Fonager, K.; Sørensen, H.T. Risk of ulcerative colitis and Crohn’s disease among offspring of patients with chronic inflammatory bowel disease. Am. J. Gastroenterol. 1999, 94, 3236–3238. [Google Scholar] [CrossRef] [PubMed]
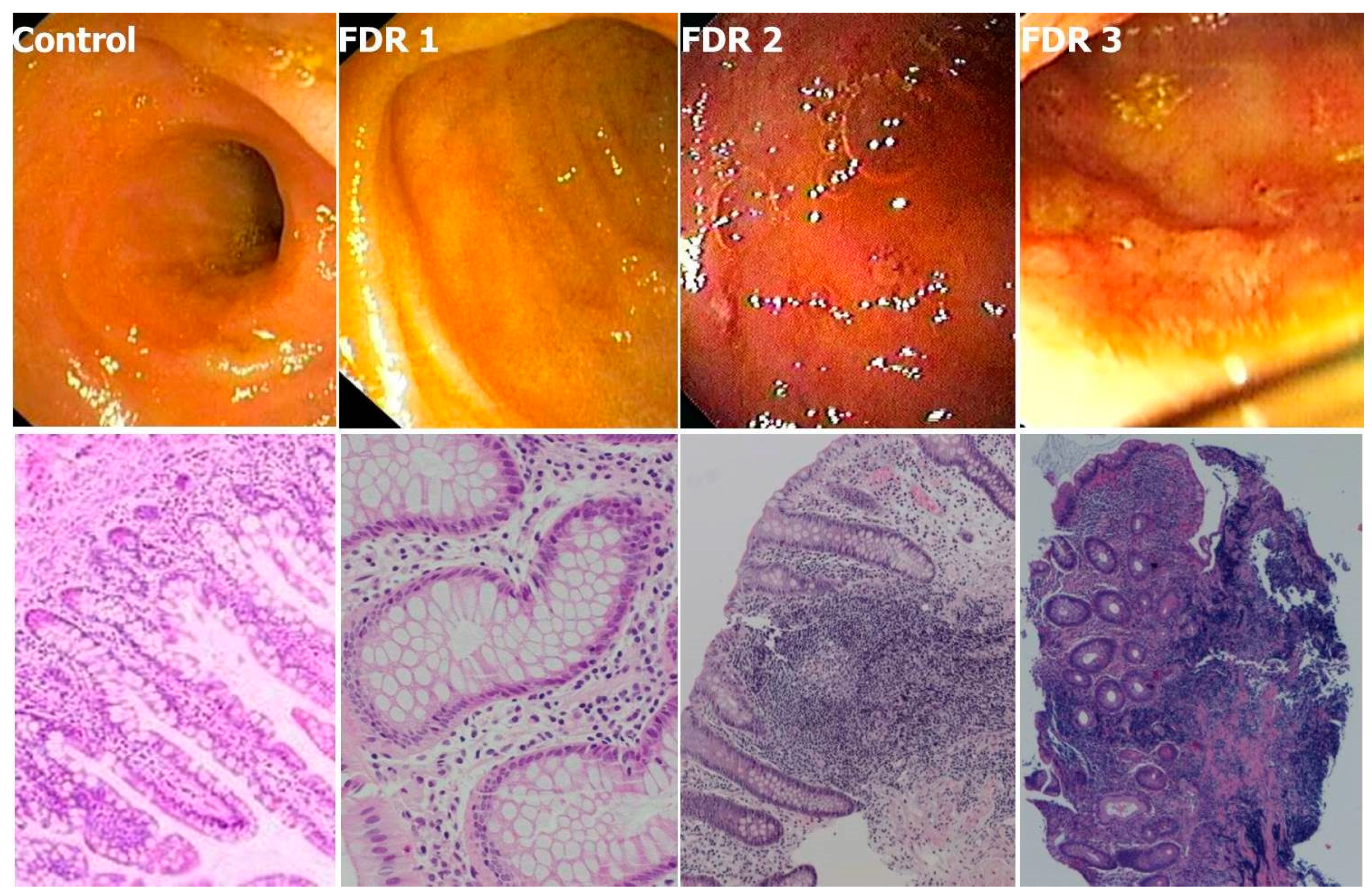
- Sorrentino, D.; Avellini, C.; Geraci, M.; Dassopoulos, T.; Zarifi, D.; Vadalaʼ di Prampero, S.F.; Benevento, G. Tissue studies in screened first-degree relatives reveal a distinct Crohn’s disease phenotype. Inflamm. Bowel Dis. 2014, 20, 1049–1056. [Google Scholar] [CrossRef]
- Thjodleifsson, B.; Sigthorsson, G.; Cariglia, N.; Reynisdottir, I.; Gudbjartsson, D.F.; Kristjansson, K.; Meddings, J.B.; Gudnason, V.; Wandall, J.H.; Andersen, L.P.; et al. Subclinical intestinal inflammation: An inherited abnormality in Crohn’s disease relatives? Gastroenterology 2003, 124, 1728–1737. [Google Scholar] [CrossRef]
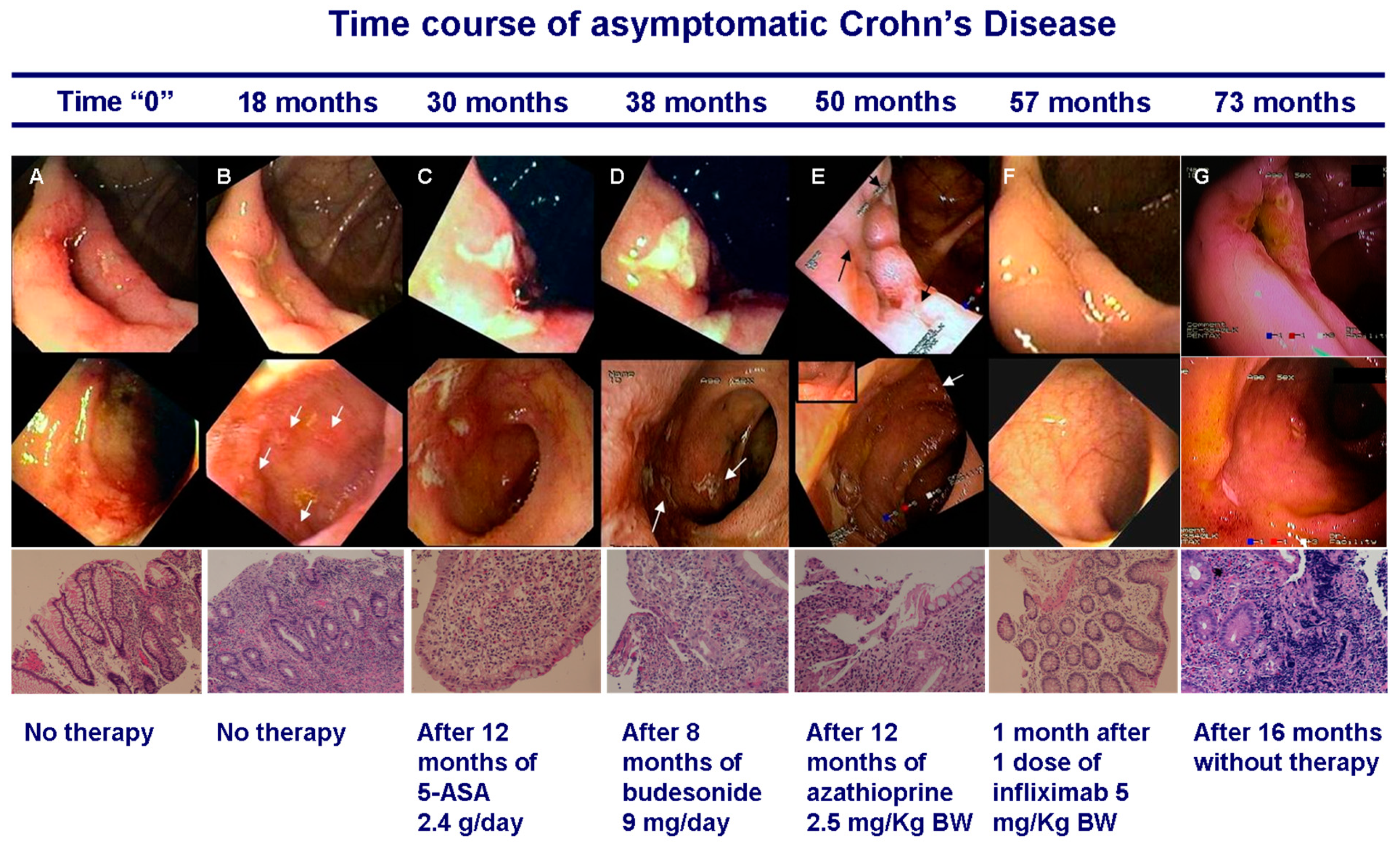
- Sorrentino, D.; Avellini, C.; Geraci, M.; Vadala, S. Pre-clinical Crohn’s disease: Diagnosis, treatment and six year follow-up. J. Crohns Colitis 2014, 8, 702–707. [Google Scholar] [CrossRef]
- Ahmad, T.; Satsangi, J.; McGovern, D.; Bunce, M.; Jewell, D.P. Review article: The genetics of inflammatory bowel disease. Aliment. Pharmacol. Ther. 2001, 15, 731–748. [Google Scholar] [CrossRef]
- Pham, M.; Leach, S.T.; Lemberg, D.A.; Day, A.S. Subclinical Intestinal Inflammation in Siblings of Children with Crohn’s Disease. Dig. Dis. Sci. 2010, 55, 3502–3507. [Google Scholar] [CrossRef]
- Montalto, M.; Curigliano, V.; Santoro, L.; Armuzzi, A.; Cammarota, G.; Covino, M.; Mentella, M.C.; Ancarani, F.; Manna, R.; Gasbarrini, A.; et al. Fecal Calprotectin in First-Degree Relatives of Patients with Ulcerative Colitis. Am. J. Gastroenterol. 2007, 102, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Tamboli, C.P.; Richard, F.; Colombel, J.-F. Fecal calprotectin in Crohn’s disease: New family ties. Gastroenterology 2003, 124, 1971–1974. [Google Scholar] [CrossRef]
- Zhulina, Y.; Shamikh, A.; Nyhlin, N.; Wickbom, A.; Bohr, J.; Hahn-Strömberg, V.; Peterson, C.G.B.; Gustavsson, A.; Bodin, L.; Tysk, C.; et al. Subclinical Inflammation with Increased Neutrophil Activity in Healthy Twin Siblings Reflect Environmental Influence in the Pathogenesis of Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2013, 19, 1725–1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keita, A.V.; Lindqvist, C.M.; Ost, A.; Magana, C.D.L.; Schoultz, I.; Halfvarson, J. OP001 Gut barrier dysfunction: A primary defect in twins with Crohn’s disease predominantly caused by genetic predisposition. J. Crohns Colitis 2018, 12, 1200–1209. [Google Scholar] [CrossRef] [PubMed]
- Hammer, T.; Lophaven, S.N.; Nielsen, K.R.; Von Euler-Chelpin, M.; Weihe, P.; Munkholm, P.; Burisch, J.; Lynge, E.; Euler-Chelpin, M. Inflammatory bowel diseases in Faroese-born Danish residents and their offspring: Further evidence of the dominant role of environmental factors in IBD development. Aliment. Pharmacol. Ther. 2017, 45, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Xavier, R.J.; Gevers, D. The Microbiome in Inflammatory Bowel Diseases: Current Status and the Future Ahead. Gastroenterology 2014, 146, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Maloy, K.J.; Powrie, F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 2011, 474, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Israeli, E.; Grotto, I.; Gilburd, B.; Balicer, R.D.; Goldin, E.; Wiik, A.; Shoenfeld, Y. Anti-Saccharomyces cerevisiae and antineutrophil cytoplasmic antibodies as predictors of inflammatory bowel disease. Gut 2005, 54, 1232–1236. [Google Scholar] [CrossRef] [PubMed]
- Van Schaik, F.D.; Oldenburg, B.; Hart, A.R.; Siersema, P.D.; Lindgren, S.; Grip, O.; Teucher, B.; Kaaks, R.; Bergmann, M.M.; Boeing, H.; et al. Serological markers predict inflammatory bowel disease years before the diagnosis. Gut 2013, 62, 683–688. [Google Scholar] [CrossRef]
- Choung, R.S.; Princen, F.; Stockfisch, T.P.; Torres, J.; Maue, A.C.; Porter, C.K.; Leon, F.; De Vroey, B.; Singh, S.; Riddle, M.S.; et al. REDICTS Study Team. Serologic microbial associated markers can predict Crohn’s disease behaviour years before disease diagnosis. Aliment. Pharmacol. Ther. 2016, 43, 1300–1310. [Google Scholar] [CrossRef] [PubMed]
- GEM Project. Crohn’s and Colitis Canada Inflammatory Bowel Disease GEM Project. Available online: http://www.gemproject.ca/ (accessed on 24 April 2019).
- Papp, M.; Lakatos, P.L. Serological studies in inflammatory bowel disease: How important are they? Curr. Opin. Gastroenterol. 2014, 30, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Lönnkvist, M.H.; Theodorsson, E.; Holst, M.; Ljung, T.; Hellström, P.M. Blood chemistry markers for evaluation of inflammatory activity in Crohn’s disease during infliximab therapy. Scand. J. Gastroenterol. 2011, 46, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Hedin, C.; van der Gast, C.J.; Rogers, G.B.; Cuthbertson, L.; McCartney, S.; Stagg, A.J.; Lindsay, J.O.; Whelan, K. Siblings of patients with Crohn’s disease exhibit a biologically relevant dysbiosis in mucosal microbial metacommunities. Gut 2016, 65, 944–953. [Google Scholar] [CrossRef] [PubMed]
- Hyams, J.; Crandall, W.; Kugathasan, S.; Griffiths, A.; Olson, A.; Johanns, J.; Liu, G.; Travers, S.; Heuschkel, R.; Markowitz, J.; et al. EACH Study Group. Induction and maintenance infliximab therapy for the treatment of moderate-to-severe Crohn’s disease in children. Gastroenterology 2007, 132, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Sipponen, T.; Kärkkäinen, P.; Savilahti, E.; Kolho, K.-L.; Nuutinen, H.; Turunen, U.; Färkkilä, M. Correlation of faecal calprotectin and lactoferrin with an endoscopic score for Crohn’s disease and histological findings. Aliment. Pharmacol. Ther. 2008, 28, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Park, S.K.; Ye, B.D.; Kim, K.O.; Park, C.H.; Lee, W.S.; Jang, B.I.; Jeen, Y.T.; Choi, M.G.; Kim, H.J.; Korean Gut Image Study Group. Guidelines for video capsule endoscopy: Emphasis on Crohn’s disease. Clin. Endosc. 2015, 48, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Teshima, C.W.; Goodman, K.J.; El-Kalla, M.; Turk, S.; El-Matary, W.; Valcheva, R.; Danchak, R.; Gordon, M.; Ho, P.; Mullins, A.; et al. Increased Intestinal Permeability in Relatives of Patients With Crohn’s Disease Is Not Associated With Small Bowel Ulcerations. Clin. Gastroenterol. Hepatol. 2017, 15, 1413–1418. [Google Scholar] [CrossRef] [PubMed]
- The Biologic Onset of Crohn’s Disease: A Screening Study in First Degree Relatives. Available online: https://clinicaltrials.gov/ct2/show/NCT03291743 (accessed on 5 June 2019).
- Nash, P.T.; Florin, T.H. Tumour necrosis factor inhibitors. Med. J. Aust. 2005, 183, 205–208. [Google Scholar] [PubMed]
- Frey, O.; Reichel, A.; Bonhagen, K.; Morawietz, L.; Rauchhaus, U.; Kamradt, T. Regulatory T cells control the transition from acute into chronic inflammation in glucose-6-phosphate isomerase-induced arthritis. Ann. Rheum. Dis. 2010, 69, 1511–1518. [Google Scholar] [CrossRef]
- Saleem, B.; Keen, H.; Goeb, V.; Parmar, R.; Nizam, S.; Hensor, E.M.; Churchman, S.M.; Quinn, M.; Wakefield, R.; Conaghan, P.G.; et al. Patients with RA in remission on TNF blockers: When and in whom can TNF blocker therapy be stopped? Ann. Rheum. Dis. 2010, 69, 1636–1642. [Google Scholar] [CrossRef]
- A Harry, R.; Anderson, A.E.; Isaacs, J.D.; Hilkens, C.M.U. Generation and characterisation of therapeutic tolerogenic dendritic cells for rheumatoid arthritis. Ann. Rheum. Dis. 2010, 69, 2042–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roord, S.T.; de Jager, W.; Boon, L.; Wulffraat, N.; Martens, A.; Prakken, B.; van Wijk, F. Autologous bone marrow transplantation in autoimmune arthritis restores immune homeostasis through CD4+CD25+Foxp3+ regulatory T cells. Blood 2008, 111, 5233–5241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radbruch, A.; Thiel, A. Cell therapy for autoimmune diseases: Does it have a future? Ann. Rheum. Dis. 2004, 63, ii96–ii101. [Google Scholar] [CrossRef] [PubMed]
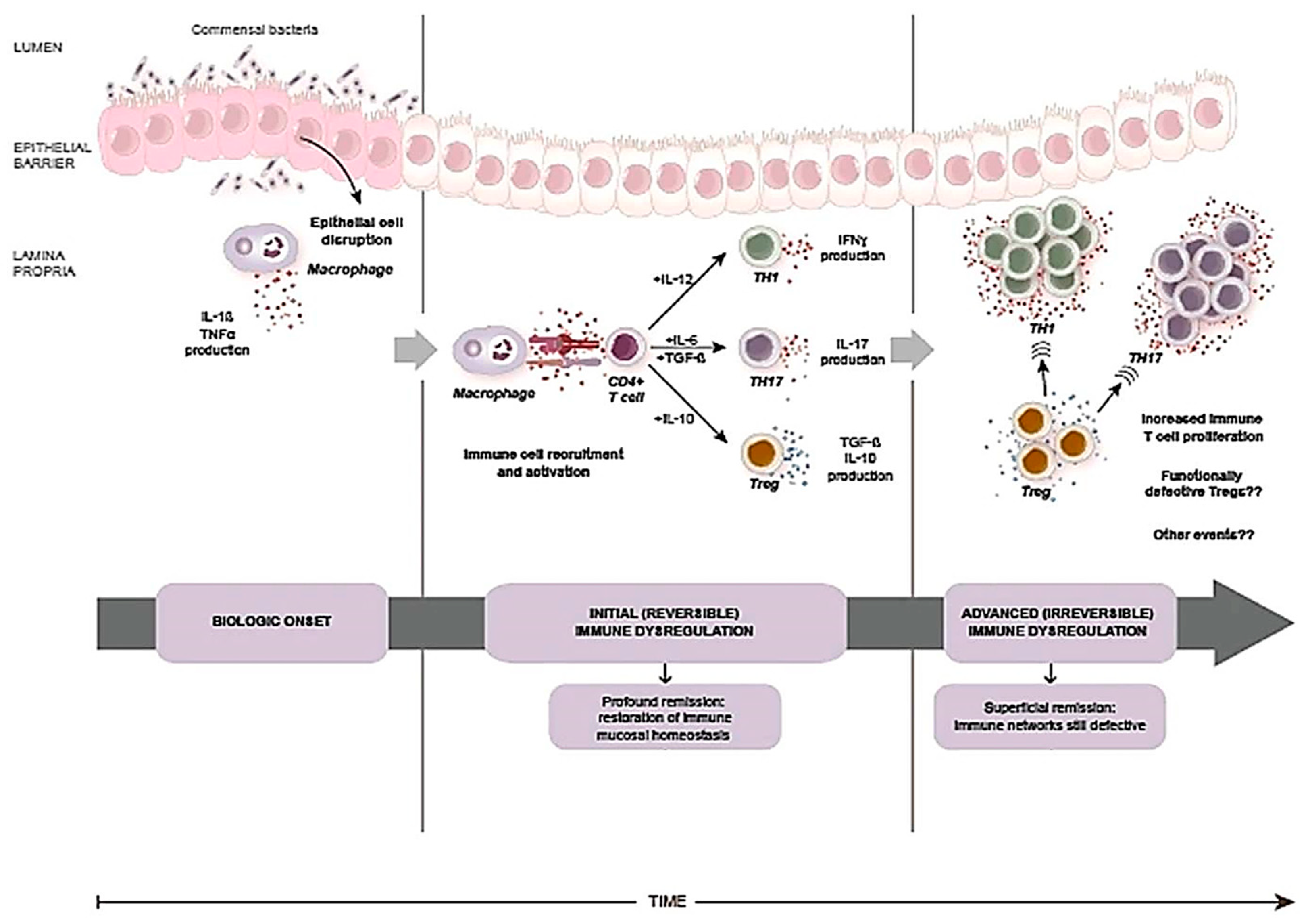
- Bouma, G.; Strober, W. The immunological and genetic basis of inflammatory bowel disease. Nat. Rev. Immunol. 2003, 3, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Barnes, M.J.; Powrie, F. Regulatory T Cells Reinforce Intestinal Homeostasis. Immunity 2009, 31, 401–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartor, R.B. Mechanisms of disease: Pathogenesis of Crohn’s disease and ulcerative colitis. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 390–407. [Google Scholar] [CrossRef] [PubMed]
- Van den Brande, J.M.; Braat, H.; van den Brink, G.R.; Versteeg, H.H.; Bauer, C.A.; Hoedemaeker, I.; van Montfrans, C.; Hommes, D.W.; Peppelenbosch, M.P.; van Deventer, S.J. Infliximab but not etanercept induces apoptosis in lamina propria T-lymphocytes from patients with Crohn’s disease. Gastroenterology 2003, 124, 1774–1785. [Google Scholar] [CrossRef]
- Peake, S.T.C.; Mann, E.R.; Al-Hassi, H.O.; Hart, A.L.; Bernardo, D.; Knight, S.C. Mechanisms of Action of Anti–tumor Necrosis Factor α Agents in Crohn’s Disease. Inflamm. Bowel Dis. 2013, 19, 1546–1555. [Google Scholar] [CrossRef]
- Danese, S. Mechanisms of action of infliximab in inflammatory bowel disease: An anti-inflammatory multitasker. Dig. Liver Dis. 2008, 40, S225–S228. [Google Scholar] [CrossRef]
- Hölttä, V.; Sipponen, T.; Westerholm-Ormio, M.; Salo, H.M.; Kolho, K.L.; Färkkilä, M.; Savilahti, E.; Vaarala, O.; Klemetti, P. In Crohn’s disease, anti-TNF-α treatment changes the balance between mucosal IL-17, FOXP3, and CD4 Cells. ISRN Gastroenterol 2012, 2012, 505432. [Google Scholar] [CrossRef]
- Suenaert, P.; Bulteel, V.; Lemmens, L.; Noman, M.; Geypens, B.; Van Assche, G.; Geboes, K.; Ceuppens, J.L.; Rutgeerts, P. Anti-tumor necrosis factor treatment restores the gut barrier in Crohn’s disease. Am. J. Gastroenterol. 2002, 97, 2000–2004. [Google Scholar] [CrossRef] [PubMed]
- Di Sabatino, A.; Pender, S.L.; Jackson, C.L.; Prothero, J.D.; Gordon, J.N.; Picariello, L.; Rovedatti, L.; Docena, G.; Monteleone, G.; Rampton, D.S. Functional modulation of Crohn’s disease myofibroblasts by anti-tumor necrosis factor antibodies. Gastroenterology 2007, 133, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Di Sabatino, A.; Biancheri, P.; Piconese, S.; Rosado, M.M.; Ardizzone, S.; Rovedatti, L.; Ubezio, C.; Massari, A.; Sampietro, G.M.; Foschi, D.; et al. Peripheral regulatory T cells and serum transforming growth factor-β: Relationship with clinical response to infliximab in Crohn’s disease. Inflamm. Bowel Dis. 2010, 16, 1891–1897. [Google Scholar] [CrossRef] [PubMed]
- Rubin, D.T.; Uluscu, O.; Sederman, R. Response to biologic therapy in Crohn’s disease is improved with early treatment: An analysis of health claims data. Inflamm. Bowel Dis. 2012, 18, 2225–2231. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Xia, X.; Wu, W.; Wu, R.; Tang, M.; Chen, T.; Xu, F.; Cong, Y.; Xu, X.; Liu, Z. Anti-tumour necrosis factor therapy enhances mucosal healing through down-regulation of interleukin-21 expression and T helper type 17 cell infiltration in Crohn’s disease. Clin. Exp. Immunol. 2013, 173, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Rismo, R.; Olsen, T.; Cui, G.; Paulssen, E.J.; Christiansen, I.; Johnsen, K.; Florholmen, J.; Goll, R. Normalization of mucosal cytokine gene expression levels predicts long-term remission after discontinuation of anti-TNF therapy in Crohn’s disease. Scand. J. Gastroenterol. 2013, 48, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Halfvarson, J.; Bodin, L.; Tysk, C.; Lindberg, E.; Järnerot, G. Inflammatory bowel disease in a Swedish twin cohort: A long-term follow-up of concordance and clinical characteristics. Gastroenterology 2003, 124, 1767–1773. [Google Scholar] [CrossRef]
- Liu, J.Z.; Anderson, C.A. Genetic studies of Crohn’s disease: Past, present and future. Best Pract. Res. Clin. Gastroenterol. 2014, 28, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Hugot, J.P.; Chamaillard, M.; Zouali, H.; Lesage, S.; Cézard, J.P.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.A.; Gassull, M.; et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001, 411, 599–603. [Google Scholar] [CrossRef]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef]
- Singh, P.; Singh, S.; Pandi-Jain, G.S. Effective heart disease prediction system using data mining techniques. Int. J. Nanomed. 2018, 13, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Weng, C.-H.; Huang, T.C.-K.; Han, R.-P. Disease prediction with different types of neural network classifiers. Telematics Informatics 2016, 33, 277–292. [Google Scholar] [CrossRef]
- Schoultz, I.; Keita, Å.V. Cellular and Molecular Therapeutic Targets in Inflammatory Bowel Disease—Focusing on Intestinal Barrier Function. Cells 2019, 8, 193. [Google Scholar] [CrossRef] [PubMed]
- Munkholm, P.; Langholz, E.; Hollander, D.; Thornberg, K.; Orholm, M.; Katz, K.D.; Binder, V. Intestinal permeability in patients with Crohn’s disease and ulcerative colitis and their first degree relatives. Gut 1994, 35, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Hollander, D.; Vadheim, C.M.; Brettholz, E.; Petersen, G.M.; Delahunty, T.; Rotter, J.I. Increased intestinal permeability in patients with Crohn’s disease and their relatives. A possible etiologic factor. Ann. Intern. Med. 1986, 105, 883–885. [Google Scholar] [CrossRef] [PubMed]
- Strober, W.; Fuss, I.J. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology 2011, 140, 1756–1767. [Google Scholar] [CrossRef] [PubMed]
- Joossens, M.; Van Steen, K.; Branche, J.; Sendid, B.; Rutgeerts, P.; Vasseur, F.; Poulain, D.; Broly, F.; Colombel, J.F.; Vermeire, S.; et al. Familial aggregation and antimicrobial response dose-dependently affect the risk for Crohn’s disease. Inflamm. Bowel Dis. 2010, 16, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Sendid, B.; Quinton, J.F.; Charrier, G.; Goulet, O.; Cortot, A.; Grandbastien, B.; Poulain, D.; Colombel, J.F. Anti-Saccharomyces cerevisiae mannan antibodies in familial Crohn’s disease. Am. J. Gastroenterol. 1998, 93, 1306–1310. [Google Scholar] [CrossRef]
- Sutton, C.L.; Yang, H.; Li, Z.; Rotter, J.I.; Targan, S.R.; Braun, J. Familial expression of anti -Saccharomyces cerevisiae mannan antibodies in affected and unaffected relatives of patients with Crohn’s disease. Gut 2000, 46, 58–63. [Google Scholar] [CrossRef]
- Annese, V.; Andreoli, A.; Andriulli, A.; Dinca, R.; Gionchetti, P.; Latiano, A.; Lombardi, G.; Piepoli, A.; Poulain, D.; Sendid, B.; et al. Familial expression of anti-saccharomyces cerevisiae mannan antibodies in Crohn’s disease and ulcerative colitis: A GISC study. Am. J. Gastroenterol. 2001, 96, 2407–2412. [Google Scholar] [CrossRef]
- Elmgreen, J.; Both, H.; Binder, V. Familial occurrence of complement dysfunction in Crohn’s disease: Correlation with intestinal symptoms and hypercatabolism of complement. Gut 1985, 26, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the Human Intestinal Microbial Flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, S.J.; Musfeldt, M.; Wenderoth, D.F.; Hampe, J.; Brant, O.; Fölsch, U.R.; Timmis, K.N.; Schreiber, S. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut 2004, 53, 685–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ananthakrishnan, A.; Luo, C.; Yajnik, V.; Khalili, H.; Garber, J.; Stevens, B.; Cleland, T.; Xavier, R.J. Gut microbiome function predicts response to anti-integrin biologic therapy in inflammatory bowel disease. Cell Host Microbe 2017, 21, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Shaw, K.A.; Bertha, M.; Hofmekler, T.; Chopra, P.; Vatanen, T.; Srivatsa, A.; Prince, J.; Kumar, A.; Sauer, C.; Zwick, M.E.; et al. Dysbiosis, inflammation, and response to treatment: A longtitudinal study of pediatric subjects with newly diagnosed inflammatory bowel disease. Genome Med. 2016, 8, 75. [Google Scholar] [CrossRef] [PubMed]
- A Commitment to Inflammatory Bowel Disease (IBD). Available online: https://www.janssen.com/immunology/ibd-commitment (accessed on 24 April 2019).
- Walker, R.W.; Clemente, J.C.; Peter, I.; Loos, R.J.F. The prenatal gut microbiome: Are we colonized with bacteria in utero? Pediatr. Obes. 2017, 12, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Sartor, R.B. Genetics and Environmental Interactions Shape the Intestinal Microbiome to Promote Inflammatory Bowel Disease Versus Mucosal Homeostasis. Gastroenterology 2010, 139, 1816–1819. [Google Scholar] [CrossRef]
- Joossens, M.; Huys, G.; Cnockaert, M.; De Preter, V.; Verbeke, K.; Rutgeerts, P.; Vandamme, P.; Vermeire, S. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 2011, 60, 631–637. [Google Scholar] [CrossRef]
- Sokol, H.; Seksik, P.; Furet, J.P.; Firmesse, O.; Nion-Larmurier, I.; Beaugerie, L.; Cosnes, J.; Corthier, G.; Marteau, P.; Doré, J. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm. Bowel Dis. 2009, 15, 1183–1189. [Google Scholar] [CrossRef]
- Varela, E.; Manichanh, C.; Gallart, M.; Torrejon, A.; Borruel, N.; Casellas, F.; Guarner, F.; Antolin, M. Colonisation by Faecalibacterium prausnitzii and maintenance of clinical remission in patients with ulcerative colitis. Aliment. Pharmacol. Ther. 2013, 38, 151–161. [Google Scholar] [CrossRef]
- Darfeuille-Michaud, A.; Neut, C.; Barnich, N.; Lederman, E.; Di Martino, P.; Desreumaux, P.; Gambiez, L.; Joly, B.; Cortot, A.; Colombel, J.F. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology 1998, 115, 1405–1413. [Google Scholar] [CrossRef]
- Palmela, C.; Chevarin, C.; Xu, Z.; Torres, J.; Sevrin, G.; Hirten, R.; Barnich, N.; Ng, S.C.; Colombel, J.F. Adherent-invasive Escherichia coli in inflammatory bowel disease. Gut 2018, 67, 574–587. [Google Scholar] [CrossRef] [PubMed]
- Ogura, Y.; Bonen, D.K.; Inohara, N.; Nicolae, D.L.; Chen, F.F.; Ramos, R.; Britton, H.; Moran, T.; Karaliuskas, R.; Duerr, R.H.; et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 2001, 411, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Hamm, C.M.; Gulati, A.S.; Sartor, R.B.; Chen, H.; Wu, X.; Zhang, T.; Rohlf, F.J.; Zhu, W.; Gu, C.; et al. Inflammatory bowel diseases phenotype, C. difficile and NOD2 genotype are associated with shifts in human ileum associated microbial composition. PLoS ONE 2012, 7, e26284. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Khosravi, A.; Kusumawardhani, I.P.; Kwon, A.H.; Vasconcelos, A.C.; Cunha, L.D.; Mayer, A.E.; Shen, Y.; Wu, W.L.; Kambal, A.; et al. Gene—Microbiota interactions contribute to the pathogenesis of inflammatory bowel disease. Science 2016, 352, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Bennet, J.; Brinkman, M. Treatment of ulcerative colitis by implantation of normal colonic flora. Lancet 1989, 333, 164. [Google Scholar] [CrossRef]
- Moayyedi, P.; Surette, M.G.; Kim, P.T.; Libertucci, J.; Wolfe, M.; Onischi, C.; Armstrong, D.; Marshall, J.K.; Kassam, Z.; Reinisch, W.; et al. Fecal Microbiota Transplantation Induces Remission in Patients With Active Ulcerative Colitis in a Randomized Controlled Trial. Gastroenterology 2015, 149, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Paramsothy, S.; A Kamm, M.; O Kaakoush, N.; Walsh, A.J.; Bogaerde, J.V.D.; Samuel, D.; Leong, R.W.L.; Connor, S.; Ng, W.; Paramsothy, R.; et al. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: A randomised placebo-controlled trial. Lancet 2017, 389, 1218–1228. [Google Scholar] [CrossRef]
- Paramsothy, S.; Paramsothy, R.; Rubin, D.T.; Kamm, M.A.; Kaakoush, N.O.; Mitchell, H.M.; Castaño-Rodríguez, N. Faecal microbiota transplantation for inflammatory bowel disease: A systematic review and meta-analysis. J. Crohns Colitis 2017, 11, 1180–1199. [Google Scholar] [CrossRef] [PubMed]
- Costello, S.P.; Soo, W.; Bryant, R.V.; Jairath, V.; Hart, A.L.; Andrews, J.M. Systematic review with meta-analysis: Faecal microbiota transplantation for the induction of remission for active ulcerative colitis. Aliment. Pharmacol. Ther. 2017, 46, 213–224. [Google Scholar] [CrossRef]
- Costello, S.P.; Hughes, P.A.; Waters, O.; Bryant, R.V.; Vincent, A.D.; Blatchford, P.; Katsikeros, R.; Makanyanga, J.; Campaniello, M.A.; Mavrangelos, C.; et al. Effect of Fecal Microbiota Transplantation on 8-Week Remission in Patients with ulcerative colitis. JAMA 2019, 321, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Imdad, A.; Nicholson, M.R.; E Tanner-Smith, E.; Zackular, J.P.; Gomez-Duarte, O.G.; Beaulieu, D.B.; Acra, S. Fecal transplantation for treatment of inflammatory bowel disease. Cochrane Database Syst. Rev. 2018, 11, CD012774. [Google Scholar] [CrossRef] [PubMed]
- Kump, P.K.; Gröchenig, H.-P.; Lackner, S.; Trajanoski, S.; Reicht, G.; Hoffmann, K.M.; Deutschmann, A.; Wenzl, H.H.; Petritsch, W.; Gorkiewicz, G.; et al. Alteration of Intestinal Dysbiosis by Fecal Microbiota Transplantation Does not Induce Remission in Patients with Chronic Active Ulcerative Colitis. Inflamm. Bowel Dis. 2013, 19, 2155–2165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paramsothy, S.; Nielsen, S.; Kamm, M.A.; Deshpande, N.P.; Faith, J.J.; Clemente, J.C.; Paramsothy, R.; Walsh, A.J.; Bogaerde, J.V.D.; Samuel, D.; et al. Specific Bacteria and Metabolites Associated With Response to Fecal Microbiota Transplantation in Patients With Ulcerative Colitis. Gastroenterology 2019, 156, 1440–1454. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.D.; Chen, E.Z.; Baldassano, R.N.; Otley, A.R.; Griffiths, A.M.; Lee, D.; Bittinger, K.; Bailey, A.; Friedman, E.S.; Hoffmann, C.; et al. Inflammation, Antibiotics, and Diet as Environmental Stressors of the Gut Microbiome in Pediatric Crohn’s Disease. Cell Host Microbe 2015, 18, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Willing, B.; Halfvarson, J.; Dicksved, J.; Rosenquist, M.; Järnerot, G.; Engstrand, L.; Tysk, C.; Jansson, J.K. Twin studies reveal specific imbalances in the mucosa-associated microbiota of patients with ileal Crohn’s disease. Inflamm. Bowel Dis. 2009, 15, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Su, J.W.; Ma, J.J.; Zhang, H.J. Use of antibiotics in patients with Crohn’s disease: A systematic review and meta-analysis. J. Dig. Dis. 2015, 16, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, D. Microbial dysbiosis in spouses of ulcerative colitis patients: Any clues to disease pathogenesis? World J. Gastroenterol. 2017, 23, 6747–6749. [Google Scholar] [CrossRef] [PubMed]
- Kuballa, A.; Geraci, M.; Johnstone, M.; Sorrentino, D. The gut microbial profile of preclinical Crohn’s disease is similar to that of healthy controls. Gastroenterology 2019, 156, S-1120. [Google Scholar]
- Buttó, L.F.; Haller, D. Dysbiosis in Crohn’s disease—Joint action of stochastic injuries and focal inflammation in the gut. Gut Microbes 2017, 8, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.N. Epidemiology and risk factors for IBD. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Andersen, V.; Chan, S.; Luben, R.; Khaw, K.T.; Olsen, A.; Tjonneland, A.; Kaaks, R.; Grip, O.; Bergmann, M.M.; Boeing, H. Fibre intake and the development of inflammatory bowel disease: A European prospective multi-centre cohort study (EPIC-IBD). J. Crohns Colitis 2018, 12, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Khalili, H.; Ananthakrishnan, A.N.; Konijeti, G.G.; Liao, X.; Higuchi, L.M.; Fuchs, C.S.; Spiegelman, D.; Richter, J.M.; Korzenik, J.R.; Chan, A.T. Physical activity and risk of inflammatory bowel disease: Prospective study from the Nurses’ Health Study cohorts. BMJ 2013, 347, f6633. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.M.; Dam, A.N.; Farraye, F.A. Environmental Influences on the Onset and Clinical Course of Crohn’s Disease—Part 2: Infections and Medication Use. Gastroenterol. Hepatol. 2013, 9, 803–810. [Google Scholar]
- Knowler, W.C.; Barrett-Connor, E.; Fowler, S.E.; Hamman, R.F.; Lachin, J.M.; Walker, E.A.; Nathan, D.M. Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N. Engl. J. Med. 2002, 346, 393–403. [Google Scholar] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sorrentino, D.; Nguyen, V.Q.; Chitnavis, M.V. Capturing the Biologic Onset of Inflammatory Bowel Diseases: Impact on Translational and Clinical Science. Cells 2019, 8, 548. https://doi.org/10.3390/cells8060548
Sorrentino D, Nguyen VQ, Chitnavis MV. Capturing the Biologic Onset of Inflammatory Bowel Diseases: Impact on Translational and Clinical Science. Cells. 2019; 8(6):548. https://doi.org/10.3390/cells8060548
Chicago/Turabian StyleSorrentino, Dario, Vu Q. Nguyen, and Maithili V. Chitnavis. 2019. "Capturing the Biologic Onset of Inflammatory Bowel Diseases: Impact on Translational and Clinical Science" Cells 8, no. 6: 548. https://doi.org/10.3390/cells8060548
APA StyleSorrentino, D., Nguyen, V. Q., & Chitnavis, M. V. (2019). Capturing the Biologic Onset of Inflammatory Bowel Diseases: Impact on Translational and Clinical Science. Cells, 8(6), 548. https://doi.org/10.3390/cells8060548