RhoA as a Key Regulator of Innate and Adaptive Immunity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
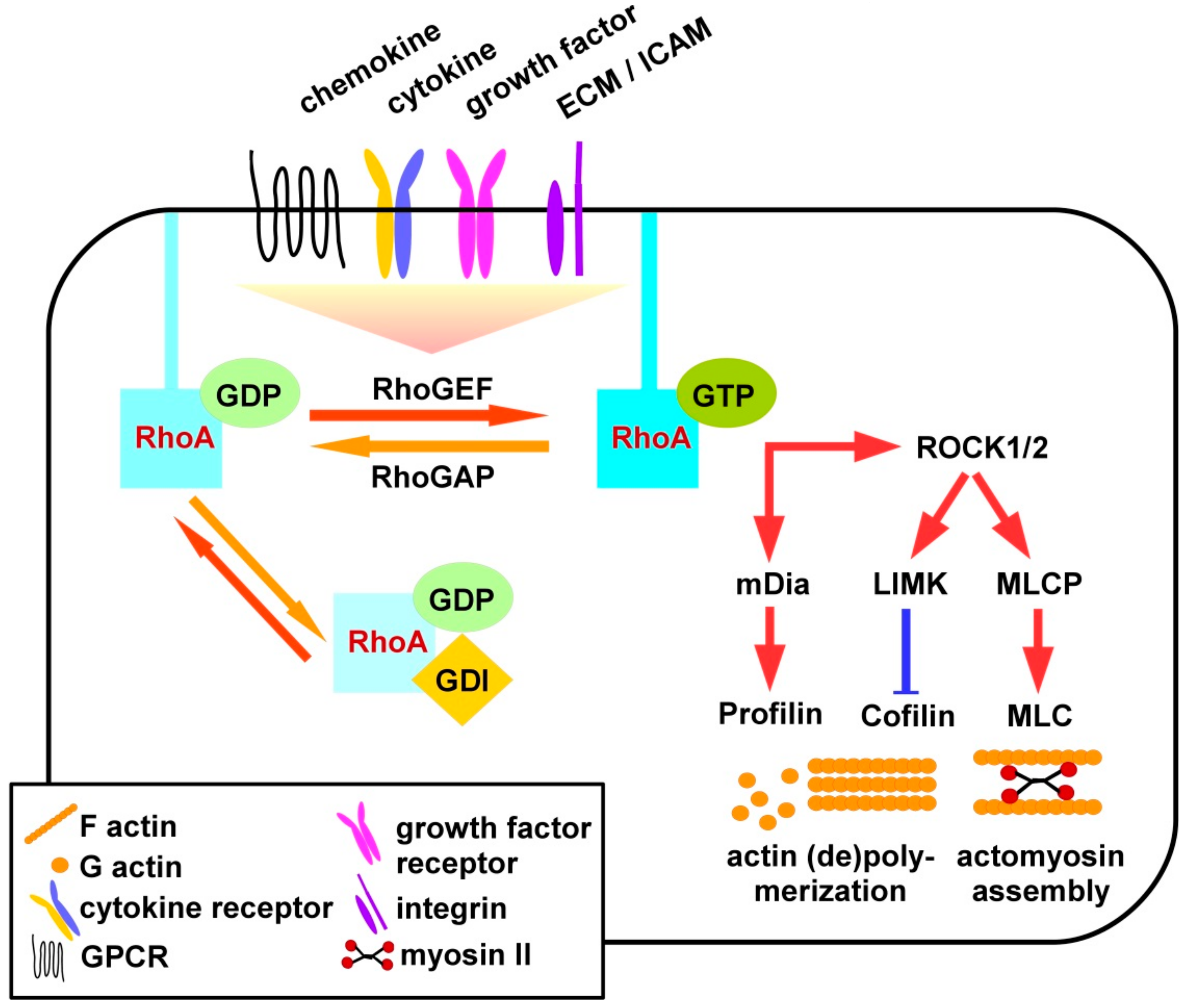
2. RhoA Signaling
3. Role of RhoA in Immune Cell Differentiation and Activity
3.1. Myeloid Innate Immune Cells
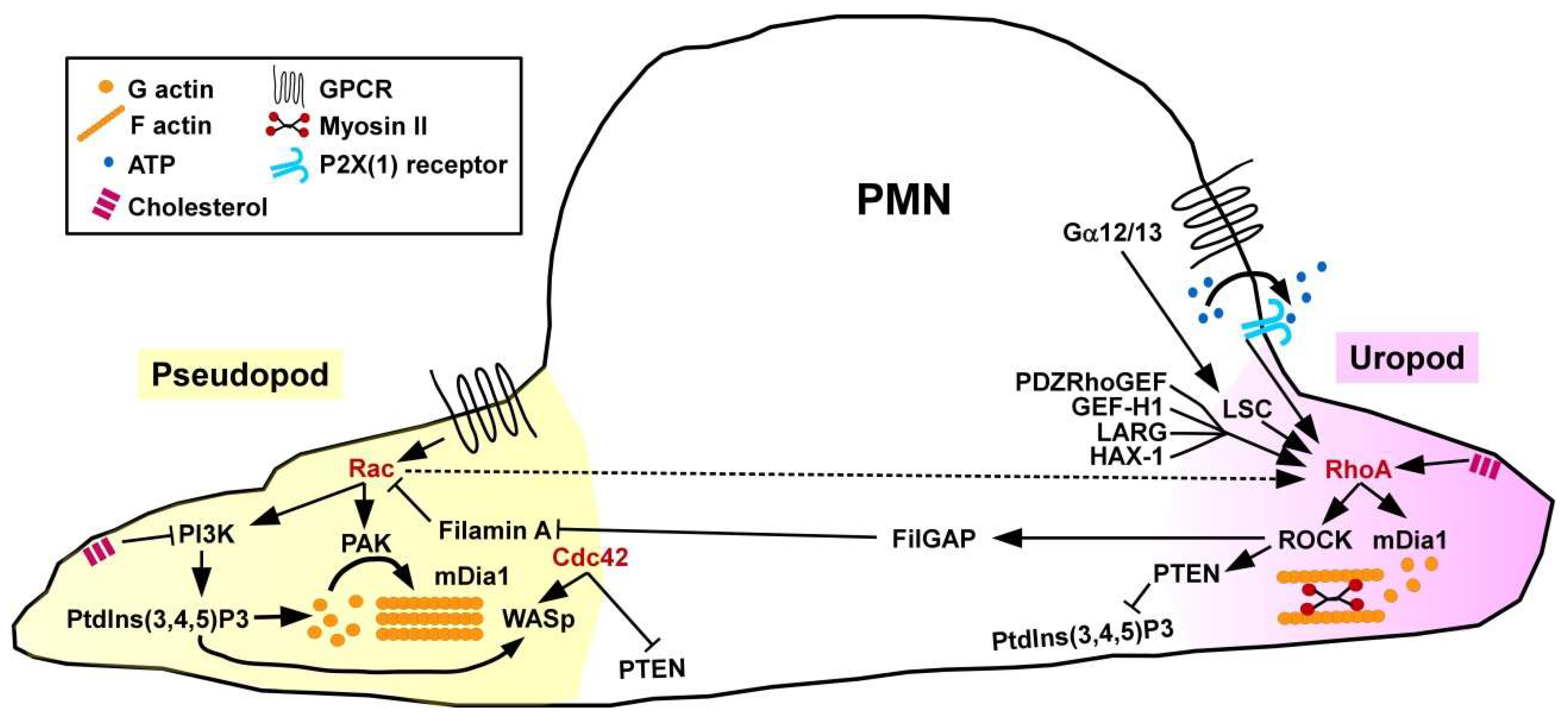
3.1.1. PMN
Migration
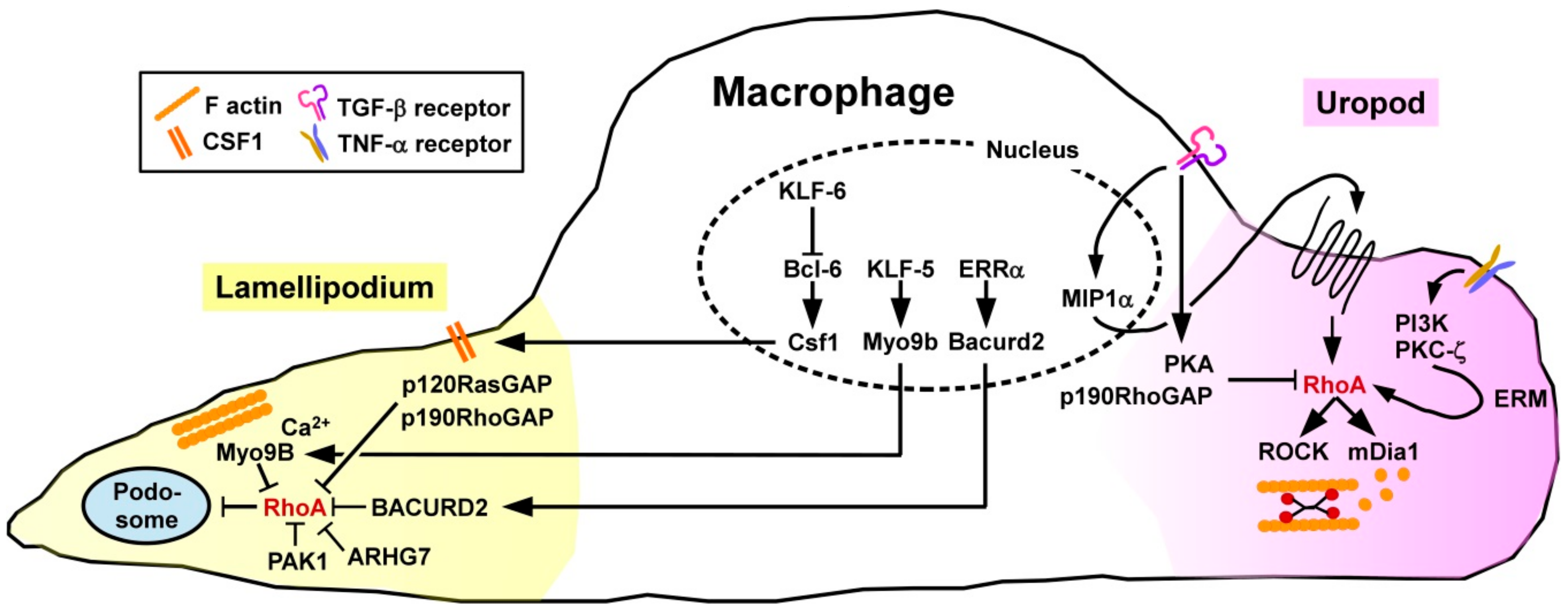
3.1.2. Macrophages
Polarization
Migration
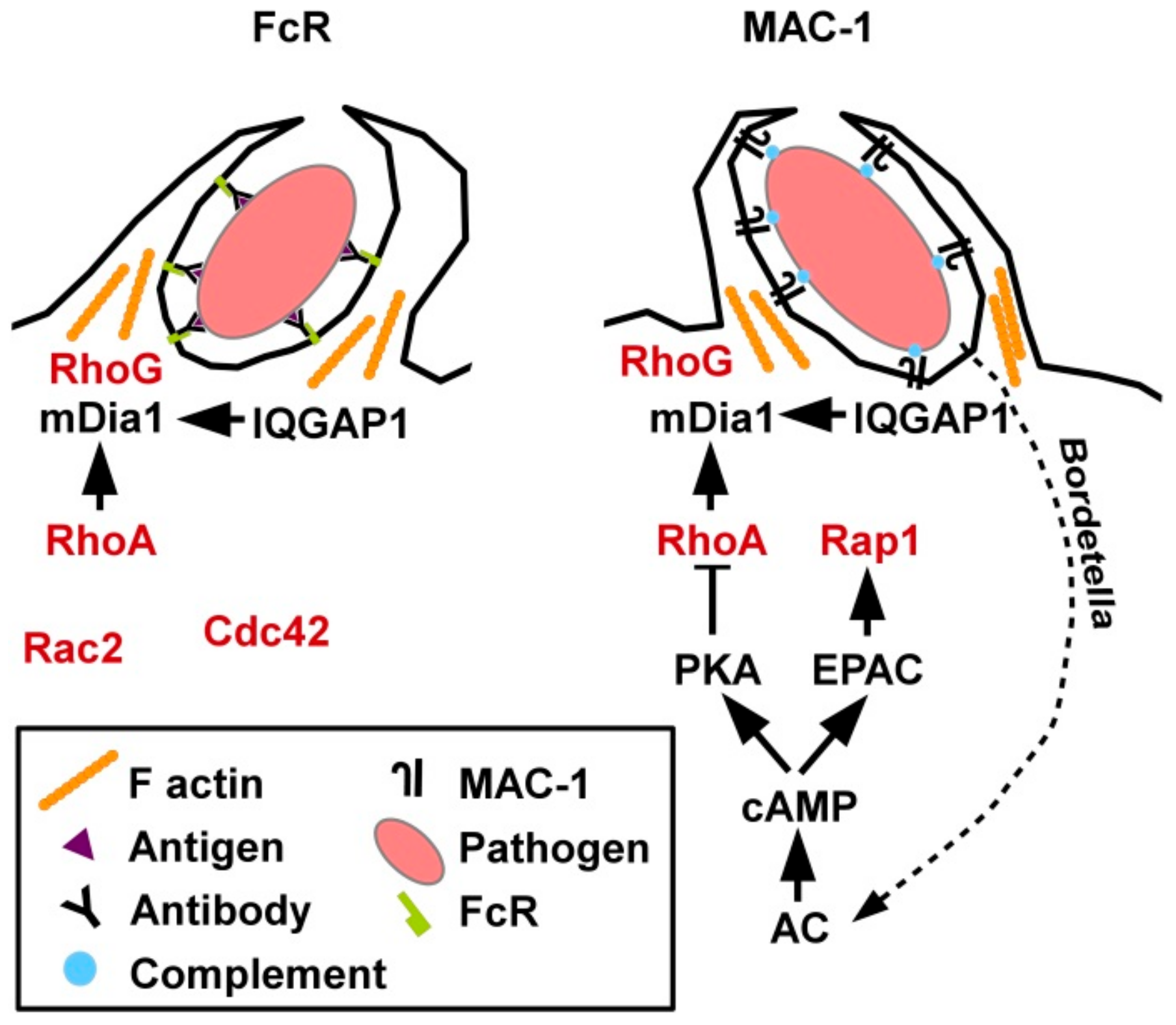
Phagocytosis
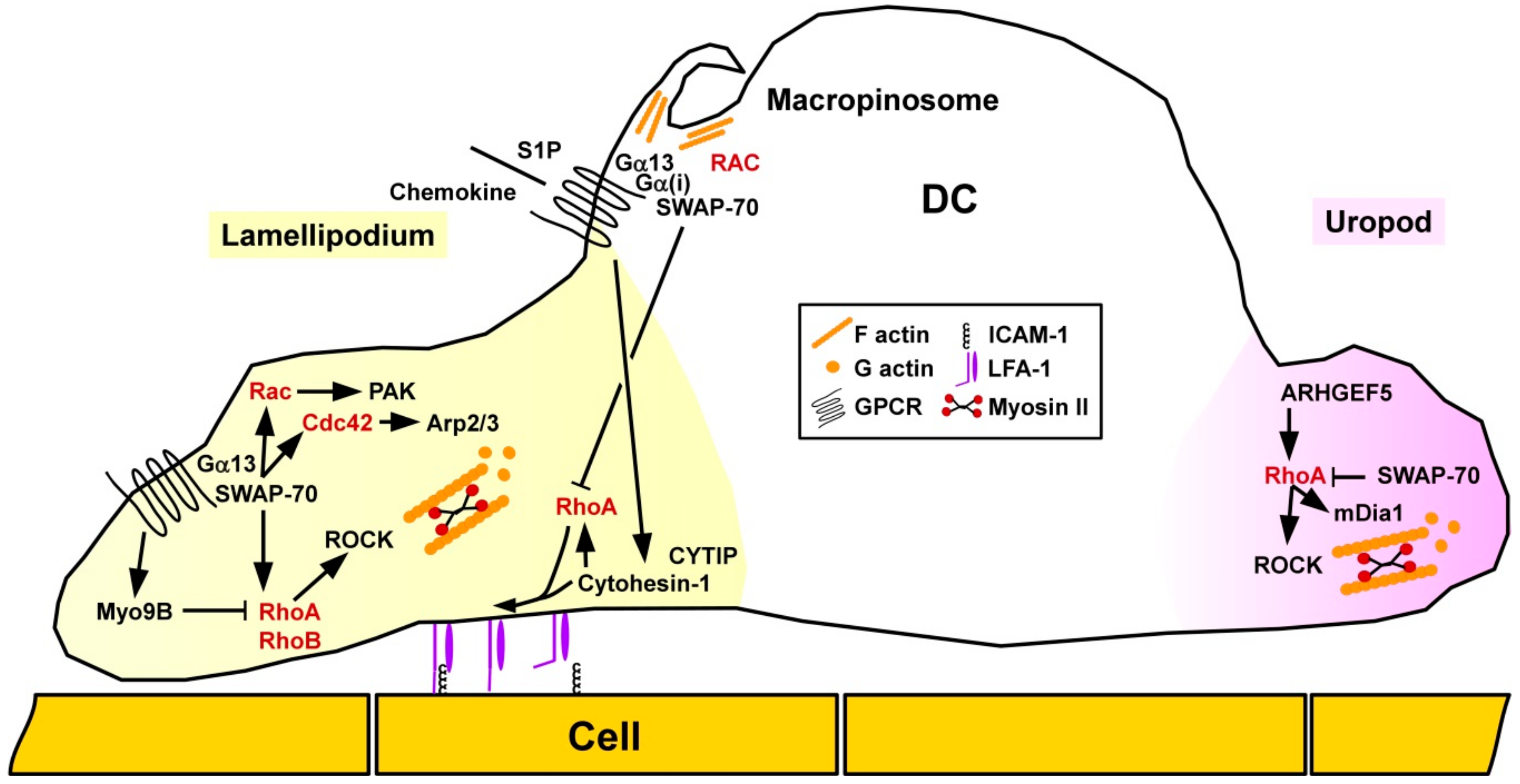
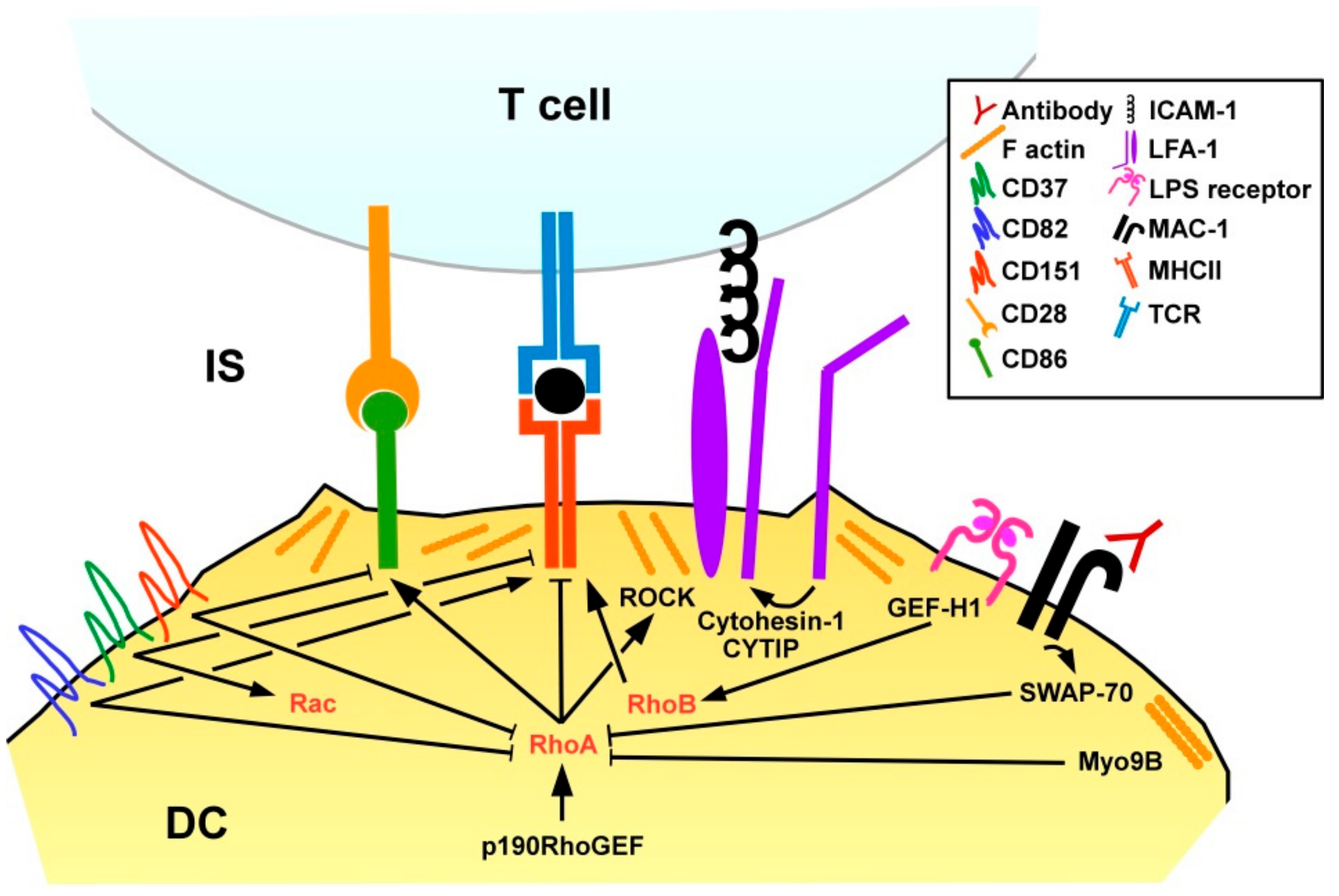
3.1.3. Dendritic Cells
Differentiation
Migration
Activation and Interaction with T Cells
3.2. Adaptive Immune Cells
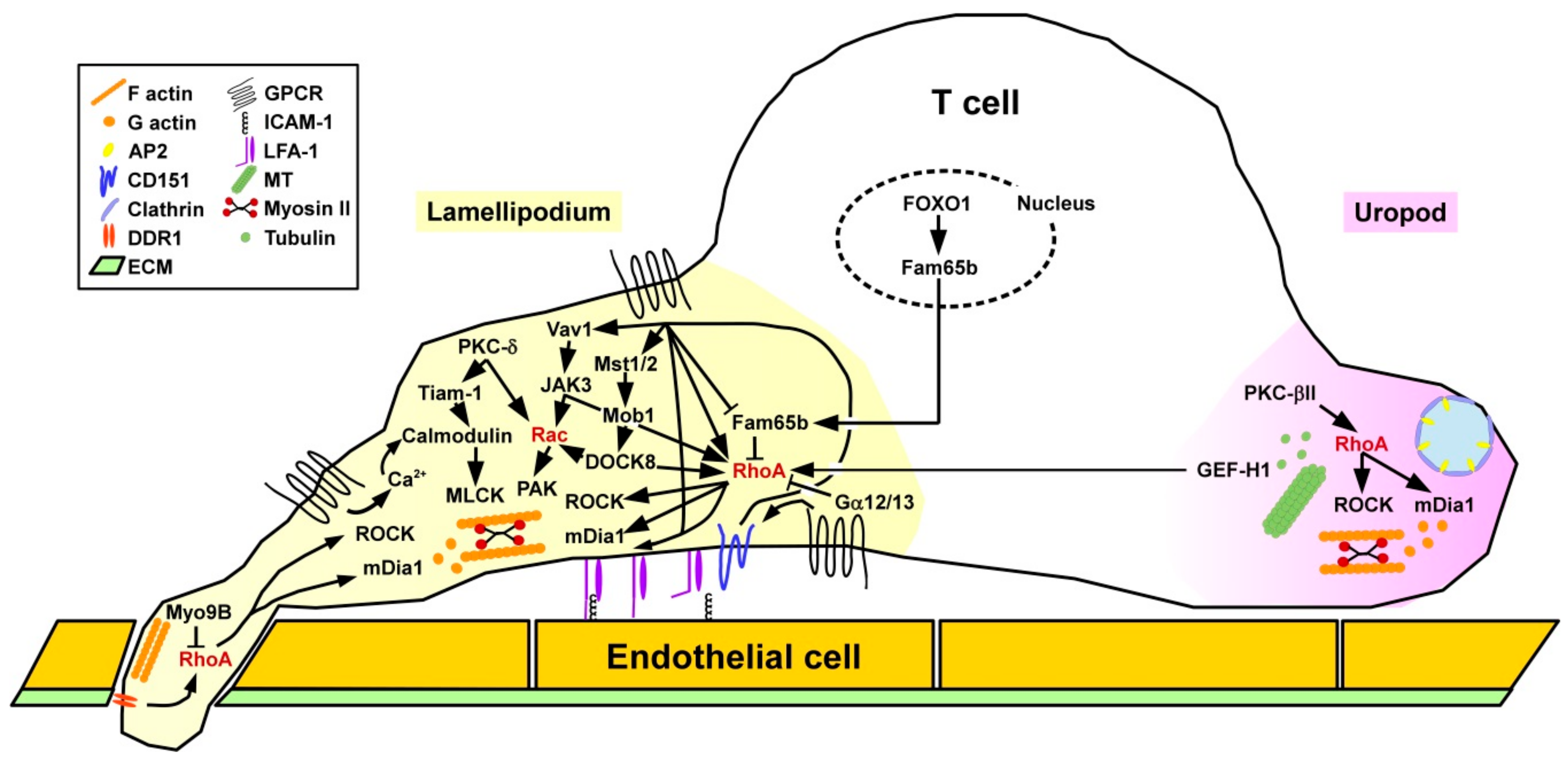
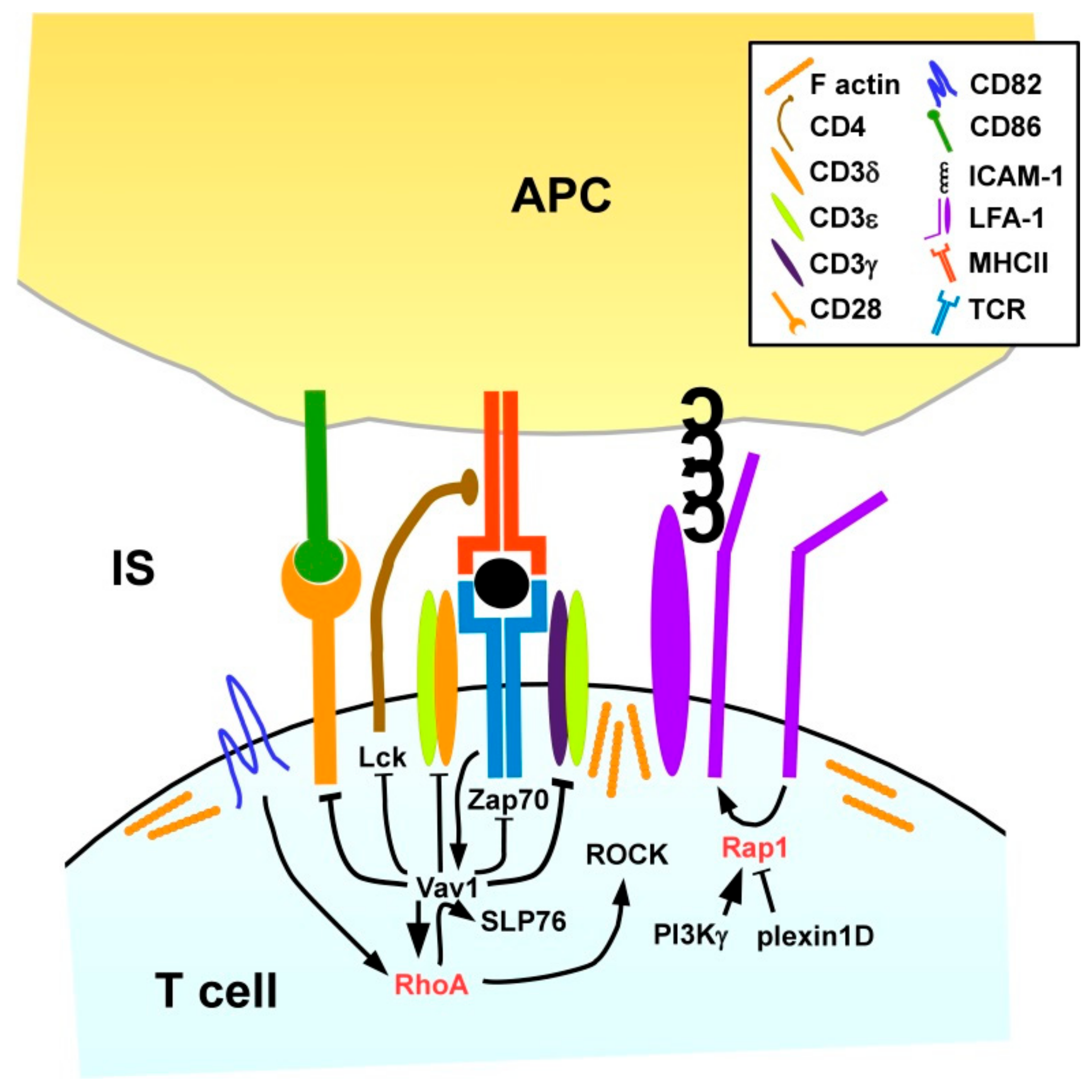
3.2.1. T Cells
Differentiation
Migration
Activation
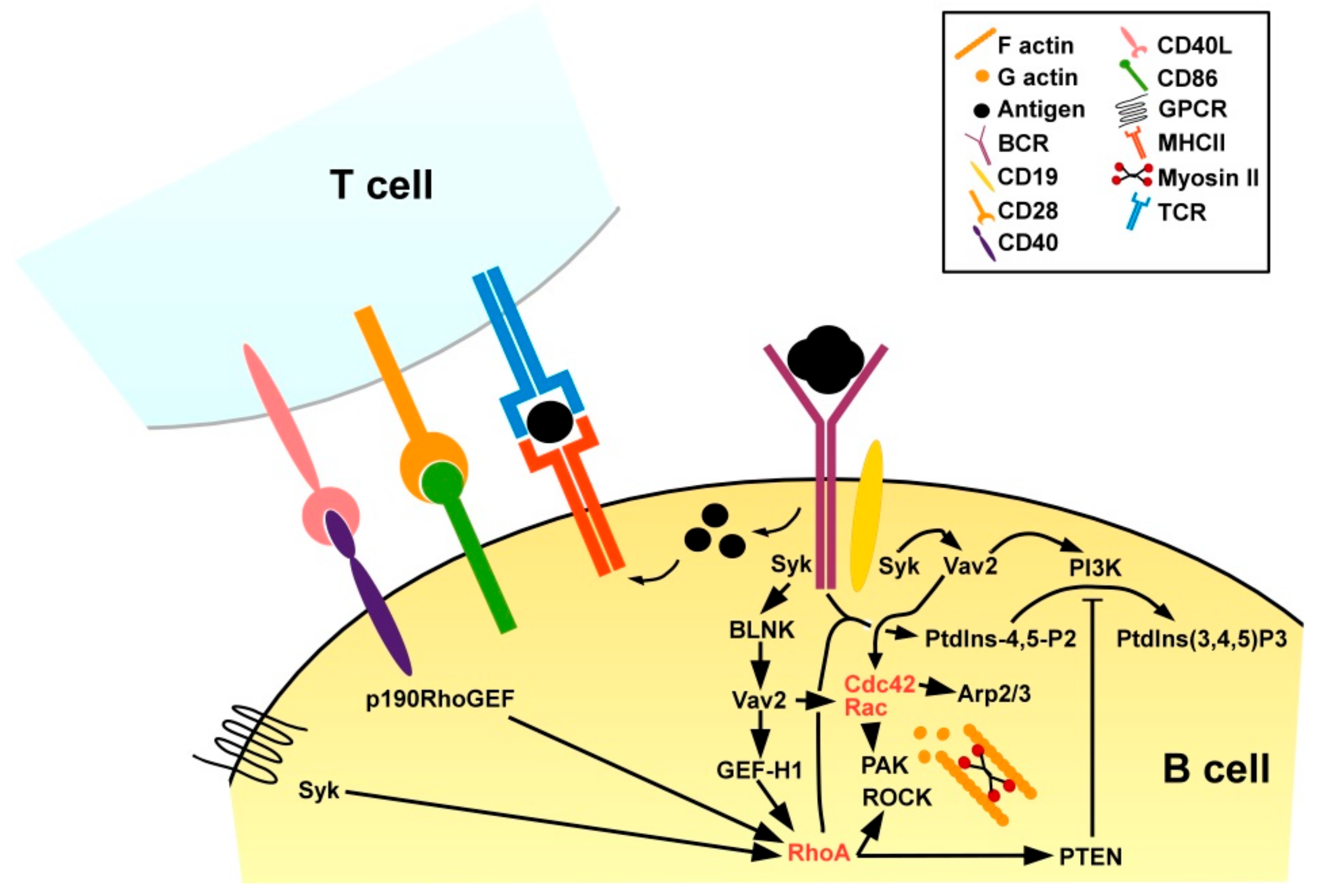
3.2.2. B Cells
Differentiation
Activation
4. Pathophysiological Role of RhoA Signaling in the Course of Immune Reactions
4.1. The State of RhoA Activity Determines both Cellular Uptake and Elimination of Pathogens
4.2. Key Factors of the RhoA Signaling Pathway Constitute Targets for the Treatment of Autoimmune Diseases
5. Mutations of RhoA Signaling Components in Immune Cells Cause Malignancies
6. Concluding Remarks
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Reiner, D.J.; Lundquist, E.A. Small GTPases. Wormbook Online Rev. C. Elegans Biol. 2018, 2018, 1–65. [Google Scholar] [CrossRef] [PubMed]
- Steffen, A.; Stradal, T.E.; Rottner, K. Signalling Pathways Controlling Cellular Actin Organization. Handb. Exp. Pharmacol. 2017, 235, 153–178. [Google Scholar] [CrossRef] [PubMed]
- Narumiya, S.; Thumkeo, D. Rho signaling research: history, current status and future directions. FEBS Lett. 2018, 592, 1763–1776. [Google Scholar] [CrossRef] [Green Version]
- Durand-Onayli, V.; Haslauer, T.; Harzschel, A.; Hartmann, T.N. Rac GTPases in Hematological Malignancies. Int. J. Mol. Sci. 2018, 19, 4041. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, B.J.; Zhou, H.; Lu, Q. Cdc42 Signaling Pathway Inhibition as a Therapeutic Target in Ras- Related Cancers. Curr. Med. Chem. 2017, 24, 3485–3507. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Mellor, H. The small Rho GTPase Rif and actin cytoskeletal remodelling. Biochem. Soc. Trans. 2012, 40, 268–272. [Google Scholar] [CrossRef]
- Pernis, A.B.; Ricker, E.; Weng, C.H.; Rozo, C.; Yi, W. Rho Kinases in Autoimmune Diseases. Ann. Rev. Med. 2016, 67, 355–374. [Google Scholar] [CrossRef]
- Ben-Shmuel, A.; Joseph, N.; Sabag, B.; Barda-Saad, M. Lymphocyte mechanotransduction: The regulatory role of cytoskeletal dynamics in signaling cascades and effector functions. J. Leukoc. Biol. 2019. [Google Scholar] [CrossRef]
- Liu, M.; Clarke, C.J.; Salama, M.F.; Choi, Y.J.; Obeid, L.M.; Hannun, Y.A. Co-ordinated activation of classical and novel PKC isoforms is required for PMA-induced mTORC1 activation. PLoS ONE 2017, 12, e0184818. [Google Scholar] [CrossRef]
- Bora, I.; Shrivastava, N. ABCs of RhoGTPases indicating potential role as oncotargets. J. Cancer Res. Ther. 2017, 13, 2–8. [Google Scholar] [CrossRef]
- Hodge, R.G.; Ridley, A.J. Regulating Rho GTPases and their regulators. Nat. Rev. Mol. Cell Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef]
- Pantarelli, C.; Welch, H.C.E. Rac-GTPases and Rac-GEFs in neutrophil adhesion, migration and recruitment. Eur. J. Clin. Investig. 2018, 48 (Suppl. 2), e12939. [Google Scholar] [CrossRef] [Green Version]
- Denk-Lobnig, M.; Martin, A.C. Modular regulation of Rho family GTPases in development. Small GTPases 2019, 10, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Shao, S.; Aziz, A.U.R.; Zhang, B.; Wang, H.; Liu, B. Role of Rho-specific guanine nucleotide dissociation inhibitor alpha regulation in cell migration. Acta Histochem. 2017, 119, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.J.; Pan, D.; Welch, H.C. Small GTPases and their guanine-nucleotide exchange factors and GTPase-activating proteins in neutrophil recruitment. Curr. Opin. Hematol. 2016, 23, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Patel, M.; Karginov, A.V. Phosphorylation-mediated regulation of GEFs for RhoA. Cell Adhes. Migr. 2014, 8, 11–18. [Google Scholar] [CrossRef]
- Swat, W.; Fujikawa, K. The Vav family: at the crossroads of signaling pathways. Immunol. Res. 2005, 32, 259–265. [Google Scholar] [CrossRef]
- Hanley, P.J.; Xu, Y.; Kronlage, M.; Grobe, K.; Schon, P.; Song, J.; Sorokin, L.; Schwab, A.; Bahler, M. Motorized RhoGAP myosin IXb (Myo9b) controls cell shape and motility. Proc. Natl. Acad. Sci. USA 2010, 107, 12145–12150. [Google Scholar] [CrossRef] [Green Version]
- Wirth, J.A.; Jensen, K.A.; Post, P.L.; Bement, W.M.; Mooseker, M.S. Human myosin-IXb, an unconventional myosin with a chimerin-like rho/rac GTPase-activating protein domain in its tail. J. Cell Sci. 1996, 109 Pt 3, 653–661. [Google Scholar]
- Reinhard, J.; Scheel, A.A.; Diekmann, D.; Hall, A.; Ruppert, C.; Bahler, M. A novel type of myosin implicated in signalling by rho family GTPases. EMBO J. 1995, 14, 697–704. [Google Scholar] [CrossRef]
- Post, P.L.; Bokoch, G.M.; Mooseker, M.S. Human myosin-IXb is a mechanochemically active motor and a GAP for rho. J. Cell Sci. 1998, 111, 941–950. [Google Scholar]
- Saeki, N.; Hiroshi, T.; Ikebe, M. BIG1 is a binding partner of myosin IXb and regulates its RhoGAP activity. J. Biol. Chem. 2005, 280, 10128–10134. [Google Scholar] [CrossRef]
- Bishop, A.L.; Hall, A. Rho GTPases and their effector proteins. Biochem. J. 2000, 348, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Dovas, A.; Couchman, J.R. RhoGDI: Multiple functions in the regulation of Rho family GTPase activities. Biochem. J. 2005, 390, 1–9. [Google Scholar] [CrossRef]
- Ricker, E.; Chowdhury, L.; Yi, W.; Pernis, A.B. The RhoA-ROCK pathway in the regulation of T and B cell responses. F1000Res 2016, 5. [Google Scholar] [CrossRef]
- Julian, L.; Olson, M.F. Rho-associated coiled-coil containing kinases (ROCK): Structure, regulation, and functions. Small GTPases 2014, 5, e29846. [Google Scholar] [CrossRef]
- Prunier, C.; Prudent, R.; Kapur, R.; Sadoul, K.; Lafanechere, L. LIM kinases: Cofilin and beyond. Oncotarget 2017, 8, 41749–41763. [Google Scholar] [CrossRef] [PubMed]
- Ostrowska, Z.; Robaszkiewicz, K.; Moraczewska, J. Regulation of actin filament turnover by cofilin-1 and cytoplasmic tropomyosin isoforms. Biochim. Et Biophys. Acta. Proteins Proteom. 2017, 1865, 88–98. [Google Scholar] [CrossRef]
- Lu, Q.; Lu, L.; Chen, W.; Chen, H.; Xu, X.; Zheng, Z. RhoA/mDia-1/profilin-1 signaling targets microvascular endothelial dysfunction in diabetic retinopathy. Graefe’s Arch. Clin. Exp. Ophthalmol. 2015, 253, 669–680. [Google Scholar] [CrossRef]
- Grabowska, J.; Lopez-Venegas, M.A.; Affandi, A.J.; den Haan, J.M.M. CD169(+) Macrophages Capture and Dendritic Cells Instruct: The Interplay of the Gatekeeper and the General of the Immune System. Front. Immunol. 2018, 9, 2472. [Google Scholar] [CrossRef]
- Steinman, R.M.; Hemmi, H. Dendritic cells: translating innate to adaptive immunity. Curr. Top. Microbiol. Immunol. 2006, 311, 17–58. [Google Scholar]
- Governa, V.; Trella, E.; Mele, V.; Tornillo, L.; Amicarella, F.; Cremonesi, E.; Muraro, M.G.; Xu, H.; Droeser, R.; Daster, S.R.; et al. The Interplay Between Neutrophils and CD8(+) T Cells Improves Survival in Human Colorectal Cancer. Clin. Cancer Res. 2017, 23, 3847–3858. [Google Scholar] [CrossRef]
- Tang-Huau, T.L.; Segura, E. Human in vivo-differentiated monocyte-derived dendritic cells. Semin. Cell Dev. Biol. 2019, 86, 44–49. [Google Scholar] [CrossRef]
- Fites, J.S.; Gui, M.; Kernien, J.F.; Negoro, P.; Dagher, Z.; Sykes, D.B.; Nett, J.E.; Mansour, M.K.; Klein, B.S. An unappreciated role for neutrophil-DC hybrids in immunity to invasive fungal infections. Plos Pathog. 2018, 14, e1007073. [Google Scholar] [CrossRef]
- Teng, T.S.; Ji, A.L.; Ji, X.Y.; Li, Y.Z. Neutrophils and Immunity: From Bactericidal Action to Being Conquered. J. Immunol. Res. 2017, 2017, 9671604. [Google Scholar] [CrossRef]
- Gazendam, R.P.; van de Geer, A.; Roos, D.; van den Berg, T.K.; Kuijpers, T.W. How neutrophils kill fungi. Immunol. Rev. 2016, 273, 299–311. [Google Scholar] [CrossRef]
- Halilovic, I.; Wu, J.; Alexander, M.; Lin, F. Neutrophil migration under spatially-varying chemoattractant gradient profiles. Biomed. Microdevices 2015, 17, 9963. [Google Scholar] [CrossRef]
- Itakura, A.; Aslan, J.E.; Kusanto, B.T.; Phillips, K.G.; Porter, J.E.; Newton, P.K.; Nan, X.; Insall, R.H.; Chernoff, J.; McCarty, O.J. p21-Activated kinase (PAK) regulates cytoskeletal reorganization and directional migration in human neutrophils. PloS ONE 2013, 8, e73063. [Google Scholar] [CrossRef]
- Cavnar, P.J.; Berthier, E.; Beebe, D.J.; Huttenlocher, A. Hax1 regulates neutrophil adhesion and motility through RhoA. J. Cell Biol. 2011, 193, 465–473. [Google Scholar] [CrossRef] [Green Version]
- Wong, K.; Van Keymeulen, A.; Bourne, H.R. PDZRhoGEF and myosin II localize RhoA activity to the back of polarizing neutrophil-like cells. J. Cell Biol. 2007, 179, 1141–1148. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Forster, C.; Nakamura, F.; Glogauer, M. Filamin-A regulates neutrophil uropod retraction through RhoA during chemotaxis. PloS ONE 2013, 8, e79009. [Google Scholar] [CrossRef]
- Xu, J.; Wang, F.; Van Keymeulen, A.; Herzmark, P.; Straight, A.; Kelly, K.; Takuwa, Y.; Sugimoto, N.; Mitchison, T.; Bourne, H.R. Divergent signals and cytoskeletal assemblies regulate self-organizing polarity in neutrophils. Cell 2003, 114, 201–214. [Google Scholar] [CrossRef]
- Siehler, S. Regulation of RhoGEF proteins by G12/13-coupled receptors. Br. J. Pharmacol. 2009, 158, 41–49. [Google Scholar] [CrossRef]
- Francis, S.A.; Shen, X.; Young, J.B.; Kaul, P.; Lerner, D.J. Rho GEF Lsc is required for normal polarization, migration, and adhesion of formyl-peptide-stimulated neutrophils. Blood 2006, 107, 1627–1635. [Google Scholar] [CrossRef]
- Fine, N.; Dimitriou, I.D.; Rottapel, R. Go with the flow: GEF-H1 mediated shear stress mechanotransduction in neutrophils. Small GTPases 2017, 10, 1–9. [Google Scholar] [CrossRef]
- Ohta, Y.; Hartwig, J.H.; Stossel, T.P. FilGAP, a Rho- and ROCK-regulated GAP for Rac binds filamin A to control actin remodelling. Nat. Cell Biol. 2006, 8, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Pestonjamasp, K.N.; Forster, C.; Sun, C.; Gardiner, E.M.; Bohl, B.; Weiner, O.; Bokoch, G.M.; Glogauer, M. Rac1 links leading edge and uropod events through Rho and myosin activation during chemotaxis. Blood 2006, 108, 2814–2820. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Dong, X.; Wang, Z.; Liu, W.; Deng, N.; Ding, Y.; Tang, L.; Hla, T.; Zeng, R.; Li, L.; et al. Regulation of PTEN by Rho small GTPases. Nat. Cell Biol. 2005, 7, 399–404. [Google Scholar] [CrossRef]
- Bodin, S.; Welch, M.D. Plasma membrane organization is essential for balancing competing pseudopod- and uropod-promoting signals during neutrophil polarization and migration. Mol. Biol. Cell 2005, 16, 5773–5783. [Google Scholar] [CrossRef]
- Lecut, C.; Frederix, K.; Johnson, D.M.; Deroanne, C.; Thiry, M.; Faccinetto, C.; Maree, R.; Evans, R.J.; Volders, P.G.; Bours, V.; et al. P2 × 1 ion channels promote neutrophil chemotaxis through Rho kinase activation. J. Immunol. 2009, 183, 2801–2809. [Google Scholar] [CrossRef]
- Vizcarra, C.L.; Bor, B.; Quinlan, M.E. The role of formin tails in actin nucleation, processive elongation, and filament bundling. J. Biol. Chem. 2014, 289, 30602–30613. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, J.; Mullin, M.; Dong, B.; Alberts, A.S.; Siminovitch, K.A. The mDial formin is required for neutrophil polarization, migration, and activation of the LARG/RhoA/ROCK signaling axis during chemotaxis. J. Immunol. 2009, 182, 3837–3845. [Google Scholar] [CrossRef]
- Theret, M.; Mounier, R.; Rossi, F. The origins and non-canonical functions of macrophages in development and regeneration. Development 2019, 146. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G. How macrophages deal with death. Nat. Rev. Immunol. 2019. [Google Scholar] [CrossRef]
- Jablonski, K.A.; Amici, S.A.; Webb, L.M.; Ruiz-Rosado Jde, D.; Popovich, P.G.; Partida-Sanchez, S.; Guerau-de-Arellano, M. Novel Markers to Delineate Murine M1 and M2 Macrophages. PloS ONE 2015, 10, e0145342. [Google Scholar] [CrossRef]
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of Human Macrophage Polarization in Inflammation during Infectious Diseases. Int. J. Mol. Sci. 2018, 19, 1801. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, R.; Shukla, N. Attributes of alternatively activated (M2) macrophages. Life Sci. 2019, 224, 222–231. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhu, L. Update on the role of alternatively activated macrophages in asthma. J. Asthma Allergy 2016, 9, 101–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, T.A. Type 2 cytokines: mechanisms and therapeutic strategies. Nat. Rev. Immunol. 2015, 15, 271–282. [Google Scholar] [CrossRef]
- Kim, S.Y.; Nair, M.G. Macrophages in wound healing: activation and plasticity. Immunol. Cell Biol. 2019, 97, 258–267. [Google Scholar] [CrossRef]
- Yahaya, M.A.F.; Lila, M.A.M.; Ismail, S.; Zainol, M.; Afizan, N. Tumour-Associated Macrophages (TAMs) in Colon Cancer and How to Reeducate Them. J. Immunol. Res. 2019, 2019, 2368249. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, W.; Minze, L.J.; Kubiak, J.Z.; Li, X.C.; Ghobrial, R.M.; Kloc, M. Dissonant response of M0/M2 and M1 bone-marrow-derived macrophages to RhoA pathway interference. Cell Tissue Res. 2016, 366, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhao, Y.; Li, X.C.; Kubiak, J.Z.; Ghobrial, R.M.; Kloc, M. Rho-specific Guanine nucleotide exchange factors (Rho-GEFs) inhibition affects macrophage phenotype and disrupts Golgi complex. Int. J. Biochem. Cell Biol. 2017, 93, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Konigs, V.; Jennings, R.; Vogl, T.; Horsthemke, M.; Bachg, A.C.; Xu, Y.; Grobe, K.; Brakebusch, C.; Schwab, A.; Bahler, M.; et al. Mouse macrophages completely lacking Rho subfamily GTPases (RhoA, RhoB, and RhoC) have severe lamellipodial retraction defects, but robust chemotactic navigation and altered motility. J. Biol. Chem. 2014, 289, 30772–30784. [Google Scholar] [CrossRef] [PubMed]
- El Azzouzi, K.; Wiesner, C.; Linder, S. Metalloproteinase MT1-MMP islets act as memory devices for podosome reemergence. J. Cell Biol. 2016, 213, 109–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Ghobrial, R.M.; Li, X.C.; Kloc, M. Inhibition of RhoA and mTORC2/Rictor by Fingolimod (FTY720) induces p21-activated kinase 1, PAK-1 and amplifies podosomes in mouse peritoneal macrophages. Immunobiology 2018, 223, 634–647. [Google Scholar] [CrossRef] [PubMed]
- Pixley, F.J.; Xiong, Y.; Yu, R.Y.; Sahai, E.A.; Stanley, E.R.; Ye, B.H. BCL6 suppresses RhoA activity to alter macrophage morphology and motility. J. Cell Sci. 2005, 118, 1873–1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welliver, T.P.; Swanson, J.A. A growth factor signaling cascade confined to circular ruffles in macrophages. Biol. Open 2012, 1, 754–760. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.D.; Das, R.; Goduni, L.; McClellan, S.; Hazlett, L.D.; Mahabeleshwar, G.H. Kruppel-like Factor 6 Promotes Macrophage-mediated Inflammation by Suppressing B Cell Leukemia/Lymphoma 6 Expression. J. Biol. Chem. 2016, 291, 21271–21282. [Google Scholar] [CrossRef] [Green Version]
- Pegoretti, V.; Baron, W.; Laman, J.D.; Eisel, U.L.M. Selective Modulation of TNF-TNFRs Signaling: Insights for Multiple Sclerosis Treatment. Front. Immunol. 2018, 9, 925. [Google Scholar] [CrossRef]
- Lim, S.; Ryu, J.; Shin, J.A.; Shin, M.J.; Ahn, Y.K.; Kim, J.J.; Han, K.H. Tumor necrosis factor-alpha potentiates RhoA-mediated monocyte transmigratory activity in vivo at a picomolar level. Arter. Thromb. Vasc. Biol. 2009, 29, 2138–2145. [Google Scholar] [CrossRef]
- Troncone, E.; Marafini, I.; Stolfi, C.; Monteleone, G. Transforming Growth Factor-beta1/Smad7 in Intestinal Immunity, Inflammation, and Cancer. Front. Immunol. 2018, 9, 1407. [Google Scholar] [CrossRef]
- Kim, J.S.; Kim, J.G.; Moon, M.Y.; Jeon, C.Y.; Won, H.Y.; Kim, H.J.; Jeon, Y.J.; Seo, J.Y.; Kim, J.I.; Kim, J.; et al. Transforming growth factor-beta1 regulates macrophage migration via RhoA. Blood 2006, 108, 1821–1829. [Google Scholar] [CrossRef]
- Liao, W.; Elfrink, K.; Bahler, M. Head of myosin IX binds calmodulin and moves processively toward the plus-end of actin filaments. J. Biol. Chem. 2010, 285, 24933–24942. [Google Scholar] [CrossRef]
- Ma, D.; Zheng, B.; Suzuki, T.; Zhang, R.; Jiang, C.; Bai, D.; Yin, W.; Yang, Z.; Zhang, X.; Hou, L.; et al. Inhibition of KLF5-Myo9b-RhoA Pathway-Mediated Podosome Formation in Macrophages Ameliorates Abdominal Aortic Aneurysm. Circ. Res. 2017, 120, 799–815. [Google Scholar] [CrossRef]
- Zuo, Y.; d’Aigle, J.; Chauhan, A.; Frost, J.A. Genetic deletion of the Rho GEF Net1 impairs mouse macrophage motility and actin cytoskeletal organization. Small GTPases 2017, 1–8. [Google Scholar] [CrossRef]
- Sailland, J.; Tribollet, V.; Forcet, C.; Billon, C.; Barenton, B.; Carnesecchi, J.; Bachmann, A.; Gauthier, K.C.; Yu, S.; Giguere, V.; et al. Estrogen-related receptor alpha decreases RHOA stability to induce orientated cell migration. Proc. Natl. Acad. Sci. USA 2014, 111, 15108–15113. [Google Scholar] [CrossRef]
- Gordan, S.; Biburger, M.; Nimmerjahn, F. bIgG time for large eaters: monocytes and macrophages as effector and target cells of antibody-mediated immune activation and repression. Immunol. Rev. 2015, 268, 52–65. [Google Scholar] [CrossRef]
- Lukacsi, S.; Nagy-Balo, Z.; Erdei, A.; Sandor, N.; Bajtay, Z. The role of CR3 (CD11b/CD18) and CR4 (CD11c/CD18) in complement-mediated phagocytosis and podosome formation by human phagocytes. Immunol. Lett. 2017, 189, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Podolnikova, N.P.; Podolnikov, A.V.; Haas, T.A.; Lishko, V.K.; Ugarova, T.P. Ligand recognition specificity of leukocyte integrin alphaMbeta2 (Mac-1, CD11b/CD18) and its functional consequences. Biochemistry 2015, 54, 1408–1420. [Google Scholar] [CrossRef]
- Tzircotis, G.; Braga, V.M.; Caron, E. RhoG is required for both FcgammaR- and CR3-mediated phagocytosis. J. Cell Sci. 2011, 124, 2897–2902. [Google Scholar] [CrossRef]
- Kim, J.G.; Moon, M.Y.; Kim, H.J.; Li, Y.; Song, D.K.; Kim, J.S.; Lee, J.Y.; Kim, J.; Kim, S.C.; Park, J.B. Ras-related GTPases Rap1 and RhoA collectively induce the phagocytosis of serum-opsonized zymosan particles in macrophages. J. Biol. Chem. 2012, 287, 5145–5155. [Google Scholar] [CrossRef]
- Kamanova, J.; Kofronova, O.; Masin, J.; Genth, H.; Vojtova, J.; Linhartova, I.; Benada, O.; Just, I.; Sebo, P. Adenylate cyclase toxin subverts phagocyte function by RhoA inhibition and unproductive ruffling. J. Immunol. 2008, 181, 5587–5597. [Google Scholar] [CrossRef]
- Banerjee, U.; Cheng, X. Exchange protein directly activated by cAMP encoded by the mammalian rapgef3 gene: Structure, function and therapeutics. Gene 2015, 570, 157–167. [Google Scholar] [CrossRef]
- Nakaya, M.; Tanaka, M.; Okabe, Y.; Hanayama, R.; Nagata, S. Opposite effects of rho family GTPases on engulfment of apoptotic cells by macrophages. J. Biol. Chem. 2006, 281, 8836–8842. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kim, S.; Bae, D.J.; Park, S.Y.; Lee, G.Y.; Park, G.M.; Kim, I.S. Coordinated balance of Rac1 and RhoA plays key roles in determining phagocytic appetite. PloS ONE 2017, 12, e0174603. [Google Scholar] [CrossRef]
- Brandt, D.T.; Marion, S.; Griffiths, G.; Watanabe, T.; Kaibuchi, K.; Grosse, R. Dia1 and IQGAP1 interact in cell migration and phagocytic cup formation. J. Cell Biol. 2007, 178, 193–200. [Google Scholar] [CrossRef]
- Macri, C.; Pang, E.S.; Patton, T.; O’Keeffe, M. Dendritic cell subsets. Semin. Cell Dev. Biol. 2018, 84, 11–21. [Google Scholar] [CrossRef]
- Li, H.; Marshall, A.J. Phosphatidylinositol (3,4) bisphosphate-specific phosphatases and effector proteins: A distinct branch of PI3K signaling. Cell. Signal. 2015, 27, 1789–1798. [Google Scholar] [CrossRef]
- Rodrigues, D.H.; Vilela, M.C.; Barcelos, L.S.; Pinho, V.; Teixeira, M.M.; Teixeira, A.L. Absence of PI3Kgamma leads to increased leukocyte apoptosis and diminished severity of experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2010, 222, 90–94. [Google Scholar] [CrossRef]
- Nitschke, M.; Aebischer, D.; Abadier, M.; Haener, S.; Lucic, M.; Vigl, B.; Luche, H.; Fehling, H.J.; Biehlmaier, O.; Lyck, R.; et al. Differential requirement for ROCK in dendritic cell migration within lymphatic capillaries in steady-state and inflammation. Blood 2012, 120, 2249–2258. [Google Scholar] [CrossRef] [Green Version]
- Vargas, P.; Maiuri, P.; Bretou, M.; Saez, P.J.; Pierobon, P.; Maurin, M.; Chabaud, M.; Lankar, D.; Obino, D.; Terriac, E.; et al. Innate control of actin nucleation determines two distinct migration behaviours in dendritic cells. Nat. Cell Biol. 2016, 18, 43–53. [Google Scholar] [CrossRef]
- Nawrotek, A.; Zeghouf, M.; Cherfils, J. Allosteric regulation of Arf GTPases and their GEFs at the membrane interface. Small GTPases 2016, 7, 283–296. [Google Scholar] [CrossRef] [Green Version]
- Quast, T.; Tappertzhofen, B.; Schild, C.; Grell, J.; Czeloth, N.; Forster, R.; Alon, R.; Fraemohs, L.; Dreck, K.; Weber, C.; et al. Cytohesin-1 controls the activation of RhoA and modulates integrin-dependent adhesion and migration of dendritic cells. Blood 2009, 113, 5801–5810. [Google Scholar] [CrossRef] [Green Version]
- Grosche, L.; Drassner, C.; Muhl-Zurbes, P.; Kamm, L.; Le-Trilling, V.T.K.; Trilling, M.; Steinkasserer, A.; Heilingloh, C.S. Human Cytomegalovirus-Induced Degradation of CYTIP Modulates Dendritic Cell Adhesion and Migration. Front. Immunol. 2017, 8, 461. [Google Scholar] [CrossRef]
- Gomez-Cambronero, J. Structure analysis between the SWAP-70 RHO-GEF and the newly described PLD2-GEF. Small GTPases 2012, 3, 202–208. [Google Scholar] [CrossRef] [Green Version]
- Chacon-Martinez, C.A.; Kiessling, N.; Winterhoff, M.; Faix, J.; Muller-Reichert, T.; Jessberger, R. The switch-associated protein 70 (SWAP-70) bundles actin filaments and contributes to the regulation of F-actin dynamics. J. Biol. Chem. 2013, 288, 28687–28703. [Google Scholar] [CrossRef]
- Ocana-Morgner, C.; Wahren, C.; Jessberger, R. SWAP-70 regulates RhoA/RhoB-dependent MHCII surface localization in dendritic cells. Blood 2009, 113, 1474–1482. [Google Scholar] [CrossRef] [Green Version]
- Arlt, O.; Schwiebs, A.; Japtok, L.; Ruger, K.; Katzy, E.; Kleuser, B.; Radeke, H.H. Sphingosine-1-phosphate modulates dendritic cell function: focus on non-migratory effects in vitro and in vivo. Cell. Physiol. Biochem. 2014, 34, 27–44. [Google Scholar] [CrossRef]
- Ocana-Morgner, C.; Reichardt, P.; Chopin, M.; Braungart, S.; Wahren, C.; Gunzer, M.; Jessberger, R. Sphingosine 1-phosphate-induced motility and endocytosis of dendritic cells is regulated by SWAP-70 through RhoA. J. Immunol. 2011, 186, 5345–5355. [Google Scholar] [CrossRef]
- Oberbanscheidt, P.; Balkow, S.; Kuhnl, J.; Grabbe, S.; Bahler, M. SWAP-70 associates transiently with macropinosomes. Eur. J. Cell Biol. 2007, 86, 13–24. [Google Scholar] [CrossRef]
- Canton, J. Macropinocytosis: New Insights Into Its Underappreciated Role in Innate Immune Cell Surveillance. Front. Immunol. 2018, 9, 2286. [Google Scholar] [CrossRef] [Green Version]
- Baranov, M.V.; Revelo, N.H.; Dingjan, I.; Maraspini, R.; Ter Beest, M.; Honigmann, A.; van den Bogaart, G. SWAP70 Organizes the Actin Cytoskeleton and Is Essential for Phagocytosis. Cell Rep. 2016, 17, 1518–1531. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Kumamoto, Y.; Wang, P.; Gan, X.; Lehmann, D.; Smrcka, A.V.; Cohn, L.; Iwasaki, A.; Li, L.; Wu, D. Regulation of immature dendritic cell migration by RhoA guanine nucleotide exchange factor Arhgef5. J. Biol. Chem. 2009, 284, 28599–28606. [Google Scholar] [CrossRef]
- Xu, Y.; Pektor, S.; Balkow, S.; Hemkemeyer, S.A.; Liu, Z.; Grobe, K.; Hanley, P.J.; Shen, L.; Bros, M.; Schmidt, T.; et al. Dendritic cell motility and T cell activation requires regulation of Rho-cofilin signaling by the Rho-GTPase activating protein myosin IXb. J. Immunol. 2014, 192, 3559–3568. [Google Scholar] [CrossRef]
- Shurin, G.V.; Tourkova, I.L.; Chatta, G.S.; Schmidt, G.; Wei, S.; Djeu, J.Y.; Shurin, M.R. Small rho GTPases regulate antigen presentation in dendritic cells. J. Immunol. 2005, 174, 3394–3400. [Google Scholar] [CrossRef]
- Reinherz, E.L.; Wang, J.H. Codification of bidentate pMHC interaction with TCR and its co-receptor. Trends Immunol. 2015, 36, 300–306. [Google Scholar] [CrossRef] [Green Version]
- Kamon, H.; Kawabe, T.; Kitamura, H.; Lee, J.; Kamimura, D.; Kaisho, T.; Akira, S.; Iwamatsu, A.; Koga, H.; Murakami, M.; et al. TRIF-GEFH1-RhoB pathway is involved in MHCII expression on dendritic cells that is critical for CD4 T-cell activation. EMBO J. 2006, 25, 4108–4119. [Google Scholar] [CrossRef]
- Ocana-Morgner, C.; Gotz, A.; Wahren, C.; Jessberger, R. SWAP-70 restricts spontaneous maturation of dendritic cells. J. Immunol. 2013, 190, 5545–5558. [Google Scholar] [CrossRef]
- Seul, H.J.; Ahn, Y.R.; Song, H.M.; Ha, Y.J.; Lee, J.R. Over-expression of a RhoA-specific guanine nucleotide exchange factor, p190RhoGEF, in mouse dendritic cells negatively regulates cellular responses to bacterial lipopolysaccharide. Mol. Cells 2012, 34, 159–164. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto-Tane, A.; Saito, T. Dynamic Regulation of TCR-Microclusters and the Microsynapse for T Cell Activation. Front. Immunol. 2016, 7, 255. [Google Scholar] [CrossRef]
- Van Deventer, S.J.; Dunlock, V.E.; van Spriel, A.B. Molecular interactions shaping the tetraspanin web. Biochem. Soc. Trans. 2017, 45, 741–750. [Google Scholar] [CrossRef]
- Sheng, K.C.; van Spriel, A.B.; Gartlan, K.H.; Sofi, M.; Apostolopoulos, V.; Ashman, L.; Wright, M.D. Tetraspanins CD37 and CD151 differentially regulate Ag presentation and T-cell co-stimulation by DC. Eur. J. Immunol. 2009, 39, 50–55. [Google Scholar] [CrossRef]
- Johnson, J.L.; Winterwood, N.; DeMali, K.A.; Stipp, C.S. Tetraspanin CD151 regulates RhoA activation and the dynamic stability of carcinoma cell-cell contacts. J. Cell Sci. 2009, 122, 2263–2273. [Google Scholar] [CrossRef] [Green Version]
- Jones, E.L.; Wee, J.L.; Demaria, M.C.; Blakeley, J.; Ho, P.K.; Vega-Ramos, J.; Villadangos, J.A.; van Spriel, A.B.; Hickey, M.J.; Hammerling, G.J.; et al. Dendritic Cell Migration and Antigen Presentation Are Coordinated by the Opposing Functions of the Tetraspanins CD82 and CD37. J. Immunol. 2016, 196, 978–987. [Google Scholar] [CrossRef] [Green Version]
- De Winde, C.M.; Matthews, A.L.; van Deventer, S.; van der Schaaf, A.; Tomlinson, N.D.; Jansen, E.; Eble, J.A.; Nieswandt, B.; McGettrick, H.M.; Figdor, C.G.; et al. C-type lectin-like receptor 2 (CLEC-2)-dependent dendritic cell migration is controlled by tetraspanin CD37. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef]
- Zuidscherwoude, M.; Worah, K.; van der Schaaf, A.; Buschow, S.I.; van Spriel, A.B. Differential expression of tetraspanin superfamily members in dendritic cell subsets. PloS ONE 2017, 12, e0184317. [Google Scholar] [CrossRef]
- Hofer, S.; Pfeil, K.; Niederegger, H.; Ebner, S.; Nguyen, V.A.; Kremmer, E.; Auffinger, M.; Neyer, S.; Furhapter, C.; Heufler, C. Dendritic cells regulate T-cell deattachment through the integrin-interacting protein CYTIP. Blood 2006, 107, 1003–1009. [Google Scholar] [CrossRef] [Green Version]
- Balkow, S.; Heinz, S.; Schmidbauer, P.; Kolanus, W.; Holzmann, B.; Grabbe, S.; Laschinger, M. LFA-1 activity state on dendritic cells regulates contact duration with T cells and promotes T-cell priming. Blood 2010, 116, 1885–1894. [Google Scholar] [CrossRef]
- Zhang, S.; Konstantinidis, D.G.; Yang, J.Q.; Mizukawa, B.; Kalim, K.; Lang, R.A.; Kalfa, T.A.; Zheng, Y.; Guo, F. Gene targeting RhoA reveals its essential role in coordinating mitochondrial function and thymocyte development. J. Immunol. 2014, 193, 5973–5982. [Google Scholar] [CrossRef]
- Zhitkovich, A. N-Acetylcysteine: Antioxidant, Aldehyde Scavenger, and More. Chem. Res. Toxicol. 2019. [Google Scholar] [CrossRef]
- David, M.D.; Petit, D.; Bertoglio, J. The RhoGAP ARHGAP19 controls cytokinesis and chromosome segregation in T lymphocytes. J. Cell Sci. 2014, 127, 400–410. [Google Scholar] [CrossRef]
- Smith, A.; Bracke, M.; Leitinger, B.; Porter, J.C.; Hogg, N. LFA-1-induced T cell migration on ICAM-1 involves regulation of MLCK-mediated attachment and ROCK-dependent detachment. J. Cell Sci. 2003, 116, 3123–3133. [Google Scholar] [CrossRef] [Green Version]
- Song, K.H.; Lee, J.; Jung, H.R.; Park, H.; Doh, J. Turning behaviors of T cells climbing up ramp-like structures are regulated by myosin light chain kinase activity and lamellipodia formation. Sci. Rep. 2017, 7, 11533. [Google Scholar] [CrossRef]
- Heasman, S.J.; Carlin, L.M.; Cox, S.; Ng, T.; Ridley, A.J. Coordinated RhoA signaling at the leading edge and uropod is required for T cell transendothelial migration. J. Cell Biol. 2010, 190, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Pollock, J.K.; Verma, N.K.; O’Boyle, N.M.; Carr, M.; Meegan, M.J.; Zisterer, D.M. Combretastatin (CA)-4 and its novel analogue CA-432 impair T-cell migration through the Rho/ROCK signalling pathway. Biochem. Pharmacol. 2014, 92, 544–557. [Google Scholar] [CrossRef]
- Pasvolsky, R.; Grabovsky, V.; Giagulli, C.; Shulman, Z.; Shamri, R.; Feigelson, S.W.; Laudanna, C.; Alon, R. RhoA is involved in LFA-1 extension triggered by CXCL12 but not in a novel outside-in LFA-1 activation facilitated by CXCL9. J. Immunol. 2008, 180, 2815–2823. [Google Scholar] [CrossRef]
- Walling, B.L.; Kim, M. LFA-1 in T Cell Migration and Differentiation. Front. Immunol. 2018, 9, 952. [Google Scholar] [CrossRef] [Green Version]
- Vielkind, S.; Gallagher-Gambarelli, M.; Gomez, M.; Hinton, H.J.; Cantrell, D.A. Integrin regulation by RhoA in thymocytes. J. Immunol. 2005, 175, 350–357. [Google Scholar] [CrossRef]
- Katakai, T.; Kondo, N.; Ueda, Y.; Kinashi, T. Autotaxin produced by stromal cells promotes LFA-1-independent and Rho-dependent interstitial T cell motility in the lymph node paracortex. J. Immunol. 2014, 193, 617–626. [Google Scholar] [CrossRef]
- Ryu, J.M.; Han, H.J. Autotaxin-LPA axis regulates hMSC migration by adherent junction disruption and cytoskeletal rearrangement via LPAR1/3-dependent PKC/GSK3beta/beta-catenin and PKC/Rho GTPase pathways. Stem Cells (Dayt. Ohio) 2015, 33, 819–832. [Google Scholar] [CrossRef]
- El Azreq, M.A.; Kadiri, M.; Boisvert, M.; Page, N.; Tessier, P.A.; Aoudjit, F. Discoidin domain receptor 1 promotes Th17 cell migration by activating the RhoA/ROCK/MAPK/ERK signaling pathway. Oncotarget 2016, 7, 44975–44990. [Google Scholar] [CrossRef] [Green Version]
- Vicente-Manzanares, M.; Rey, M.; Perez-Martinez, M.; Yanez-Mo, M.; Sancho, D.; Cabrero, J.R.; Barreiro, O.; de la Fuente, H.; Itoh, K.; Sanchez-Madrid, F. The RhoA effector mDia is induced during T cell activation and regulates actin polymerization and cell migration in T lymphocytes. J. Immunol. 2003, 171, 1023–1034. [Google Scholar] [CrossRef]
- Montresor, A.; Bolomini-Vittori, M.; Toffali, L.; Rossi, B.; Constantin, G.; Laudanna, C. JAK tyrosine kinases promote hierarchical activation of Rho and Rap modules of integrin activation. J. Cell Biol. 2013, 203, 1003–1019. [Google Scholar] [CrossRef] [Green Version]
- Ambriz-Pena, X.; Garcia-Zepeda, E.A.; Meza, I.; Soldevila, G. Jak3 enables chemokine-dependent actin cytoskeleton reorganization by regulating cofilin and Rac/Rhoa GTPases activation. PLoS ONE 2014, 9, e88014. [Google Scholar] [CrossRef]
- Galan, J.A.; Avruch, J. MST1/MST2 Protein Kinases: Regulation and Physiologic Roles. Biochemistry 2016, 55, 5507–5519. [Google Scholar] [CrossRef] [Green Version]
- Mou, F.; Praskova, M.; Xia, F.; Van Buren, D.; Hock, H.; Avruch, J.; Zhou, D. The Mst1 and Mst2 kinases control activation of rho family GTPases and thymic egress of mature thymocytes. J. Exp. Med. 2012, 209, 741–759. [Google Scholar] [CrossRef]
- Samaniego, R.; Sanchez-Martin, L.; Estecha, A.; Sanchez-Mateos, P. Rho/ROCK and myosin II control the polarized distribution of endocytic clathrin structures at the uropod of moving T lymphocytes. J. Cell Sci. 2007, 120, 3534–3543. [Google Scholar] [CrossRef] [Green Version]
- Marei, H.; Malliri, A. GEFs: Dual regulation of Rac1 signaling. Small GTPases 2017, 8, 90–99. [Google Scholar] [CrossRef]
- Wei, S.Y.; Lin, T.E.; Wang, W.L.; Lee, P.L.; Tsai, M.C.; Chiu, J.J. Protein kinase C-delta and -beta coordinate flow-induced directionality and deformation of migratory human blood T-lymphocytes. J. Mol. Cell Biol. 2014, 6, 458–472. [Google Scholar] [CrossRef]
- Herroeder, S.; Reichardt, P.; Sassmann, A.; Zimmermann, B.; Jaeneke, D.; Hoeckner, J.; Hollmann, M.W.; Fischer, K.D.; Vogt, S.; Grosse, R.; et al. Guanine nucleotide-binding proteins of the G12 family shape immune functions by controlling CD4+ T cell adhesiveness and motility. Immunity 2009, 30, 708–720. [Google Scholar] [CrossRef]
- Moalli, F.; Ficht, X.; Germann, P.; Vladymyrov, M.; Stolp, B.; de Vries, I.; Lyck, R.; Balmer, J.; Fiocchi, A.; Kreutzfeldt, M.; et al. The Rho regulator Myosin IXb enables nonlymphoid tissue seeding of protective CD8(+) T cells. J. Exp. Med. 2018, 215, 1869–1890. [Google Scholar] [CrossRef]
- Rougerie, P.; Largeteau, Q.; Megrelis, L.; Carrette, F.; Lejeune, T.; Toffali, L.; Rossi, B.; Zeghouf, M.; Cherfils, J.; Constantin, G.; et al. Fam65b is a new transcriptional target of FOXO1 that regulates RhoA signaling for T lymphocyte migration. J. Immunol. 2013, 190, 748–755. [Google Scholar] [CrossRef]
- Megrelis, L.; El Ghoul, E.; Moalli, F.; Versapuech, M.; Cassim, S.; Ruef, N.; Stein, J.V.; Mangeney, M.; Delon, J. Fam65b Phosphorylation Relieves Tonic RhoA Inhibition During T Cell Migration. Front. Immunol. 2018, 9, 2001. [Google Scholar] [CrossRef]
- Cabrera-Ortega, A.A.; Feinberg, D.; Liang, Y.; Rossa, C., Jr.; Graves, D.T. The Role of Forkhead Box 1 (FOXO1) in the Immune System: Dendritic Cells, T Cells, B Cells, and Hematopoietic Stem Cells. Crit. Rev. Immunol. 2017, 37, 1–13. [Google Scholar] [CrossRef]
- Ramsay, A.G.; Evans, R.; Kiaii, S.; Svensson, L.; Hogg, N.; Gribben, J.G. Chronic lymphocytic leukemia cells induce defective LFA-1-directed T-cell motility by altering Rho GTPase signaling that is reversible with lenalidomide. Blood 2013, 121, 2704–2714. [Google Scholar] [CrossRef]
- Hammer, J.A.; Wang, J.C.; Saeed, M.; Pedrosa, A.T. Origin, Organization, Dynamics, and Function of Actin and Actomyosin Networks at the T Cell Immunological Synapse. Annu. Rev. Immunol. 2019, 37, 201–224. [Google Scholar] [CrossRef]
- Thauland, T.J.; Hu, K.H.; Bruce, M.A.; Butte, M.J. Cytoskeletal adaptivity regulates T cell receptor signaling. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef]
- Croft, M.; Dubey, C. Accessory Molecule and Co-stimulation Requirements for CD4 T Cell Response. Crit. Rev. Immunol. 2017, 37, 261–290. [Google Scholar] [CrossRef]
- Varga, G.; Nippe, N.; Balkow, S.; Peters, T.; Wild, M.K.; Seeliger, S.; Beissert, S.; Krummen, M.; Roth, J.; Sunderkotter, C.; et al. LFA-1 contributes to signal I of T-cell activation and to the production of T(h)1 cytokines. J. Invest. Derm. 2010, 130, 1005–1012. [Google Scholar] [CrossRef]
- Katagiri, K.; Hattori, M.; Minato, N.; Kinashi, T. Rap1 functions as a key regulator of T-cell and antigen-presenting cell interactions and modulates T-cell responses. Mol. Cell Biol. 2002, 22, 1001–1015. [Google Scholar] [CrossRef]
- Moon, M.Y.; Kim, H.J.; Kim, J.G.; Lee, J.Y.; Kim, J.; Kim, S.C.; Choi, I.G.; Kim, P.H.; Park, J.B. Small GTPase Rap1 regulates cell migration through regulation of small GTPase RhoA activity in response to transforming growth factor-beta1. J. Cell. Physiol. 2013, 228, 2119–2126. [Google Scholar] [CrossRef]
- Garcon, F.; Okkenhaug, K. PI3Kdelta promotes CD4(+) T-cell interactions with antigen-presenting cells by increasing LFA-1 binding to ICAM-1. Immunol. Cell Biol. 2016, 94, 486–495. [Google Scholar] [CrossRef]
- Ueda, Y.; Kondo, N.; Ozawa, M.; Yasuda, K.; Tomiyama, T.; Kinashi, T. Sema3e/Plexin D1 Modulates Immunological Synapse and Migration of Thymocytes by Rap1 Inhibition. J. Immunol. 2016, 196, 3019–3031. [Google Scholar] [CrossRef] [Green Version]
- Delaguillaumie, A.; Lagaudriere-Gesbert, C.; Popoff, M.R.; Conjeaud, H. Rho GTPases link cytoskeletal rearrangements and activation processes induced via the tetraspanin CD82 in T lymphocytes. J. Cell Sci. 2002, 115, 433–443. [Google Scholar]
- Helou, Y.A.; Petrashen, A.P.; Salomon, A.R. Vav1 Regulates T-Cell Activation through a Feedback Mechanism and Crosstalk between the T-Cell Receptor and CD28. J. Proteome Res. 2015, 14, 2963–2975. [Google Scholar] [CrossRef] [Green Version]
- Goodfellow, H.S.; Frushicheva, M.P.; Ji, Q.; Cheng, D.A.; Kadlecek, T.A.; Cantor, A.J.; Kuriyan, J.; Chakraborty, A.K.; Salomon, A.; Weiss, A. The catalytic activity of the kinase ZAP-70 mediates basal signaling and negative feedback of the T cell receptor pathway. Sci. Signal. 2015, 8, ra49. [Google Scholar] [CrossRef]
- Kim, J.G.; Islam, R.; Cho, J.Y.; Jeong, H.; Cap, K.C.; Park, Y.; Hossain, A.J.; Park, J.B. Regulation of RhoA GTPase and various transcription factors in the RhoA pathway. J. Cell. Physiol. 2018, 233, 6381–6392. [Google Scholar] [CrossRef]
- Helms, W.S.; Jeffrey, J.L.; Holmes, D.A.; Townsend, M.B.; Clipstone, N.A.; Su, L. Modulation of NFAT-dependent gene expression by the RhoA signaling pathway in T cells. J. Leukoc. Biol. 2007, 82, 361–369. [Google Scholar] [CrossRef]
- Zhang, S.; Zhou, X.; Lang, R.A.; Guo, F. RhoA of the Rho family small GTPases is essential for B lymphocyte development. PloS ONE 2012, 7, e33773. [Google Scholar] [CrossRef]
- Ishizaki, H.; Togawa, A.; Tanaka-Okamoto, M.; Hori, K.; Nishimura, M.; Hamaguchi, A.; Imai, T.; Takai, Y.; Miyoshi, J. Defective chemokine-directed lymphocyte migration and development in the absence of Rho guanosine diphosphate-dissociation inhibitors alpha and beta. J. Immunol. 2006, 177, 8512–8521. [Google Scholar] [CrossRef]
- Sun, Y.; Thapa, N.; Hedman, A.C.; Anderson, R.A. Phosphatidylinositol 4,5-bisphosphate: targeted production and signaling. Bioessays 2013, 35, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Saci, A.; Carpenter, C.L. RhoA GTPase regulates B cell receptor signaling. Mol. Cell 2005, 17, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Tolar, P. Cytoskeletal control of B cell responses to antigens. Nat. Rev. Immunol. 2017, 17, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Weng, W.K.; Jarvis, L.; LeBien, T.W. Signaling through CD19 activates Vav/mitogen-activated protein kinase pathway and induces formation of a CD19/Vav/phosphatidylinositol 3-kinase complex in human B cell precursors. J. Biol. Chem. 1994, 269, 32514–32521. [Google Scholar] [PubMed]
- Li, G.; Liu, L.; Shan, C.; Cheng, Q.; Budhraja, A.; Zhou, T.; Cui, H.; Gao, N. RhoA/ROCK/PTEN signaling is involved in AT-101-mediated apoptosis in human leukemia cells in vitro and in vivo. Cell Death Dis. 2014, 5, e998. [Google Scholar] [CrossRef] [PubMed]
- Getahun, A.; Wemlinger, S.M.; Rudra, P.; Santiago, M.L.; van Dyk, L.F.; Cambier, J.C. Impaired B cell function during viral infections due to PTEN-mediated inhibition of the PI3K pathway. J. Exp. Med. 2017, 214, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Rubin, S.J.S.; Bloom, M.S.; Robinson, W.H. B cell checkpoints in autoimmune rheumatic diseases. Nat. Rev. Rheumatol. 2019, 15, 303–315. [Google Scholar] [CrossRef]
- Laman, J.D.; Claassen, E.; Noelle, R.J. Functions of CD40 and Its Ligand, gp39 (CD40L). Crit. Rev. Immunol. 2017, 37, 371–420. [Google Scholar] [CrossRef]
- Lee, J.R.; Ha, Y.J.; Kim, H.J. Cutting edge: induced expression of a RhoA-specific guanine nucleotide exchange factor, p190RhoGEF, following CD40 stimulation and WEHI 231 B cell activation. J. Immunol. 2003, 170, 19–23. [Google Scholar] [CrossRef]
- Ha, Y.J.; Jeong, J.H.; Park, Y.; Lee, J.R. Increased p190RhoGEF expression in activated B cells correlates with the induction of the plasma cell differentiation. Exp. Mol. Med. 2012, 44, 138–148. [Google Scholar] [CrossRef] [Green Version]
- Badr, G.; Borhis, G.; Treton, D.; Richard, Y. IFN{alpha} enhances human B-cell chemotaxis by modulating ligand-induced chemokine receptor signaling and internalization. Int. Immunol. 2005, 17, 459–467. [Google Scholar] [CrossRef]
- Matsusaka, S.; Tohyama, Y.; He, J.; Shi, Y.; Hazama, R.; Kadono, T.; Kurihara, R.; Tohyama, K.; Yamamura, H. Protein-tyrosine kinase, Syk, is required for CXCL12-induced polarization of B cells. Biochem. Biophys. Res. Commun. 2005, 328, 1163–1169. [Google Scholar] [CrossRef]
- Alto, N.M.; Shao, F.; Lazar, C.S.; Brost, R.L.; Chua, G.; Mattoo, S.; McMahon, S.A.; Ghosh, P.; Hughes, T.R.; Boone, C.; et al. Identification of a bacterial type III effector family with G protein mimicry functions. Cell 2006, 124, 133–145. [Google Scholar] [CrossRef]
- Heilig, R.; Broz, P. Function and mechanism of the pyrin inflammasome. Eur. J. Immunol. 2018, 48, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Yang, J.; Gao, W.; Li, L.; Li, P.; Zhang, L.; Gong, Y.N.; Peng, X.; Xi, J.J.; Chen, S.; et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 2014, 513, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.H.; Wood, G.; Kastner, D.L.; Chae, J.J. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat. Immunol. 2016, 17, 914–921. [Google Scholar] [CrossRef]
- Hachani, A.; Biskri, L.; Rossi, G.; Marty, A.; Menard, R.; Sansonetti, P.; Parsot, C.; Van Nhieu, G.T.; Bernardini, M.L.; Allaoui, A. IpgB1 and IpgB2, two homologous effectors secreted via the Mxi-Spa type III secretion apparatus, cooperate to mediate polarized cell invasion and inflammatory potential of Shigella flexenri. Microbes Infect. 2008, 10, 260–268. [Google Scholar] [CrossRef]
- Tsoi, L.C.; Spain, S.L.; Knight, J.; Ellinghaus, E.; Stuart, P.E.; Capon, F.; Ding, J.; Li, Y.; Tejasvi, T.; Gudjonsson, J.E.; et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat. Genet. 2012, 44, 1341–1348. [Google Scholar] [CrossRef] [Green Version]
- Raychaudhuri, S.; Thomson, B.P.; Remmers, E.F.; Eyre, S.; Hinks, A.; Guiducci, C.; Catanese, J.J.; Xie, G.; Stahl, E.A.; Chen, R.; et al. Genetic variants at CD28, PRDM1 and CD2/CD58 are associated with rheumatoid arthritis risk. Nat. Genet. 2009, 41, 1313–1318. [Google Scholar] [CrossRef] [Green Version]
- Franke, A.; McGovern, D.P.; Barrett, J.C.; Wang, K.; Radford-Smith, G.L.; Ahmad, T.; Lees, C.W.; Balschun, T.; Lee, J.; Roberts, R.; et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet. 2010, 42, 1118–1125. [Google Scholar] [CrossRef]
- Hunt, K.A.; Zhernakova, A.; Turner, G.; Heap, G.A.; Franke, L.; Bruinenberg, M.; Romanos, J.; Dinesen, L.C.; Ryan, A.W.; Panesar, D.; et al. Newly identified genetic risk variants for celiac disease related to the immune response. Nat. Genet. 2008, 40, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Smyth, D.J.; Plagnol, V.; Walker, N.M.; Cooper, J.D.; Downes, K.; Yang, J.H.; Howson, J.M.; Stevens, H.; McManus, R.; Wijmenga, C.; et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease. N. Engl. J. Med. 2008, 359, 2767–2777. [Google Scholar] [CrossRef]
- Santiago, J.L.; Martinez, A.; Nunez, C.; de la Calle, H.; Fernandez-Arquero, M.; de la Concha, E.G.; Urcelay, E. Association of MYO9B haplotype with type 1 diabetes. Hum. Immunol. 2008, 69, 112–115. [Google Scholar] [CrossRef]
- Sanchez, E.; Alizadeh, B.Z.; Valdigem, G.; Ortego-Centeno, N.; Jimenez-Alonso, J.; de Ramon, E.; Garcia, A.; Lopez-Nevot, M.A.; Wijmenga, C.; Martin, J.; et al. MYO9B gene polymorphisms are associated with autoimmune diseases in Spanish population. Hum. Immunol. 2007, 68, 610–615. [Google Scholar] [CrossRef]
- Paintlia, A.S.; Paintlia, M.K.; Hollis, B.W.; Singh, A.K.; Singh, I. Interference with RhoA-ROCK signaling mechanism in autoreactive CD4+ T cells enhances the bioavailability of 1,25-dihydroxyvitamin D3 in experimental autoimmune encephalomyelitis. Am. J. Pathol. 2012, 181, 993–1006. [Google Scholar] [CrossRef]
- Liu, Z.; Xu, Y.; Zhang, X.; Song, J.; Sorokin, L.; Bahler, M. The motorized RhoGAP myosin IXb (Myo9b) in leukocytes regulates experimental autoimmune encephalomyelitis induction and recovery. J. Neuroimmunol. 2015, 282, 25–32. [Google Scholar] [CrossRef]
- Manresa-Arraut, A.; Johansen, F.F.; Brakebusch, C.; Issazadeh-Navikas, S.; Hasseldam, H. RhoA Drives T-Cell Activation and Encephalitogenic Potential in an Animal Model of Multiple Sclerosis. Front. Immunol. 2018, 9, 1235. [Google Scholar] [CrossRef] [Green Version]
- Tamehiro, N.; Nishida, K.; Yanobu-Takanashi, R.; Goto, M.; Okamura, T.; Suzuki, H. T-cell activation RhoGTPase-activating protein plays an important role in TH17-cell differentiation. Immunol. Cell Biol. 2017, 95, 729–735. [Google Scholar] [CrossRef]
- Patsopoulos, N.A.; the Bayer Pharma MS Genetics Working Group; the Steering Committees of Studies Evaluating IFNβ-1b and a CCR1-Antagonist; ANZgene Consortium; GeneMSA; International Multiple Sclerosis Genetics Consortium; de Bakker, P.I.W. Genome-wide meta-analysis identifies novel multiple sclerosis susceptibility loci. Ann. Neurol. 2011, 70, 897–912. [Google Scholar] [CrossRef]
- Bertuzzi, M.; Hayes, G.E.; Bignell, E.M. Microbial uptake by the respiratory epithelium: outcomes for host and pathogen. Fems Microbiol. Rev. 2019, 43, 145–161. [Google Scholar] [CrossRef]
- Paulsson, M.; Riesbeck, K. How bacteria hack the matrix and dodge the bullets of immunity. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2018, 27. [Google Scholar] [CrossRef]
- Jamilloux, Y.; Magnotti, F.; Belot, A.; Henry, T. The pyrin inflammasome: from sensing RhoA GTPases-inhibiting toxins to triggering autoinflammatory syndromes. Pathog. Dis. 2018, 76. [Google Scholar] [CrossRef] [Green Version]
- Randow, F.; MacMicking, J.D.; James, L.C. Cellular self-defense: how cell-autonomous immunity protects against pathogens. Science 2013, 340, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Mostowy, S.; Shenoy, A.R. The cytoskeleton in cell-autonomous immunity: structural determinants of host defence. Nat. Rev.Immunol. 2015, 15, 559–573. [Google Scholar] [CrossRef] [Green Version]
- Bent, R.; Moll, L.; Grabbe, S.; Bros, M. Interleukin-1 Beta-A Friend or Foe in Malignancies? Int. J. Mol. Sci. 2018, 19, 2155. [Google Scholar] [CrossRef]
- Wang, L.; Wang, F.S.; Gershwin, M.E. Human autoimmune diseases: A comprehensive update. J. Intern. Med. 2015, 278, 369–395. [Google Scholar] [CrossRef]
- Zhang, Z.; Schittenhelm, J.; Meyermann, R.; Schluesener, H.J. Lesional accumulation of RhoA(+) cells in brains of experimental autoimmune encephalomyelitis and multiple sclerosis. Neuropathol. Appl. Neurobiol. 2008, 34, 231–240. [Google Scholar] [CrossRef]
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef]
- Trapp, B.D.; Nave, K.A. Multiple sclerosis: An immune or neurodegenerative disorder? Annu. Rev. Neurosci. 2008, 31, 247–269. [Google Scholar] [CrossRef]
- Ramagopalan, S.V.; Sadovnick, A.D. Epidemiology of multiple sclerosis. Neurol. Clin. 2011, 29, 207–217. [Google Scholar] [CrossRef]
- Cahalan, M.D.; Gutman, G.A. The sense of place in the immune system. Nat. Immunol. 2006, 7, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Hoebe, K.J.E.; Beutler, B. The interface between innate and adaptive immunity. Nat. Immunol. 2004, 5, 971–974. [Google Scholar] [CrossRef]
- Makaroff, L.E.; Hendricks, D.W.; Niec, R.E.; Fink, P.J. Postthymic maturation influences the CD8 T cell response to antigen. Proc. Natl. Acad. Sci. USA 2009, 106, 4799–4804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicola Galiani, S.H. Basic Aspects of T Helper Cell Differentiation. Methods Mol. Biol. 2016, 19–30. [Google Scholar]
- Steinman, R.M.; Hawiger, D.; Liu, K.; Bonifaz, L.; Bonnyay, D.; Mahnke, K.; Iyoda, T.; Ravetch, J.; Dhodapkar, M.; Inaba, K.; et al. Dendritic cell function in vivo during the steady state: A role in peripheral tolerance. Ann. N Y Acad. Sci. 2003, 987, 15–25. [Google Scholar] [CrossRef]
- Bedoya, S.K.; Lam, B.; Lau, K.; Larkin, J., III. Th17 cells in immunity and autoimmunity. Clin. Dev. Immunol. 2013, 2013, 986789. [Google Scholar] [CrossRef]
- Yang, J.; Sundrud, M.S.; Skepner, J.; Yamagata, T. Targeting Th17 cells in autoimmune diseases. Trends Pharm. Sci. 2014, 35, 493–500. [Google Scholar] [CrossRef]
- Tak, W.; Mak, M.E.S. 16- Immune Tolerance in the Periphery. Immune Response Basic Clin. Princ. 2006, 433–462. [Google Scholar]
- Tak, W.; Mak, M.E.S.; Jett, B.D. Chapter 10—Regulation of Immune Responses in the Periphery. In Primer to the Immune Response, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 227–246. [Google Scholar]
- Asao, H. Interleukin-2. Encycl. Endocr. Dis. 2004, 60–63. [Google Scholar]
- Zhu, J.; Paul, W.E. Peripheral CD4+ T-cell differentiation regulated by networks of cytokines and transcription factors. Immunol. Rev. 2010, 238, 247–262. [Google Scholar] [CrossRef]
- Singh, R.P.; Hasan, S.; Sharma, S.; Nagra, S.; Yamaguchi, D.T.; Wong, D.T.; Hahn, B.H.; Hossain, A. Th17 cells in inflammation and autoimmunity. Autoimmun. Rev. 2014, 13, 1174–1181. [Google Scholar] [CrossRef]
- Komatsu, N.; Okamoto, K.; Sawa, S.; Nakashima, T.; Oh-hora, M.; Kodama, T.; Tanaka, S.; Bluestone, J.A.; Takayanagi, H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat. Med. 2014, 20, 62–68. [Google Scholar] [CrossRef]
- Cantorna, M.T.; Hayes, C.E.; DeLuca, H.F. 1,25-Dihydroxyvitamin D3 reversibly blocks the progression of relapsing encephalomyelitis, a model of multiple sclerosis. Proc. Natl. Acad. Sci. USA 1996, 93, 7861–7864. [Google Scholar] [CrossRef]
- Spach, K.M.; Pedersen, L.B.; Nashold, F.E.; Kayo, T.; Yandell, B.S.; Prolla, T.A.; Hayes, C.E. Gene expression analysis suggests that 1,25-dihydroxyvitamin D3 reverses experimental autoimmune encephalomyelitis by stimulating inflammatory cell apoptosis. Physiol. Genom. 2004, 18, 141–151. [Google Scholar] [CrossRef]
- Joshi, S.; Pantalena, L.C.; Liu, X.K.; Gaffen, S.L.; Liu, H.; Rohowsky-Kochan, C.; Ichiyama, K.; Yoshimura, A.; Steinman, L.; Christakos, S.; et al. 1,25-dihydroxyvitamin D(3) ameliorates Th17 autoimmunity via transcriptional modulation of interleukin-17A. Mol. Cell Biol. 2011, 31, 3653–3669. [Google Scholar] [CrossRef]
- Mayne, C.G.; Spanier, J.A.; Relland, L.M.; Williams, C.B.; Hayes, C.E. 1,25-Dihydroxyvitamin D3 acts directly on the T lymphocyte vitamin D receptor to inhibit experimental autoimmune encephalomyelitis. Eur. J. Immunol. 2011, 41, 822–832. [Google Scholar] [CrossRef]
- Spach, K.M.; Hayes, C.E. Vitamin D3 confers protection from autoimmune encephalomyelitis only in female mice. J. Immunol. 2005, 175, 4119–4126. [Google Scholar] [CrossRef]
- Cortes, J.R.; Ambesi-Impiombato, A.; Couronne, L.; Quinn, S.A.; Kim, C.S.; da Silva Almeida, A.C.; West, Z.; Belver, L.; Martin, M.S.; Scourzic, L.; et al. RHOA G17V Induces T Follicular Helper Cell Specification and Promotes Lymphomagenesis. Cancer Cell 2018, 33, 259–273.e257. [Google Scholar] [CrossRef]
- Ng, S.Y.; Brown, L.; Stevenson, K.; deSouza, T.; Aster, J.C.; Louissaint, A., Jr.; Weinstock, D.M. RhoA G17V is sufficient to induce autoimmunity and promotes T-cell lymphomagenesis in mice. Blood 2018, 132, 935–947. [Google Scholar] [CrossRef] [Green Version]
- Fujisawa, M.; Sakata-Yanagimoto, M.; Nishizawa, S.; Komori, D.; Gershon, P.; Kiryu, M.; Tanzima, S.; Fukumoto, K.; Enami, T.; Muratani, M.; et al. Activation of RHOA-VAV1 signaling in angioimmunoblastic T-cell lymphoma. Leukemia 2018, 32, 694–702. [Google Scholar] [CrossRef]
- Keating, G.M. Dasatinib: A Review in Chronic Myeloid Leukaemia and Ph+ Acute Lymphoblastic Leukaemia. Drugs 2017, 77, 85–96. [Google Scholar] [CrossRef]
- Nagata, Y.; Kontani, K.; Enami, T.; Kataoka, K.; Ishii, R.; Totoki, Y.; Kataoka, T.R.; Hirata, M.; Aoki, K.; Nakano, K.; et al. Variegated RHOA mutations in adult T-cell leukemia/lymphoma. Blood 2016, 127, 596–604. [Google Scholar] [CrossRef] [Green Version]
- Morin, R.D.; Mungall, K.; Pleasance, E.; Mungall, A.J.; Goya, R.; Huff, R.D.; Scott, D.W.; Ding, J.; Roth, A.; Chiu, R.; et al. Mutational and structural analysis of diffuse large B-cell lymphoma using whole-genome sequencing. Blood 2013, 122, 1256–1265. [Google Scholar] [CrossRef] [Green Version]
- O’Hayre, M.; Inoue, A.; Kufareva, I.; Wang, Z.; Mikelis, C.M.; Drummond, R.A.; Avino, S.; Finkel, K.; Kalim, K.W.; DiPasquale, G.; et al. Inactivating mutations in GNA13 and RHOA in Burkitt’s lymphoma and diffuse large B-cell lymphoma: A tumor suppressor function for the Galpha13/RhoA axis in B cells. Oncogene 2016, 35, 3771–3780. [Google Scholar] [CrossRef]
- Muppidi, J.R.; Schmitz, R.; Green, J.A.; Xiao, W.; Larsen, A.B.; Braun, S.E.; An, J.; Xu, Y.; Rosenwald, A.; Ott, G.; et al. Loss of signalling via Galpha13 in germinal centre B-cell-derived lymphoma. Nature 2014, 516, 254–258. [Google Scholar] [CrossRef]
- Healy, J.A.; Nugent, A.; Rempel, R.E.; Moffitt, A.B.; Davis, N.S.; Jiang, X.; Shingleton, J.R.; Zhang, J.; Love, C.; Datta, J.; et al. GNA13 loss in germinal center B cells leads to impaired apoptosis and promotes lymphoma in vivo. Blood 2016, 127, 2723–2731. [Google Scholar] [CrossRef] [Green Version]
- Bouafia, A.; Lofek, S.; Bruneau, J.; Chentout, L.; Lamrini, H.; Trinquand, A.; Deau, M.C.; Heurtier, L.; Meignin, V.; Picard, C.; et al. Loss of ARHGEF1 causes a human primary antibody deficiency. J. Clin. Invest 2019, 129, 1047–1060. [Google Scholar] [CrossRef] [Green Version]
- Schittenhelm, L.; Hilkens, C.M.; Morrison, V.L. beta2 Integrins As Regulators of Dendritic Cell, Monocyte, and Macrophage Function. Front. Immunol. 2017, 8, 1866. [Google Scholar] [CrossRef]
- Rozo, C.; Chinenov, Y.; Maharaj, R.K.; Gupta, S.; Leuenberger, L.; Kirou, K.A.; Bykerk, V.P.; Goodman, S.M.; Salmon, J.E.; Pernis, A.B. Targeting the RhoA-ROCK pathway to reverse T-cell dysfunction in SLE. Ann. Rheum. Dis. 2017, 76, 740–747. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bros, M.; Haas, K.; Moll, L.; Grabbe, S. RhoA as a Key Regulator of Innate and Adaptive Immunity. Cells 2019, 8, 733. https://doi.org/10.3390/cells8070733
Bros M, Haas K, Moll L, Grabbe S. RhoA as a Key Regulator of Innate and Adaptive Immunity. Cells. 2019; 8(7):733. https://doi.org/10.3390/cells8070733
Chicago/Turabian StyleBros, Matthias, Katharina Haas, Lorna Moll, and Stephan Grabbe. 2019. "RhoA as a Key Regulator of Innate and Adaptive Immunity" Cells 8, no. 7: 733. https://doi.org/10.3390/cells8070733
APA StyleBros, M., Haas, K., Moll, L., & Grabbe, S. (2019). RhoA as a Key Regulator of Innate and Adaptive Immunity. Cells, 8(7), 733. https://doi.org/10.3390/cells8070733