Regulation of Acetylcholine Quantal Release by Coupled Thrombin/BDNF Signaling in Mouse Motor Synapses

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Neuromuscular Preparations
2.2. Electrophysiology
2.3. Drugs
2.4. Data Processing and Statistical Analysis
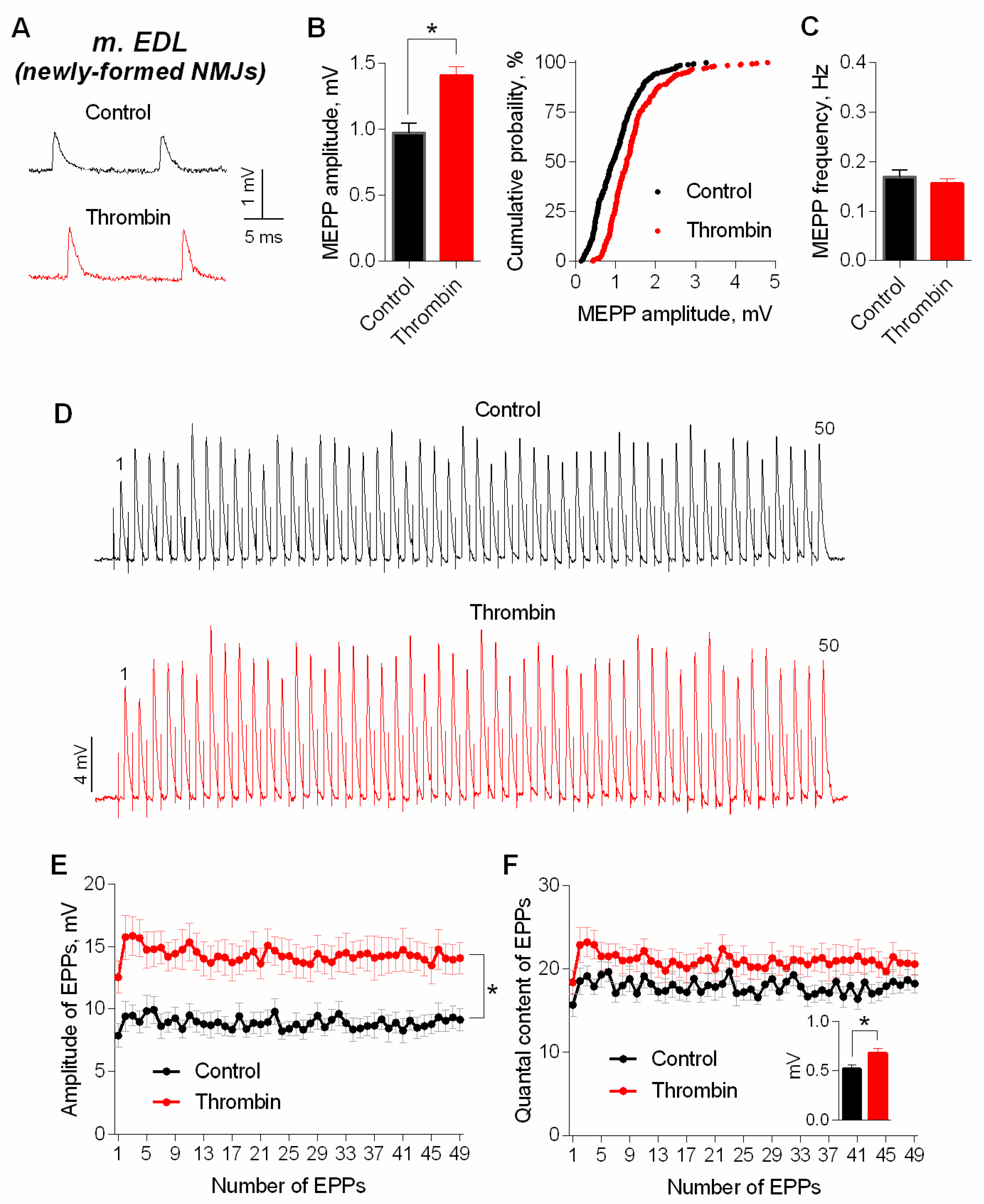
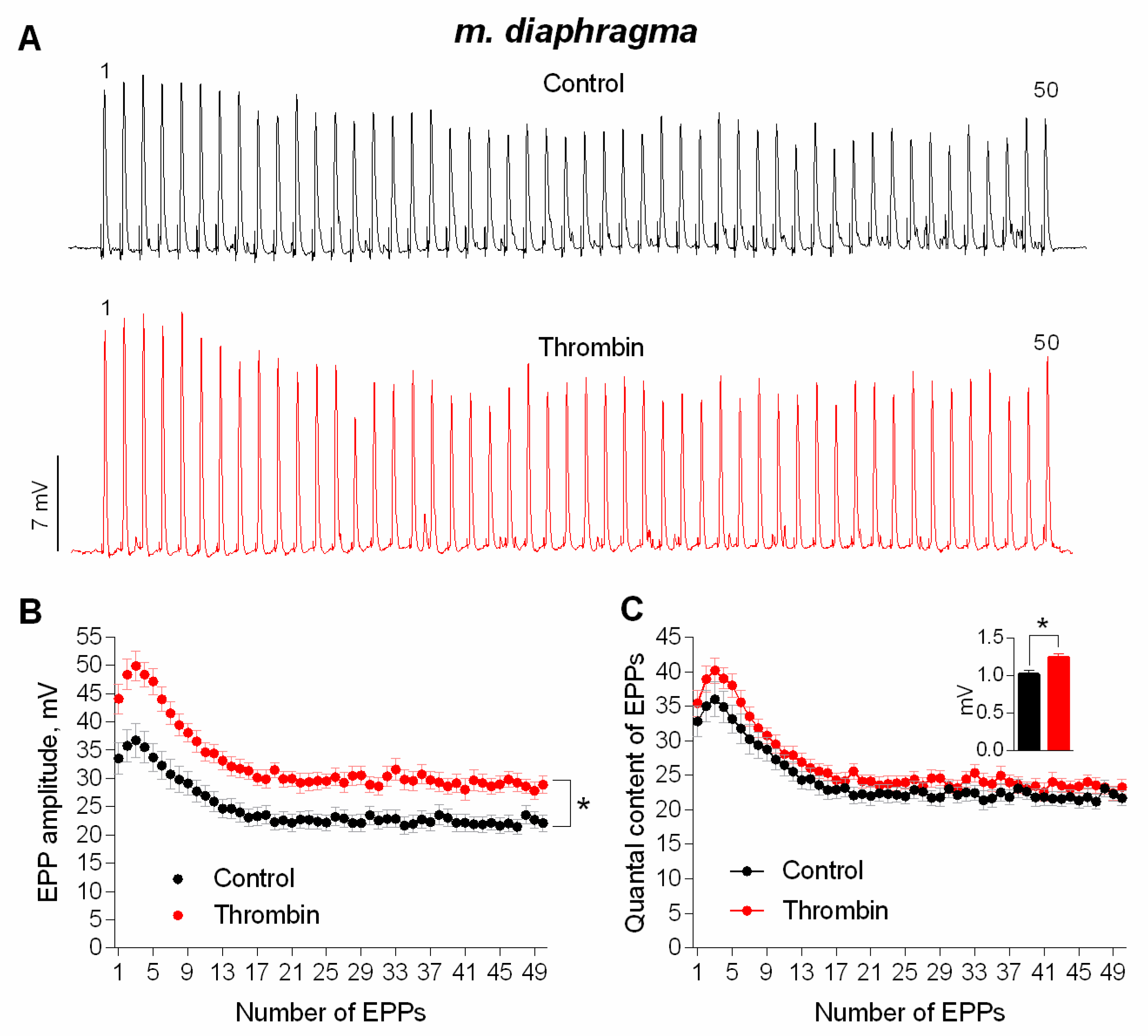
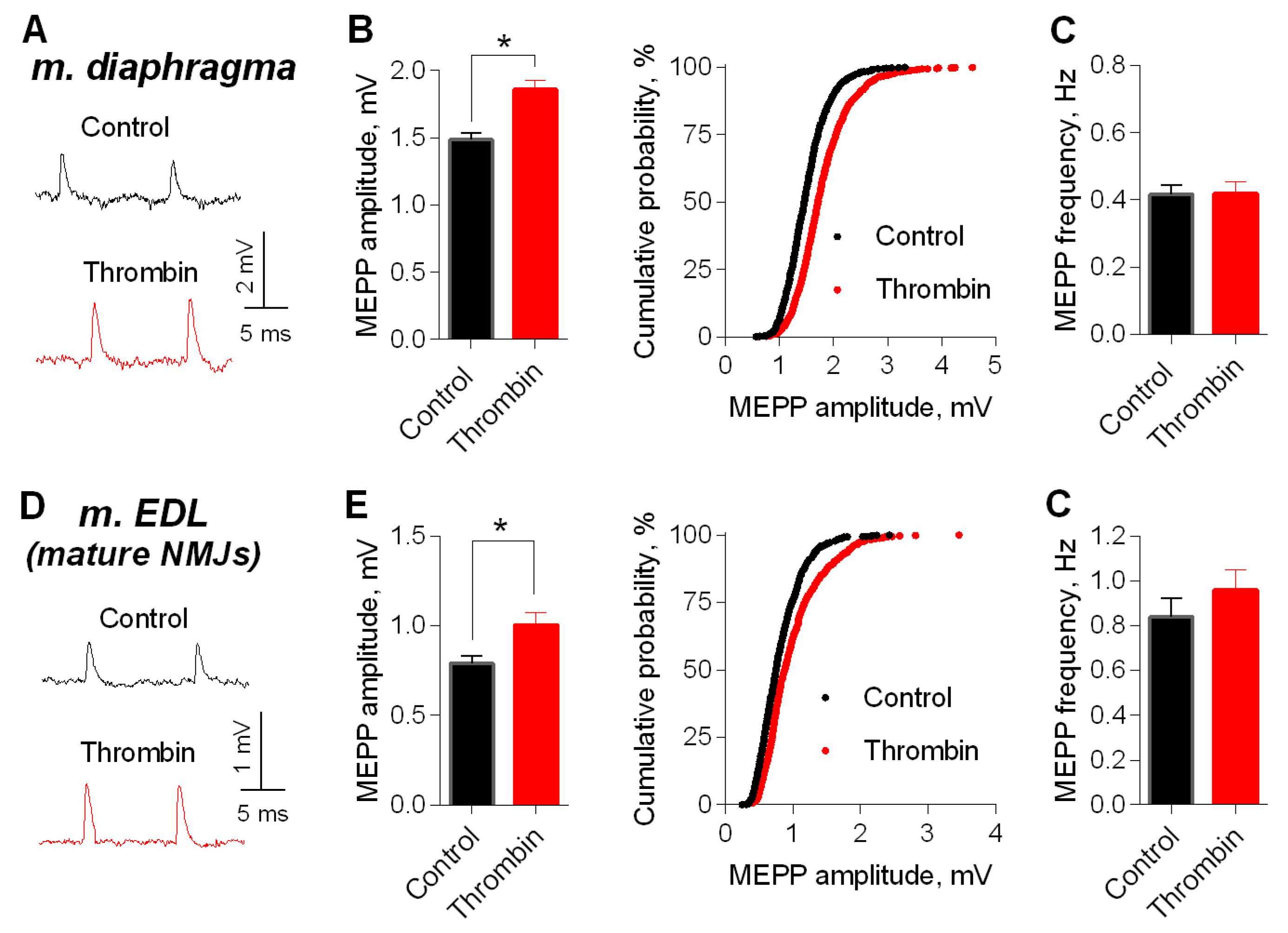
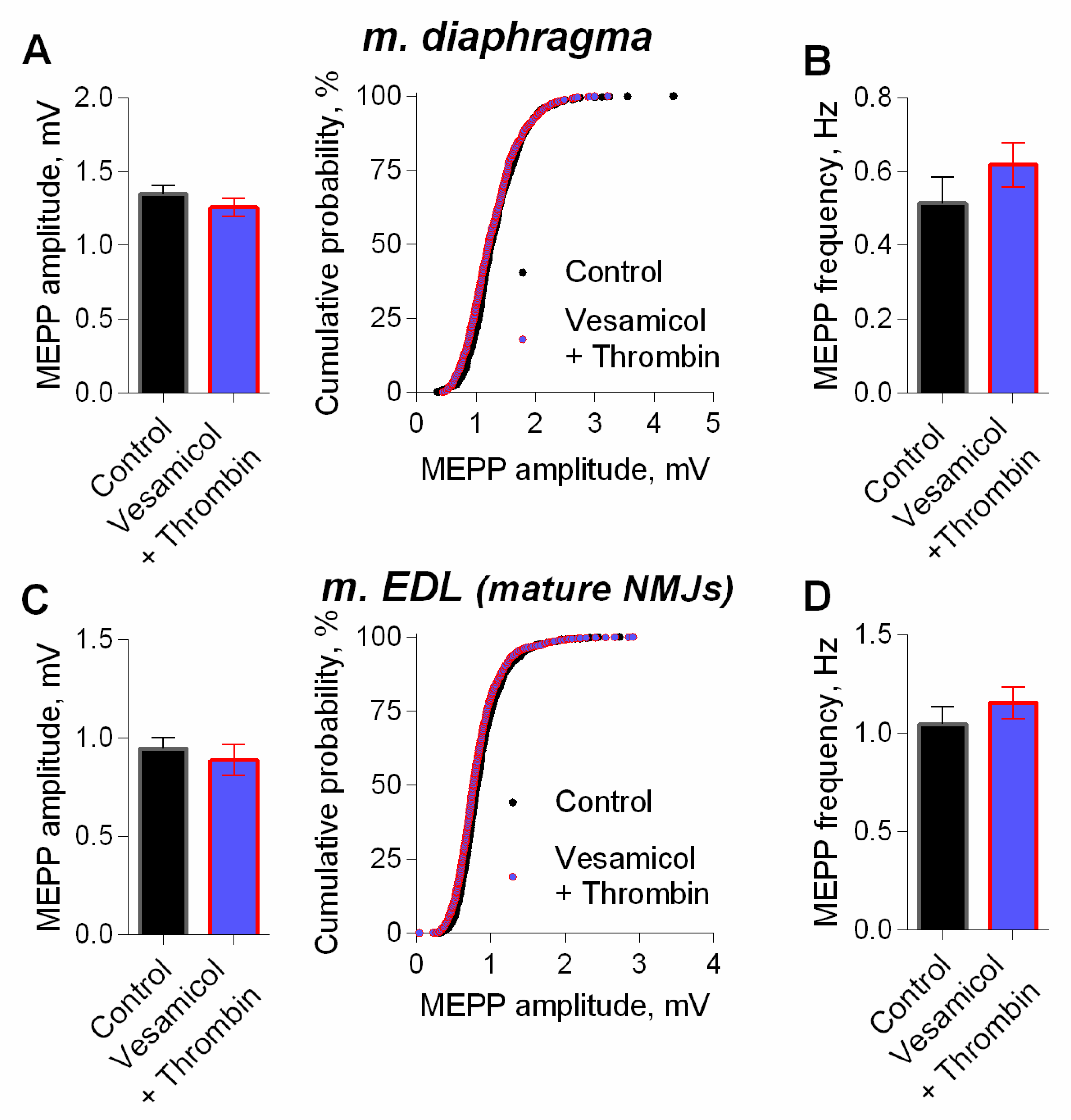
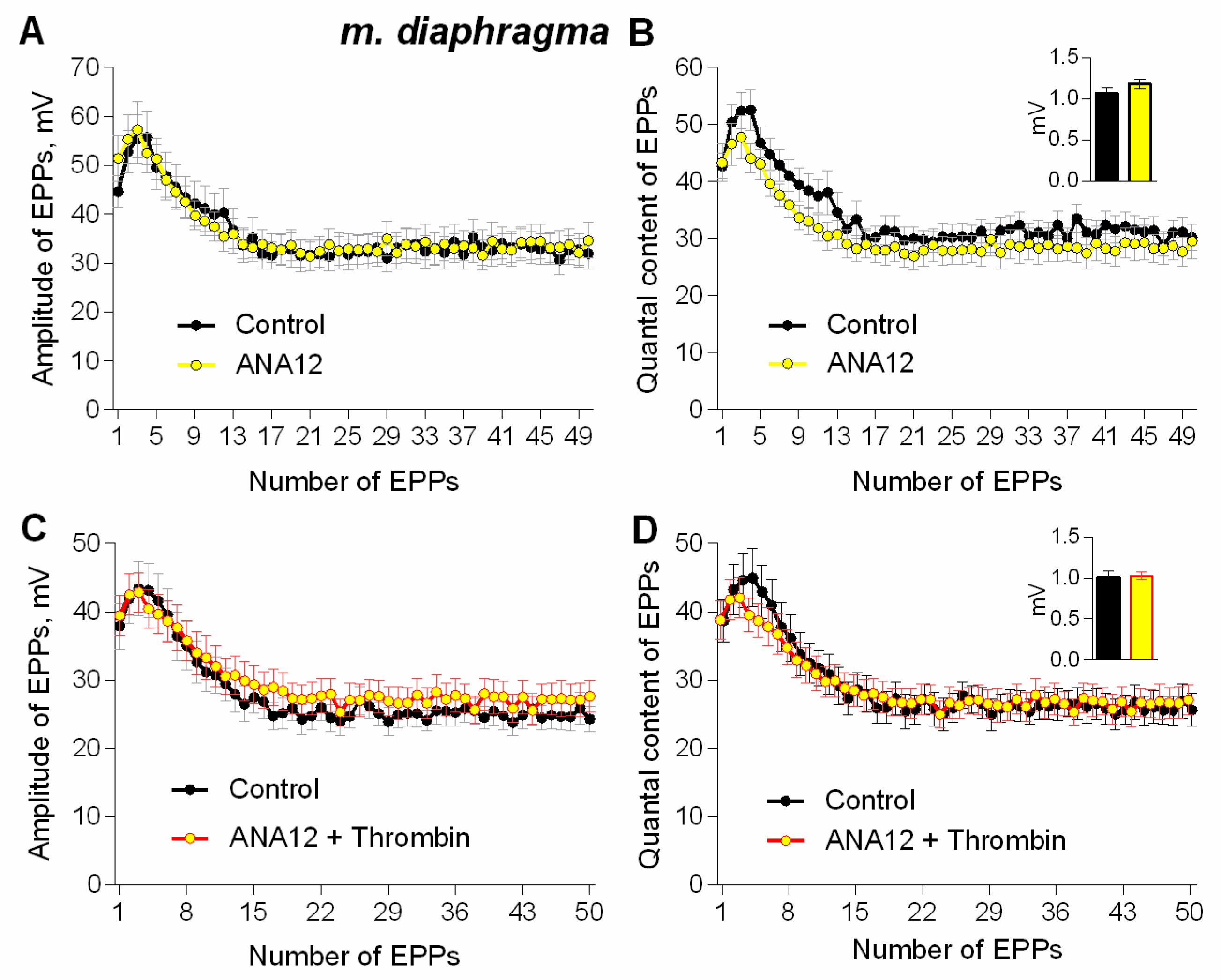
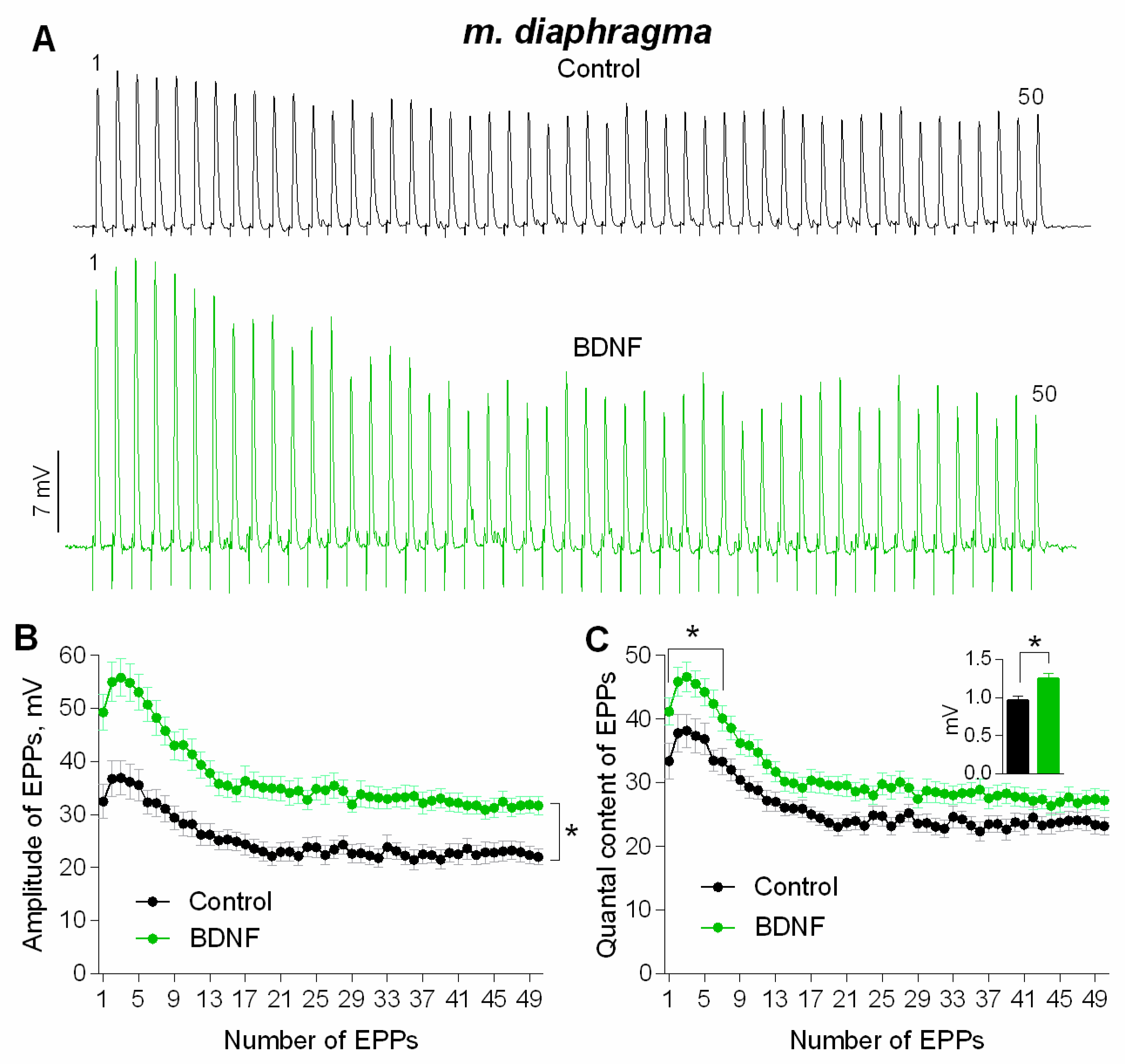
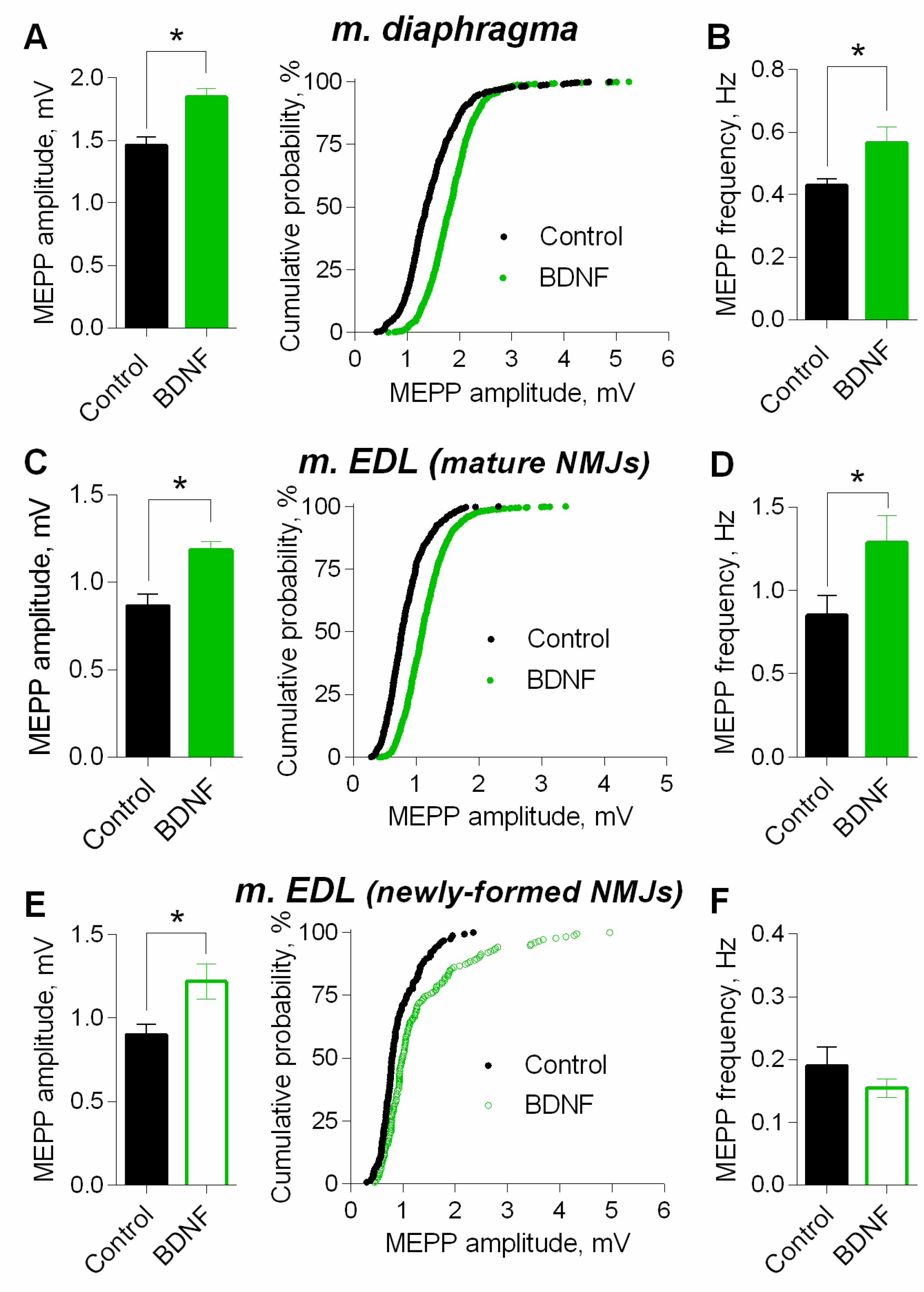
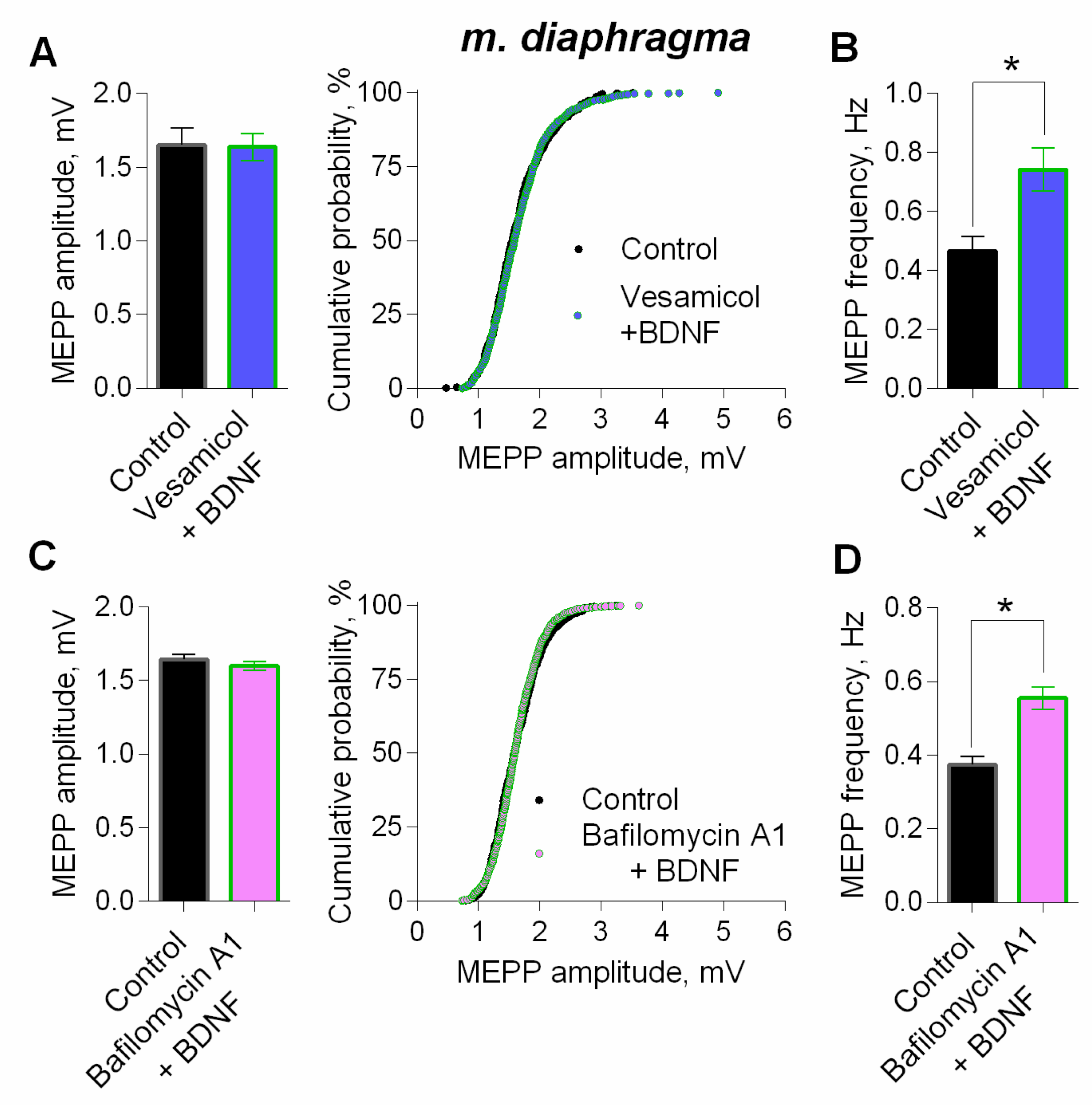
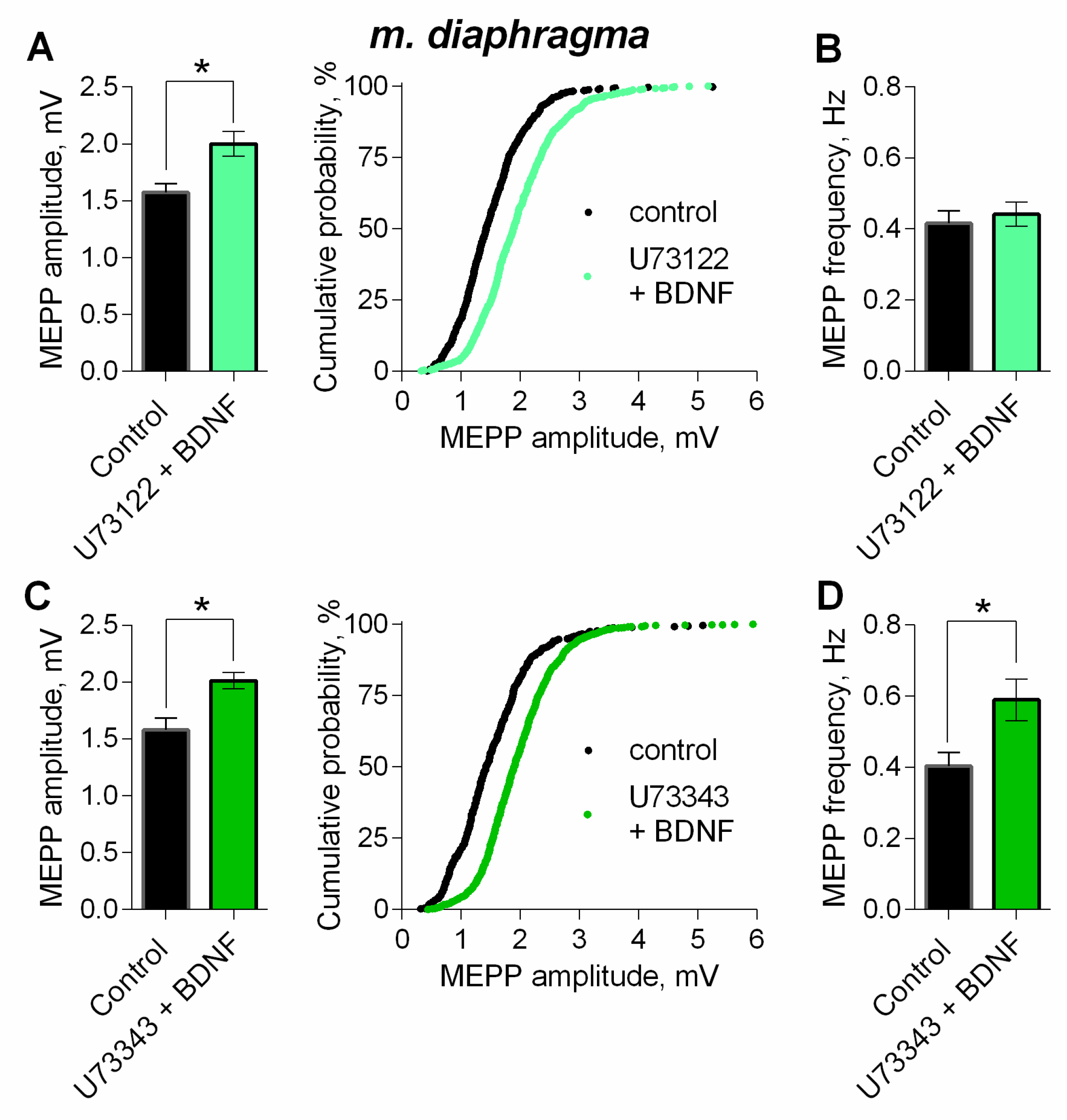
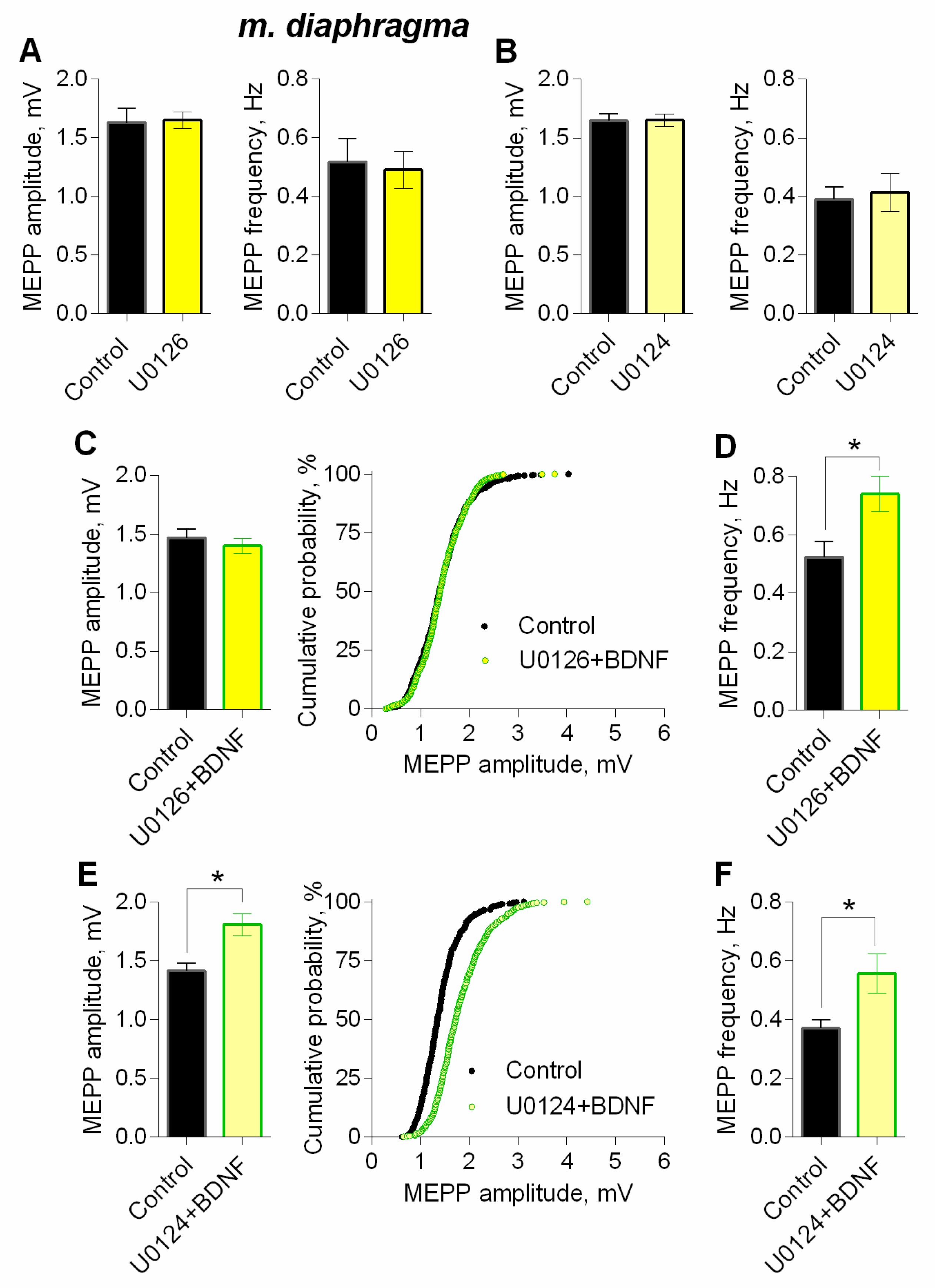
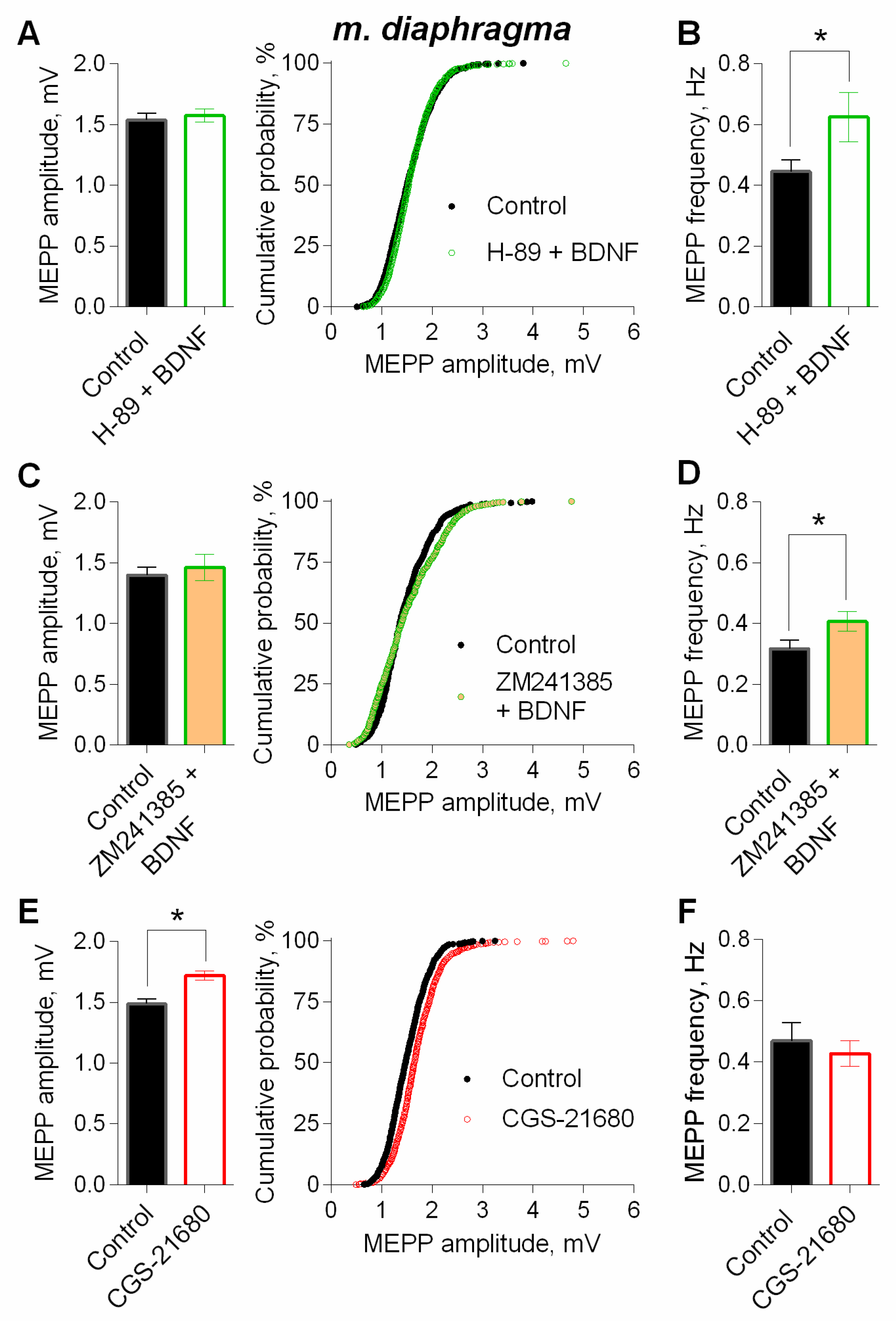
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pedersen, B.K. Muscle as a Secretory Organ. In Comprehensive Physiology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; Volume 3, pp. 1337–1362. [Google Scholar]
- Giudice, J.; Taylor, J.M. Muscle as a paracrine and endocrine organ. Curr. Opin. Pharmacol. 2017, 34, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Delezie, J.; Handschin, C. Endocrine Crosstalk Between Skeletal Muscle and the Brain. Front. Neurol. 2018, 9, 698. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, C.; Weigert, C. Skeletal Muscle as an Endocrine Organ: The Role of Myokines in Exercise Adaptations. Cold Spring Harb. Perspect. Med. 2017, 7, a029793. [Google Scholar] [CrossRef] [PubMed]
- Matthews, V.B.; Åström, M.-B.; Chan, M.H.S.; Bruce, C.R.; Krabbe, K.S.; Prelovsek, O.; Åkerström, T.; Yfanti, C.; Broholm, C.; Mortensen, O.H.; et al. Brain-derived neurotrophic factor is produced by skeletal muscle cells in response to contraction and enhances fat oxidation via activation of AMP-activated protein kinase. Diabetologia 2009, 52, 1409–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liem, R.S.; Brouwer, N.; Copray, J.C. Ultrastructural localisation of intramuscular expression of BDNF mRNA by silver-gold intensified non-radioactive in situ hybridisation. Histochem. Cell Biol. 2001, 116, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Pitts, E.V.; Potluri, S.; Hess, D.M.; Balice-Gordon, R.J. Neurotrophin and Trk-mediated signaling in the neuromuscular system. Int. Anesthesiol. Clin. 2006, 44, 21–76. [Google Scholar] [CrossRef] [PubMed]
- Zoubine, M.N.; Ma, J.Y.; Smirnova, I.V.; Citron, B.A.; Festoff, B.W. A Molecular Mechanism for Synapse Elimination: Novel Inhibition of Locally Generated Thrombin Delays Synapse Loss in Neonatal Mouse Muscle. Dev. Biol. 1996, 179, 447–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glazner, G.W.; Yadav, K.; Fitzgerald, S.; Coven, E.; Brenneman, D.E.; Nelson, P.G. Cholinergic stimulation increases thrombin activity and gene expression in cultured mouse muscle. Brain Res. Dev. Brain Res. 1997, 99, 148–154. [Google Scholar] [CrossRef]
- Citron, B.A.; Smirnova, I.V.; Zoubine, M.N.; Festoff, B.W. Quantitative PCR analysis reveals novel expression of prothrombin mRNA and regulation of its levels in developing mouse muscle. Thromb. Res. 1997, 87, 303–313. [Google Scholar] [CrossRef]
- Faraut, B.; Barbier, J.; Ravel-Chapuis, A.; Doyennette, M.-A.; Jandrot-Perrus, M.; Verdière-Sahuqué, M.; Schaeffer, L.; Koenig, J.; Hantaï, D. Thrombin downregulates muscle acetylcholine receptors via an IP3 signaling pathway by activating its G-protein-coupled protease-activated receptor-1. J. Cell. Physiol. 2003, 196, 105–112. [Google Scholar] [CrossRef]
- Lanuza, M.A.; Garcia, N.; González, C.M.; Santafé, M.M.; Nelson, P.G.; Tomas, J. Role and expression of thrombin receptor PAR-1 in muscle cells and neuromuscular junctions during the synapse elimination period in the neonatal rat. J. Neurosci. Res. 2003, 73, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Suidan, H.S.; Stone, S.R.; Hemmings, B.A.; Monard, D. Thrombin causes neurite retraction in neuronal cells through activation of cell surface receptors. Neuron 1992, 8, 363–375. [Google Scholar] [CrossRef]
- Gaydukov, A.E.; Akutin, I.A.; Bogacheva, P.O.; Balezina, O.P. Changes in the Parameters of Quantal Acetylcholine Release after Activation of PAR1-Type Thrombin Receptors at the Mouse Neuromuscular Junctions. Biochem. (Moscow), Suppl. Ser. A Membr. Cell Biol. 2018, 12, 33–42. [Google Scholar] [CrossRef]
- Lanuza, M.A.; Besalduch, N.; Garcia, N.; Sabaté, M.; Santafé, M.M.; Tomàs, J. Plastic-embedded semithin cross-sections as a tool for high-resolution immunofluorescence analysis of the neuromuscular junction molecules: Specific cellular location of protease-activated receptor-1. J. Neurosci. Res. 2007, 85, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Garcia, N.; Tomàs, M.; Santafe, M.M.; Lanuza, M.A.; Besalduch, N.; Tomàs, J. Localization of brain-derived neurotrophic factor, neurotrophin-4, tropomyosin-related kinase b receptor, and p75 NTR receptor by high-resolution immunohistochemistry on the adult mouse neuromuscular junction. J. Peripher. Nerv. Syst. 2010, 15, 40–49. [Google Scholar] [CrossRef]
- Barstad, J.A.; Lilleheil, G. Transversaly cut diaphragm preparation from rat. An adjuvant tool in the study of the physiology and pbarmacology of the myoneural junction. Arch. Int. Pharmacodyn. Ther. 1968, 175, 373–390. [Google Scholar] [PubMed]
- McArdle, J.J.; Albuquerque, E.X. A study of the reinnervation of fast and slow mammalian muscles. J. Gen. Physiol. 1973, 61, 1–23. [Google Scholar] [CrossRef]
- Argentieri, T.M.; Aiken, S.P.; Laxminarayan, S.; McArdle, J.J. Characteristics of synaptic transmission in reinnervating rat skeletal muscle. Pflugers Arch. 1992, 421, 256–261. [Google Scholar] [CrossRef]
- Balezina, O.P.; Bogacheva, P.O.; Orlova, T.Y. Effect of L-type calcium channel blockers on activity of newly formed synapses in mice. Bull. Exp. Biol. Med. 2007, 143, 171–174. [Google Scholar] [CrossRef]
- Bogatcheva, P.O.; Balezina, O.P. Multidirectional effects of calmodulin kinase II on transmitter release in mature and newly formed mouse motor synapses. Bull. Exp. Biol. Med. 2013, 154, 316–319. [Google Scholar] [CrossRef]
- Flink, M.T.; Atchison, W.D. Iberiotoxin-Induced Block of Ca2+-Activated K+ Channels Induces Dihydropyridine Sensitivity of ACh Release from Mammalian Motor Nerve Terminals. J. Pharmacol. Exp. Ther. 2003, 305, 646–652. [Google Scholar] [CrossRef] [PubMed]
- McLachlan, E.M.; Martin, A.R. Non-linear summation of end-plate potentials in the frog and mouse. J. Physiol. 1981, 311, 307–324. [Google Scholar] [CrossRef] [PubMed]
- Bogacheva, P.O.; Golikova, E.A.; Balezina, O.P. The Role of Endogenous Calcitonin Gene-Related Peptide in the Neurotransmitter Quantal Size Increase in Mouse Neuromuscular Junctions. Biochem. (Moscow) Suppl. Ser. A Membr. Cell Biol. 2018, 12, 268–277. [Google Scholar] [CrossRef]
- Gaydukov, A.E.; Bogacheva, P.O.; Balezina, O.P. Calcitonin gene-related peptide increases acetylcholine quantal size in neuromuscular junctions of mice. Neurosci. Lett. 2016, 628, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Santafé, M.M.; Garcia, N.; Tomàs, M.; Obis, T.; Lanuza, M.A.; Besalduch, N.; Tomàs, J. The interaction between tropomyosin-related kinase B receptors and serine kinases modulates acetylcholine release in adult neuromuscular junctions. Neurosci. Lett. 2014, 561, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.J. Reduction of quantal size and inhibition of neuromuscular transmission by bafilomycin A. Neuropharmacology 2001, 41, 609–617. [Google Scholar] [CrossRef]
- Fong, S.W.; McLennan, I.S.; McIntyre, A.; Reid, J.; Shennan, K.I.J.; Bewick, G.S. TGF-beta2 alters the characteristics of the neuromuscular junction by regulating presynaptic quantal size. Proc. Natl. Acad. Sci. USA 2010, 107, 13515–13519. [Google Scholar] [CrossRef]
- Kaplan, D.R.; Miller, F.D. Neurotrophin signal transduction in the nervous system. Curr. Opin. Neurobiol. 2000, 10, 381–391. [Google Scholar] [CrossRef]
- Minichiello, L. TrkB signalling pathways in LTP and learning. Nat. Rev. Neurosci. 2009, 10, 850–860. [Google Scholar] [CrossRef]
- Sebastião, A.M.; Assaife-Lopes, N.; Diógenes, M.J.; Vaz, S.H.; Ribeiro, J.A. Modulation of brain-derived neurotrophic factor (BDNF) actions in the nervous system by adenosine A(2A) receptors and the role of lipid rafts. Biochim. Biophys. Acta 2011, 1808, 1340–1349. [Google Scholar] [CrossRef]
- Park, H.; Poo, M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2013, 14, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Van der Kloot, W.; Benjamin, W.B.; Balezina, O.P. Calcitonin gene-related peptide acts presynaptically to increase quantal size and output at frog neuromuscular junctions. J. Physiol. 1998, 507 Pt 3, 689–695. [Google Scholar] [CrossRef]
- Correia-de-Sá, P.; Ribeiro, J.A. Evidence that the presynaptic A2a-adenosine receptor of the rat motor nerve endings is positively coupled to adenylate cyclase. Naunyn. Schmiedebergs. Arch. Pharmacol. 1994, 350, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, L.; Correia-de-Sá, P. Protein kinase A and Ca(v)1 (L-Type) channels are common targets to facilitatory adenosine A2A and muscarinic M1 receptors on rat motoneurons. Neurosignals. 2005, 14, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Pousinha, P.A.; José Diogenes, M.; Alexandre Ribeiro, J.; Sebastião, A.M. Triggering of BDNF facilitatory action on neuromuscular transmission by adenosine A2A receptors. Neurosci. Lett. 2006, 404, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Tarasova, E.O.; Miteva, A.S.; Gaidukov, A.E.; Balezina, O.P. The role of adenosine receptors and L-type calcium channels in the regulation of the mediator secretion in mouse motor synapses. Biochem. Suppl. Ser. A Membr. Cell Biol. 2015, 9, 318–328. [Google Scholar] [CrossRef]
- Kim, S.; Buonanno, A.; Nelson, P.G. Regulation of prothrombin, thrombin receptor, and protease nexin-1 expression during development and after denervation in muscle. J. Neurosci. Res. 1998, 53, 304–311. [Google Scholar] [CrossRef]
- Gould, T.W.; Dominguez, B.; de Winter, F.; Yeo, G.W.; Liu, P.; Sundararaman, B.; Stark, T.; Vu, A.; Degen, J.L.; Lin, W.; et al. Glial cells maintain synapses by inhibiting an activity-dependent retrograde protease signal. PLoS Genet. 2019, 15, e1007948. [Google Scholar] [CrossRef]
- Hurtado, E.; Cilleros, V.; Nadal, L.; Simó, A.; Obis, T.; Garcia, N.; Santafé, M.M.; Tomàs, M.; Halievski, K.; Jordan, C.L.; et al. Muscle Contraction Regulates BDNF/TrkB Signaling to Modulate Synaptic Function through Presynaptic cPKCα and cPKCβI. Front. Mol. Neurosci. 2017, 10, 147. [Google Scholar] [CrossRef]
- Tamura, S.; Suzuki, H.; Hirowatari, Y.; Hatase, M.; Nagasawa, A.; Matsuno, K.; Kobayashi, S.; Moriyama, T. Release reaction of brain-derived neurotrophic factor (BDNF) through PAR1 activation and its two distinct pools in human platelets. Thromb. Res. 2011, 128, e55–e61. [Google Scholar] [CrossRef]
- Mantilla, C.B.; Zhan, W.-Z.; Sieck, G.C. Neurotrophins improve neuromuscular transmission in the adult rat diaphragm. Muscle Nerve 2004, 29, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Garcia, N.; Tomas, M.; Santafe, M.M.; Besalduch, N.; Lanuza, M.A.; Tomas, J. The Interaction between Tropomyosin-Related Kinase B Receptors and Presynaptic Muscarinic Receptors Modulates Transmitter Release in Adult Rodent Motor Nerve Terminals. J. Neurosci. 2010, 30, 16514–16522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohof, A.M.; Ip, N.Y.; Poo, M. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature 1993, 363, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, L.M.; Poo, M.M. Presynaptic depolarization facilitates neurotrophin-induced synaptic potentiation. Nat. Neurosci. 1999, 2, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, J.N.; Benfenati, F.; Siow, Y.L.; Sihra, T.S.; Sanghera, J.S.; Pelech, S.L.; Greengard, P.; Czernik, A.J. Neurotrophins stimulate phosphorylation of synapsin I by MAP kinase and regulate synapsin I-actin interactions. Proc. Natl. Acad. Sci. USA 1996, 93, 3679–3683. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, J.N.; Czernik, A.J.; Fienberg, A.A.; Greengard, P.; Sihra, T.S. Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nat. Neurosci. 2000, 3, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Song, S.-H.; Augustine, G.J. Calcium-Dependent and Synapsin-Dependent Pathways for the Presynaptic Actions of BDNF. Front. Cell. Neurosci. 2017, 11, 75. [Google Scholar] [CrossRef] [Green Version]
- Sweatt, J.D. The neuronal MAP kinase cascade: A biochemical signal integration system subserving synaptic plasticity and memory. J. Neurochem. 2001, 76, 1–10. [Google Scholar] [CrossRef]
- Wortzel, I.; Seger, R. The ERK Cascade: Distinct Functions within Various Subcellular Organelles. Genes Cancer 2011, 2, 195–209. [Google Scholar] [CrossRef]
- Soliman, M.; Seo, J.-Y.; Kim, D.-S.; Kim, J.-Y.; Park, J.-G.; Alfajaro, M.M.; Baek, Y.-B.; Cho, E.-H.; Kwon, J.; Choi, J.-S.; et al. Activation of PI3K, Akt, and ERK during early rotavirus infection leads to V-ATPase-dependent endosomal acidification required for uncoating. PLoS Pathog. 2018, 14, e1006820. [Google Scholar] [CrossRef]
- Milosevic, I. Revisiting the Role of Clathrin-Mediated Endoytosis in Synaptic Vesicle Recycling. Front. Cell. Neurosci. 2018, 12, 27. [Google Scholar] [CrossRef] [PubMed]
- Gaydukov, A.E.; Balezina, O.P. Ryanodine- and CaMKII-dependent release of endogenous CGRP induces an increase in acetylcholine quantal size in neuromuscular junctions of mice. Brain Behav. 2018, 8, e01058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachmann, V.A.; Riml, A.; Huber, R.G.; Baillie, G.S.; Liedl, K.R.; Valovka, T.; Stefan, E. Reciprocal regulation of PKA and Rac signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 8531–8536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell. Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Amaral, M.D.; Pozzo-Miller, L. Intracellular Ca2+ Stores and Ca 2+ Influx Are Both Required for BDNF to Rapidly Increase Quantal Vesicular Transmitter Release. Neural Plast. 2012, 2012, 1–10. [Google Scholar] [CrossRef]
- Diógenes, M.J.; Fernandes, C.C.; Sebastião, A.M.; Ribeiro, J.A. Activation of adenosine A2A receptor facilitates brain-derived neurotrophic factor modulation of synaptic transmission in hippocampal slices. J. Neurosci. 2004, 24, 2905–2913. [Google Scholar] [CrossRef]
- Colino-Oliveira, M.; Rombo, D.M.; Dias, R.B.; Ribeiro, J.A.; Sebastião, A.M. BDNF-induced presynaptic facilitation of GABAergic transmission in the hippocampus of young adults is dependent of TrkB and adenosine A2A receptors. Purinergic Signal. 2016, 12, 283–294. [Google Scholar] [CrossRef]
- Shinoda, Y.; Ahmed, S.; Ramachandran, B.; Bharat, V.; Brockelt, D.; Altas, B.; Dean, C. BDNF enhances spontaneous and activity-dependent neurotransmitter release at excitatory terminals but not at inhibitory terminals in hippocampal neurons. Front. Synaptic Neurosci. 2014, 6, 27. [Google Scholar] [CrossRef]
- Festoff, B.W.; Suo, Z.; Citron, B.A. Plasticity and stabilization of neuromuscular and CNS synapses: Interactions between thrombin protease signaling pathways and tissue transglutaminase. Int. Rev. Cytol. 2001, 211, 153–177. [Google Scholar]
- Ben Shimon, M.; Lenz, M.; Ikenberg, B.; Becker, D.; Shavit Stein, E.; Chapman, J.; Tanne, D.; Pick, C.G.; Blatt, I.; Neufeld, M.; et al. Thrombin regulation of synaptic transmission and plasticity: Implications for health and disease. Front. Cell. Neurosci. 2015, 9, 151. [Google Scholar] [CrossRef]
- Isermann, B. Homeostatic effects of coagulation protease-dependent signaling and protease activated receptors. J. Thromb. Haemost. 2017, 15, 1273–1284. [Google Scholar] [CrossRef] [PubMed]
- Mbebi, C.; Rohn, T.; Doyennette, M.A.; Chevessier, F.; Jandrot-Perrus, M.; Hantaï, D.; Verdière-Sahuqué, M. Thrombin receptor induction by injury-related factors in human skeletal muscle cells. Exp. Cell Res. 2001, 263, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, M.H.; Nagai, A.; Terashima, M.; Sheikh, A.; Murakawa, Y.; Kobayashi, S.; Yamaguchi, S. Chemokine-like factor expression in the idiopathic inflammatory myopathies. Acta Neurol. Scand. 2008, 118, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Chang, Y.; Gao, X.; Li, H.; Zhao, P. Dynamic Expression and the Role of BDNF in Exercise-induced Skeletal Muscle Regeneration. Int. J. Sports Med. 2017, 38, 959–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]













© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaydukov, A.; Bogacheva, P.; Tarasova, E.; Molchanova, A.; Miteva, A.; Pravdivceva, E.; Balezina, O. Regulation of Acetylcholine Quantal Release by Coupled Thrombin/BDNF Signaling in Mouse Motor Synapses. Cells 2019, 8, 762. https://doi.org/10.3390/cells8070762
Gaydukov A, Bogacheva P, Tarasova E, Molchanova A, Miteva A, Pravdivceva E, Balezina O. Regulation of Acetylcholine Quantal Release by Coupled Thrombin/BDNF Signaling in Mouse Motor Synapses. Cells. 2019; 8(7):762. https://doi.org/10.3390/cells8070762
Chicago/Turabian StyleGaydukov, Alexander, Polina Bogacheva, Ekaterina Tarasova, Anastasia Molchanova, Anna Miteva, Ekaterina Pravdivceva, and Olga Balezina. 2019. "Regulation of Acetylcholine Quantal Release by Coupled Thrombin/BDNF Signaling in Mouse Motor Synapses" Cells 8, no. 7: 762. https://doi.org/10.3390/cells8070762
APA StyleGaydukov, A., Bogacheva, P., Tarasova, E., Molchanova, A., Miteva, A., Pravdivceva, E., & Balezina, O. (2019). Regulation of Acetylcholine Quantal Release by Coupled Thrombin/BDNF Signaling in Mouse Motor Synapses. Cells, 8(7), 762. https://doi.org/10.3390/cells8070762