Highlights in Resistance Mechanism Pathways for Combination Therapy


Abstract
:1. Introduction
2. Search Strategy
3. Resistance Mechanisms and Signaling Pathways
4. ABC Transporters
5. Enzymatic Detoxification
6. DNA Homeostasis-Related Signaling Pathways and Cytoskeletal Disruptors
7. Activation of NF-κB
8. Increased Levels of ALDH1
9. Signaling Pathways Associated with Targeted Therapies
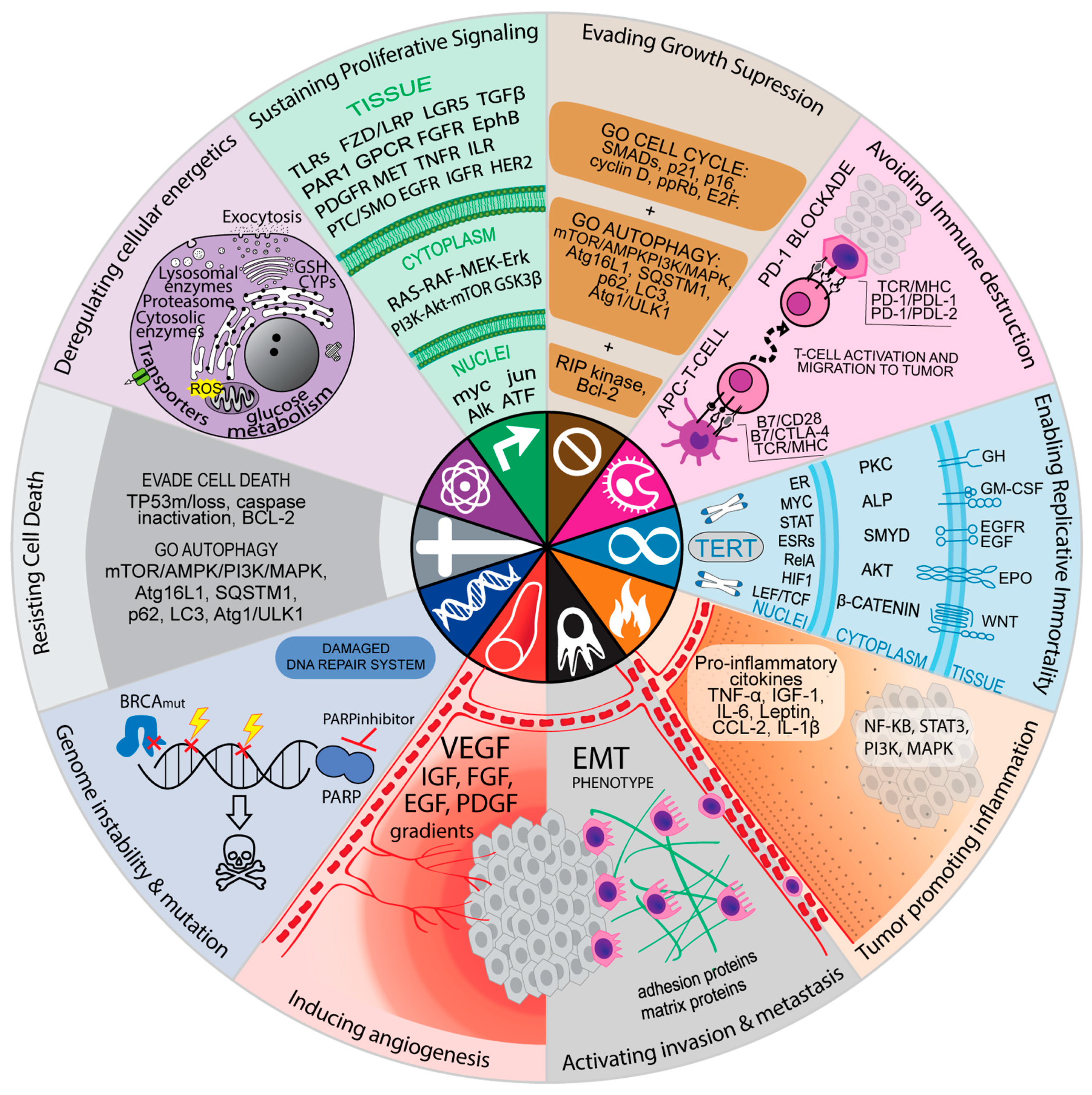
10. Resistance Mechanisms can be Associated with the Hallmarks of Cancer Cells
11. Signaling Pathways Related to the Hallmark Evasion of Growth Suppression
12. CDK Inhibitors
13. Signaling Pathways Related to Cell Death
14. Induction of Autophagy is a Common Cellular Phenomenon associated with Cell Death Resistance
15. Clinical Trials with Combination Regimens Containing Inhibitors of Signaling Pathways Related to Drug Resistance
16. Intrinsic Toxicity and Compensatory Mechanism of Inhibitors of Key Signaling Pathways of the Resistance Mechanisms
17. Synthetic Lethality
18. Drug Repurposing
19. Conclusion and Final Considerations
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gilman, A. The initial clinical trial of nitrogen mustard. Am. J. Surg. 1963, 105, 574–578. [Google Scholar] [CrossRef]
- DeVita, V.T.; Chu, E. A History of Cancer Chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2015, 8, 38022–38043. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Wu, W.; Wang, Y.; Alexander, P.B.; Sun, C.; Gong, Z.; Cheng, J.-N.; Sun, H.; Guan, Y.; Xia, X.; et al. Local mutational diversity drives intratumoral immune heterogeneity in non-small cell lung cancer. Nat. Commun. 2018, 9, 5361. [Google Scholar] [CrossRef]
- Bruce, W.R.; Meeker, B.E. Comparison of the sensitivity of hematopoietic colony-forming cells in different proliferative states to 5-fluorouracil. J. Natl. Cancer Inst. 1967, 38, 401–405. [Google Scholar]
- Hill, B.T.; Baserga, R. The cell cycle and its significance for cancer treatment. Cancer Treat. Rev. 1975, 2, 159–175. [Google Scholar] [CrossRef]
- Espinosa, E.; Zamora, P.; Feliu, J.; González Barón, M. Classification of anticancer drugs—A new system based on therapeutic targets. Cancer Treat. Rev. 2003, 29, 515–523. [Google Scholar] [CrossRef]
- Wu, X.-Z. A new classification system of anticancer drugs—Based on cell biological mechanisms. Med. Hypotheses 2006, 66, 883–887. [Google Scholar] [CrossRef]
- Dutt, R.; Madan, A.K. Classification Models for Anticancer Activity. Curr. Top. Med. Chem. 2013, 12, 2705–2726. [Google Scholar] [CrossRef]
- Pritchard, J.R.; Bruno, P.M.; Gilbert, L.A.; Capron, K.L.; Lauffenburger, D.A.; Hemann, M.T. Defining principles of combination drug mechanisms of action. Proc. Natl. Acad. Sci. 2013, 110, E170–E179. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Preissner, S.; Kroll, K.; Dunkel, M.; Senger, C.; Goldsobel, G.; Kuzman, D.; Guenther, S.; Winnenburg, R.; Schroeder, M.; Preissner, R. SuperCYP: a comprehensive database on Cytochrome P450 enzymes including a tool for analysis of CYP-drug interactions. Nucleic Acids Res. 2010, 38, D237–D243. [Google Scholar] [CrossRef] [PubMed]
- Mechanism matters. Nat. Med. 2010, 16, 347. [CrossRef] [PubMed]
- Juliano, R.L.L.; Ling, V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. Acta - Biomembr. 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: role of ATP–dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.; Stenke, L.; Varma, S.; Lindberg, M.L.; Björkholm, M.; Sjöberg, J.; Viktorsson, K.; Lewensohn, R.; Landgren, O.; Gottesman, M.M.; et al. Multidrug resistance in relapsed acute myeloid leukemia: Evidence of biological heterogeneity. Cancer 2013, 119, 3076–3083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartholomae, S.; Gruhn, B.; Debatin, K.-M.; Zimmermann, M.; Creutzig, U.; Reinhardt, D.; Steinbach, D. Coexpression of Multiple ABC-Transporters is Strongly Associated with Treatment Response in Childhood Acute Myeloid Leukemia. Pediatr. Blood Cancer 2016, 63, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Marzac, C.; Garrido, E.; Tang, R.; Fava, F.; Hirsch, P.; De Benedictis, C.; Corre, E.; Lapusan, S.; Lallemand, J.-Y.; Marie, J.-P.; et al. ATP Binding Cassette transporters associated with chemoresistance: transcriptional profiling in extreme cohorts and their prognostic impact in a cohort of 281 acute myeloid leukemia patients. Haematologica 2011, 96, 1293–1301. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.; Zhang, A.; Chen, M.; Liu, J. ABC Transporter Inhibitors in Reversing Multidrug Resistance to Chemotherapy. Curr. Drug Targets 2015, 16, 1356–1371. [Google Scholar] [CrossRef]
- Karthikeyan, S.; Hoti, S.L. Development of Fourth Generation ABC Inhibitors from Natural Products: A Novel Approach to Overcome Cancer Multidrug Resistance. Anticancer. Agents Med. Chem. 2015, 15, 605–615. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute Bleomycin Sulfate. Available online: https://www.cancer.gov/about-cancer/treatment/drugs/bleomycin (accessed on 15 June 2019).
- Zheng, W.; Johnston, S.A. The Nucleic Acid Binding Activity of Bleomycin Hydrolase Is Involved in Bleomycin Detoxification. Mol. Cell. Biol. 1998, 18, 3580–3585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazo, J.S.; Boland, C.J.; Schwartz, P.E. Bleomycin hydrolase activity and cytotoxicity in human tumors. Cancer Res. 1982, 42, 4026–4031. [Google Scholar] [PubMed]
- Lefterov, I.M.; Koldamova, R.P.; King, J.; Lazo, J.S. The C-terminus of human bleomycin hydrolase is required for protection against bleomycin-induced chromosomal damage. Mutat. Res. Mol. Mech. Mutagen. 1998, 421, 1–7. [Google Scholar] [CrossRef]
- Dortet, L.; Girlich, D.; Virlouvet, A.-L.; Poirel, L.; Nordmann, P.; Iorga, B.I.; Naas, T. Characterization of BRP MBL, the Bleomycin Resistance Protein Associated with the Carbapenemase NDM. Antimicrob. Agents Chemother. 2017, 61, e02413–e02416. [Google Scholar] [CrossRef]
- Ishikawa, T. ATP/Mg2+-dependent cardiac transport system for glutathione S-conjugates. A study using rat heart sarcolemma vesicles. J. Biol. Chem. 1989, 264, 17343–17348. [Google Scholar]
- Muller, M.; Meijer, C.; Zaman, G.J.; Borst, P.; Scheper, R.J.; Mulder, N.H.; de Vries, E.G.; Jansen, P.L. Overexpression of the gene encoding the multidrug resistance-associated protein results in increased ATP-dependent glutathione S-conjugate transport. Proc. Natl. Acad. Sci. 1994, 91, 13033–13037. [Google Scholar] [CrossRef]
- Brechbuhl, H.M.; Gould, N.; Kachadourian, R.; Riekhof, W.R.; Voelker, D.R.; Day, B.J. Glutathione Transport Is a Unique Function of the ATP-binding Cassette Protein ABCG2. J. Biol. Chem. 2010, 285, 16582–16587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.-L.; Li, B.-Y.; Yang, R.; Xia, L.-Y.; Fan, A.-L.; Chu, Y.-C.; Wang, L.-J.; Wang, Z.-C.; Jiang, A.-Q.; Zhu, H.-L. A class of novel tubulin polymerization inhibitors exert effective anti-tumor activity via mitotic catastrophe. Eur. J. Med. Chem. 2019, 163, 896–910. [Google Scholar] [CrossRef] [PubMed]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, C.H.A.; Wu, S.-Y.; Lee, T.-R.; Chang, C.-Y.; Wu, J.-S.; Hsieh, H.-P.; Chang, J.-Y. Cancer Cells Acquire Mitotic Drug Resistance Properties Through Beta I-Tubulin Mutations and Alterations in the Expression of Beta-Tubulin Isotypes. PLoS ONE 2010, 5, e12564. [Google Scholar] [CrossRef] [PubMed]
- Hari, M.; Wang, Y.; Veeraraghavan, S.; Cabral, F. Mutations in alpha- and beta-tubulin that stabilize microtubules and confer resistance to colcemid and vinblastine. Mol. Cancer Ther. 2003, 2, 597–605. [Google Scholar] [PubMed]
- Yin, S.; Bhattacharya, R.; Cabral, F. Human Mutations That Confer Paclitaxel Resistance. Mol. Cancer Ther. 2010, 9, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Orr, G.A.; Verdier-Pinard, P.; McDaid, H.; Horwitz, S.B. Mechanisms of Taxol resistance related to microtubules. Oncogene 2003, 22, 7280–7295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binaschi, M.; Zunino, F.; Capranico, G. Mechanism of action of DNA topoisomerase inhibitors. Stem Cells 1995, 13, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Hevener, K.; Verstak, T.A.; Lutat, K.E.; Riggsbee, D.L.; Mooney, J.W. Recent developments in topoisomerase-targeted cancer chemotherapy. Acta Pharm. Sin. B 2018, 8, 844–861. [Google Scholar] [CrossRef] [PubMed]
- Sabini, E.; Hazra, S.; Konrad, M.; Lavie, A. Elucidation of Different Binding Modes of Purine Nucleosides to Human Deoxycytidine Kinase. J. Med. Chem. 2008, 51, 4219–4225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathanson, D.A.; Armijo, A.L.; Tom, M.; Li, Z.; Dimitrova, E.; Austin, W.R.; Nomme, J.; Campbell, D.O.; Ta, L.; Le, T.M.; et al. Co-targeting of convergent nucleotide biosynthetic pathways for leukemia eradication. J. Exp. Med. 2014, 211, 473–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkaria, J.N.; Kitange, G.J.; James, C.D.; Plummer, R.; Calvert, H.; Weller, M.; Wick, W. Mechanisms of Chemoresistance to Alkylating Agents in Malignant Glioma. Clin. Cancer Res. 2008, 14, 2900–2908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Hientz, K.; Mohr, A.; Bhakta-Guha, D.; Efferth, T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2017, 8, 8921–8946. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Tan, S.; Zhou, Y.; Lin, J.; Wang, H.; Oyang, L.; Tian, Y.; Liu, L.; Su, M.; Wang, H.; et al. Role of the NF-kappa;B-signaling pathway in cancer. Onco. Targets. Ther. 2018, 11, 2063–2073. [Google Scholar] [CrossRef] [PubMed]
- Tanei, T.; Morimoto, K.; Shimazu, K.; Kim, S.J.; Tanji, Y.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Association of Breast Cancer Stem Cells Identified by Aldehyde Dehydrogenase 1 Expression with Resistance to Sequential Paclitaxel and Epirubicin-Based Chemotherapy for Breast Cancers. Clin. Cancer Res. 2009, 15, 4234–4241. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Lee, S.-H.; Hong, D.; Lee, J.-S.; Ahn, H.-S.; Ahn, J.-H.; Seong, T.W.; Lee, C.-H.; Jang, H.; Hong, K.M.; et al. Aldehyde dehydrogenase is used by cancer cells for energy metabolism. Exp. Mol. Med. 2016, 48, e272. [Google Scholar] [CrossRef]
- Koppaka, V.; Thompson, D.C.; Chen, Y.; Ellermann, M.; Nicolaou, K.C.; Juvonen, R.O.; Petersen, D.; Deitrich, R.A.; Hurley, T.D.; Vasiliou, V. Aldehyde Dehydrogenase Inhibitors: a Comprehensive Review of the Pharmacology, Mechanism of Action, Substrate Specificity, and Clinical Application. Pharmacol. Rev. 2012, 64, 520–539. [Google Scholar] [CrossRef] [Green Version]
- Venkatesan, S.; Swanton, C.; Taylor, B.S.; Costello, J.F. Treatment-Induced Mutagenesis and Selective Pressures Sculpt Cancer Evolution. Cold Spring Harb. Perspect. Med. 2017, 7, a026617. [Google Scholar] [CrossRef]
- Sever, R.; Brugge, J.S. Signal Transduction in Cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef]
- Katoh, M. Genomic testing, tumor microenvironment and targeted therapy of Hedgehog-related human cancers. Clin. Sci. 2019, 133, 953–970. [Google Scholar] [CrossRef] [PubMed]
- Rothenstein, J.M.; Chooback, N. ALK inhibitors, resistance development, clinical trials. Curr. Oncol. 2018, 25, 59. [Google Scholar] [CrossRef] [PubMed]
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov. 2016, 6, 1118–1133. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Dalin, S.; Hemann, M.T.; Lauffenburger, D.A.; Zhao, B. Differential selective pressure alters rate of drug resistance acquisition in heterogeneous tumor populations. Sci. Rep. 2016, 6, 36198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amirouchene-Angelozzi, N.; Swanton, C.; Bardelli, A. Tumor Evolution as a Therapeutic Target. Cancer Discov. 2017, 7, 805–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riviere, M.-K.; Le Tourneau, C.; Paoletti, X.; Dubois, F.; Zohar, S. Designs of drug-combination phase I trials in oncology: a systematic review of the literature. Ann. Oncol. 2015, 26, 669–674. [Google Scholar] [CrossRef]
- Day, D.; Siu, L.L. Approaches to modernize the combination drug development paradigm. Genome Med. 2016, 8, 115. [Google Scholar] [CrossRef]
- Mozgunov, P.; Jaki, T.; Paoletti, X. Randomized dose-escalation designs for drug combination cancer trials with immunotherapy. J. Biopharm. Stat. 2019, 29, 359–377. [Google Scholar] [CrossRef]
- Paller, C.J.; Bradbury, P.A.; Ivy, S.P.; Seymour, L.; LoRusso, P.M.; Baker, L.; Rubinstein, L.; Huang, E.; Collyar, D.; Groshen, S.; et al. Design of Phase I Combination Trials: Recommendations of the Clinical Trial Design Task Force of the NCI Investigational Drug Steering Committee. Clin. Cancer Res. 2014, 20, 4210–4217. [Google Scholar] [CrossRef]
- Milella, M.; Falcone, I.; Conciatori, F.; Cesta Incani, U.; Del Curatolo, A.; Inzerilli, N.; Nuzzo, C.M.A.; Vaccaro, V.; Vari, S.; Cognetti, F.; et al. PTEN: Multiple Functions in Human Malignant Tumors. Front. Oncol. 2015, 5. [Google Scholar] [CrossRef]
- Singh, S.S.; Vats, S.; Chia, A.Y.-Q.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G.; et al. Dual role of autophagy in hallmarks of cancer. Oncogene 2018, 37, 1142–1158. [Google Scholar] [CrossRef]
- Huang, T.; Song, X.; Yang, Y.; Wan, X.; Alvarez, A.A.; Sastry, N.; Feng, H.; Hu, B.; Cheng, S.-Y. Autophagy and Hallmarks of Cancer. Crit. Rev. Oncog. 2018, 23, 247–267. [Google Scholar] [CrossRef]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.-K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [Green Version]
- Brandmaier, A.; Hou, S.-Q.; Shen, W.H. Cell Cycle Control by PTEN. J. Mol. Biol. 2017, 429, 2265–2277. [Google Scholar] [CrossRef]
- Paramio, J.M.; Navarro, M.; Segrelles, C.; Gómez-Casero, E.; Jorcano, J.L. PTEN tumour suppressor is linked to the cell cycle control through the retinoblastoma protein. Oncogene 1999, 18, 7462–7468. [Google Scholar] [CrossRef] [Green Version]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017. [Google Scholar] [CrossRef]
- Dick, F.A.; Goodrich, D.W.; Sage, J.; Dyson, N.J. Non-canonical functions of the RB protein in cancer. Nat. Rev. Cancer 2018, 18, 442–451. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: a changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Sherr, C.J.; Beach, D.; Shapiro, G.I. Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov. 2016, 6, 353–367. [Google Scholar] [CrossRef]
- Quereda, V.; Porlan, E.; Cañamero, M.; Dubus, P.; Malumbres, M. An essential role for Ink4 and Cip/Kip cell-cycle inhibitors in preventing replicative stress. Cell Death Differ. 2016, 23, 430–441. [Google Scholar] [CrossRef]
- Sanidas, I.; Morris, R.; Fella, K.A.; Rumde, P.H.; Boukhali, M.; Tai, E.C.; Ting, D.T.; Lawrence, M.S.; Haas, W.; Dyson, N.J. A Code of Mono-phosphorylation Modulates the Function of RB. Mol. Cell 2019, 73, 985–1000.e6. [Google Scholar] [CrossRef] [Green Version]
- Rigberg, D.A.; Kim, F.S.; Sebastian, J.L.; Kazanjian, K.K.; McFadden, D.W. Hypophosphorylated Retinoblastoma Protein Is Associated with G2Arrest in Esophageal Squamous Cell Carcinoma. J. Surg. Res. 1999, 84, 101–105. [Google Scholar] [CrossRef]
- Roesch, A.; Becker, B.; Meyer, S.; Hafner, C.; Wild, P.J.; Landthaler, M.; Vogt, T. Overexpression and hyperphosphorylation of retinoblastoma protein in the progression of malignant melanoma. Mod. Pathol. 2005, 18, 565–572. [Google Scholar] [CrossRef]
- Senderowicz, A.M. Flavopiridol: the first cyclin-dependent kinase inhibitor in human clinical trials. Invest. New Drugs 1999, 17, 313–320. [Google Scholar] [CrossRef]
- Deep, A.; Marwaha, R.K.; Marwaha, M.G.; Jyoti, J.; Nandal, R.; Sharma, A.K. Flavopiridol as cyclin dependent kinase (CDK) inhibitor: a review. New J. Chem. 2018, 42, 18500–18507. [Google Scholar] [CrossRef]
- Byrd, J.C.; Shinn, C.; Waselenko, J.K.; Fuchs, E.J.; Lehman, T.A.; Nguyen, P.L.; Flinn, I.W.; Diehl, L.F.; Sausville, E.; Grever, M.R. Flavopiridol induces apoptosis in chronic lymphocytic leukemia cells via activation of caspase-3 without evidence of bcl-2 modulation or dependence on functional p53. Blood 1998, 92, 3804–3816. [Google Scholar]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [Green Version]
- Aklilu, M. Phase II study of flavopiridol in patients with advanced colorectal cancer. Ann. Oncol. 2003, 14, 1270–1273. [Google Scholar] [CrossRef]
- Burdette-Radoux, S.; Tozer, R.G.; Lohmann, R.C.; Quirt, I.; Ernst, D.S.; Walsh, W.; Wainman, N.; Colevas, A.D.; Eisenhauer, E.A. Phase II trial of flavopiridol, a cyclin dependent kinase inhibitor, in untreated metastatic malignant melanoma. Invest. New Drugs 2004, 22, 315–322. [Google Scholar] [CrossRef]
- Lin, T.S.; Howard, O.M.; Neuberg, D.S.; Kim, H.H.; Shipp, M.A. Seventy-Two Hour Continuous Infusion Flavopiridol in Relapsed and Refractory Mantle Cell Lymphoma. Leuk. Lymphoma 2002, 43, 793–797. [Google Scholar] [CrossRef]
- Kouroukis, C.T.; Belch, A.; Crump, M.; Eisenhauer, E.; Gascoyne, R.D.; Meyer, R.; Lohmann, R.; Lopez, P.; Powers, J.; Turner, R.; et al. Flavopiridol in Untreated or Relapsed Mantle-Cell Lymphoma: Results of a Phase II Study of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2003, 21, 1740–1745. [Google Scholar] [CrossRef]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef]
- Ibrance (palbociclib) FDA Approval History - Drugs.com. Available online: https://www.drugs.com/history/ibrance.html (accessed on 7 August 2019).
- Sobhani , N.; D’Angelo, A.; Pittacolo , M.; Roviello , G.; Miccoli , A.; Corona, S.P.; Bernocchi, O.; Generali, D.; Otto, T. Updates on the CDK4/6 Inhibitory Strategy and Combinations in Breast Cancer. Cells 2019, 8, 321. [Google Scholar] [CrossRef]
- Shah, M.; Nunes, M.R.; Stearns, V. CDK4/6 Inhibitors: Game Changers in the Management of Hormone Receptor–Positive Advanced Breast Cancer? Oncology (Williston Park). 2018, 32, 216–222. [Google Scholar] [PubMed]
- Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N. Engl. J. Med. 2018, 379, 2582. [CrossRef]
- Goetz, M.P.; Toi, M.; Campone, M.; Sohn, J.; Paluch-Shimon, S.; Huober, J.; Park, I.H.; Trédan, O.; Chen, S.-C.; Manso, L.; et al. MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. J. Clin. Oncol. 2017, 35, 3638–3646. [Google Scholar] [CrossRef]
- Portman, N.; Alexandrou, S.; Carson, E.; Wang, S.; Lim, E.; Caldon, C.E. Overcoming CDK4/6 inhibitor resistance in ER-positive breast cancer. Endocr. Relat. Cancer 2019, R15–R30. [Google Scholar] [CrossRef]
- Jabbour-Leung, N.A.; Chen, X.; Bui, T.; Jiang, Y.; Yang, D.; Vijayaraghavan, S.; McArthur, M.J.; Hunt, K.K.; Keyomarsi, K. Sequential Combination Therapy of CDK Inhibition and Doxorubicin Is Synthetically Lethal in p53-Mutant Triple-Negative Breast Cancer. Mol. Cancer Ther. 2016, 15, 593–607. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.M.; Cory, S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2018, 25, 27–36. [Google Scholar] [CrossRef]
- Stegh, A.H. Targeting the p53 signaling pathway in cancer therapy—The promises, challenges and perils. Expert Opin. Ther. Targets 2012, 16, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Vessoni, A.T.; Filippi-Chiela, E.C.; Menck, C.F.; Lenz, G. Autophagy and genomic integrity. Cell Death Differ. 2013, 20, 1444–1454. [Google Scholar] [CrossRef]
- Hewitt, G.; Korolchuk, V.I. Repair, Reuse, Recycle: The Expanding Role of Autophagy in Genome Maintenance. Trends Cell Biol. 2017, 27, 340–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroemer, G.; Levine, B. Autophagic cell death: the story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef]
- Kondapuram, S.K.; Sarvagalla, S.; Coumar, M.S. Targeting autophagy with small molecules for cancer therapy. J. Cancer Metastasis Treat. 2019, 2019. [Google Scholar] [CrossRef]
- Delou, J.M.A.; Biasoli, D.; Borges, H.L. The Complex Link between Apoptosis and Autophagy: a Promising New Role for RB. An. Acad. Bras. Cienc. 2016, 88, 2257–2275. [Google Scholar] [CrossRef] [Green Version]
- Morselli, E.; Galluzzi, L.; Kepp, O.; Vicencio, J.-M.; Criollo, A.; Maiuri, M.C.; Kroemer, G. Anti- and pro-tumor functions of autophagy. Biochim. Biophys. Acta - Mol. Cell Res. 2009, 1793, 1524–1532. [Google Scholar] [CrossRef] [Green Version]
- Shang, L.; Wang, X. AMPK and mTOR coordinate the regulation of Ulk1 and mammalian autophagy initiation. Autophagy 2011, 7, 924–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, L.; Shi, Z.; Zhao, S.; Wang, F.-T.; Zhou, T.-T.; Liu, B.; Bao, J.-K. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012, 45, 487–498. [Google Scholar] [CrossRef]
- Biasoli, D.; Kahn, S.A.; Cornélio, T.A.; Furtado, M.; Campanati, L.; Chneiweiss, H.; Moura-Neto, V.; Borges, H.L. Retinoblastoma protein regulates the crosstalk between autophagy and apoptosis, and favors glioblastoma resistance to etoposide. Cell Death Dis. 2013, 4, e767. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Soletti, R.C.; Sadarangani, A.; Sridevi, P.; Ramirez, M.E.; Eckmann, L.; Borges, H.L.; Wang, J.Y.J. Nuclear Expression of β-Catenin Promotes RB Stability and Resistance to TNF-Induced Apoptosis in Colon Cancer Cells. Mol. Cancer Res. 2013, 11, 207–218. [Google Scholar] [CrossRef] [PubMed]
- LEE011 Plus Everolimus in Patients With Metastatic Pancreatic Adenocarcinoma Refractory to Chemotherapy - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02985125?term=NCT02985125&rank=1 (accessed on 14 July 2019).
- Michaloglou, C.; Crafter, C.; Siersbaek, R.; Delpuech, O.; Curwen, J.O.; Carnevalli, L.S.; Staniszewska, A.D.; Polanska, U.M.; Cheraghchi-Bashi, A.; Lawson, M.; et al. Combined Inhibition of mTOR and CDK4/6 Is Required for Optimal Blockade of E2F Function and Long-term Growth Inhibition in Estrogen Receptor–positive Breast Cancer. Mol. Cancer Ther. 2018, 17, 908–920. [Google Scholar] [CrossRef] [PubMed]
- Berrak, O.; Arisan, E.D.; Obakan-Yerlikaya, P.; Coker-Gürkan, A.; Palavan-Unsal, N. mTOR is a fine tuning molecule in CDK inhibitors-induced distinct cell death mechanisms via PI3K/AKT/mTOR signaling axis in prostate cancer cells. Apoptosis 2016, 21, 1158–1178. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Nowsheen, S.; Aziz, K.; Georgakilas, A.G. Toxicity and adverse effects of Tamoxifen and other anti-estrogen drugs. Pharmacol. Ther. 2013, 139, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Bosutinib in Combination With Pemetrexed in Patients With Selected Metastatic Solid Tumors - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03023319?term=bosutinib&draw=2&rank=2 (accessed on 15 July 2019).
- Secord, A.A.; Teoh, D.K.; Barry, W.T.; Yu, M.; Broadwater, G.; Havrilesky, L.J.; Lee, P.S.; Berchuck, A.; Lancaster, J.; Wenham, R.M. A Phase I Trial of Dasatinib, an Src-Family Kinase Inhibitor, in Combination with Paclitaxel and Carboplatin in Patients with Advanced or Recurrent Ovarian Cancer. Clin. Cancer Res. 2012, 18, 5489–5498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azacitidine and Venetoclax as Induction Therapy With Venetoclax Maintenance in the Elderly With AML. Available online: https://clinicaltrials.gov/show/NCT03466294 (accessed on 15 June 2019).
- Venetoclax and Chemotherapy as Frontline Therapy in Older Patients and Patients With Relapsed/Refractory ALL. Available online: https://clinicaltrials.gov/show/NCT03319901 (accessed on 15 June 2019).
- A Study of Abemaciclib (LY2835219) in Combination With Another Anti-cancer Drug in Participants With Lung Cancer (NSCLC). Available online: https://clinicaltrials.gov/show/NCT02079636 (accessed on 15 June 2019).
- INdividualized Screening Trial of Innovative Glioblastoma Therapy (INSIGhT). Available online: https://clinicaltrials.gov/show/NCT02977780 (accessed on 15 June 2019).
- Phase II Trial Evaluating the Efficacy of Palbociclib in Combination With Carboplatin for the Treatment of Unresectable Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma. Available online: https://clinicaltrials.gov/show/NCT03194373 (accessed on 15 June 2019).
- Dose-Escalation Study Of Palbociclib + Nab-Paclitaxel In mPDAC. Available online: https://clinicaltrials.gov/show/NCT02501902 (accessed on 15 June 2019).
- Study Of Palbociclib Combined With Chemotherapy In Pediatric Patients With Recurrent/Refractory Solid Tumors. Available online: https://clinicaltrials.gov/show/NCT03709680 (accessed on 15 June 2019).
- LEE011 (Ribociclib) in Combination With Docetaxel Plus Prednisone in mCRPC. Available online: https://clinicaltrials.gov/show/NCT02494921 (accessed on 15 June 2019).
- SJDAWN: St. Jude Children’s Research Hospital Phase 1 Study Evaluating Molecularly-Driven Doublet Therapies for Children and Young Adults With Recurrent Brain Tumors. Available online: https://clinicaltrials.gov/show/NCT03434262 (accessed on 15 June 2019).
- Ribociclib (Ribociclib (LEE-011)) With Platinum-based Chemotherapy in Recurrent Platinum Sensitive Ovarian Cancer. Available online: https://clinicaltrials.gov/show/NCT03056833 (accessed on 15 June 2019).
- Niraparib and Temozolomide in Treating Patients With Extensive-Stage Small Cell Lung Cancer With a Complete or Partial Response to Platinum-Based First-Line Chemotherapy. Available online: https://clinicaltrials.gov/show/NCT03830918 (accessed on 15 June 2019).
- Olaparib Dose Escalating Trial + Concurrent RT With or Without Cisplatin in Locally Advanced NSCLC. Available online: https://clinicaltrials.gov/show/NCT01562210 (accessed on 15 June 2019).
- Rucaparib and Irinotecan in Cancers With Mutations in DNA Repair. Available online: https://clinicaltrials.gov/show/NCT03318445 (accessed on 15 June 2019).
- Berlin, J.; Ramanathan, R.K.; Strickler, J.H.; Subramaniam, D.S.; Marshall, J.; Kang, Y.-K.; Hetman, R.; Dudley, M.W.; Zeng, J.; Nickner, C.; et al. A phase 1 dose-escalation study of veliparib with bimonthly FOLFIRI in patients with advanced solid tumours. Br. J. Cancer 2018, 118, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Pommier, Y. Classification of PARP Inhibitors Based on PARP Trapping and Catalytic Inhibition, and Rationale for Combinations with Topoisomerase I Inhibitors and Alkylating Agents. In PARP Inhibitors for Cancer Therapy; Springer: Berlin, Germany, 2015; pp. 261–274. [Google Scholar]
- Veliparib and Temozolomide in Treating Patients With Acute Leukemia - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01139970 (accessed on 1 August 2019).
- Pishvaian, M.J.; Slack, R.S.; Jiang, W.; He, A.R.; Hwang, J.J.; Hankin, A.; Dorsch-Vogel, K.; Kukadiya, D.; Weiner, L.M.; Marshall, J.L.; et al. A phase 2 study of the PARP inhibitor veliparib plus temozolomide in patients with heavily pretreated metastatic colorectal cancer. Cancer 2018, 124, 2337–2346. [Google Scholar] [CrossRef] [PubMed]
- Veliparib and Topotecan With or Without Carboplatin in Treating Patients With Relapsed or Refractory Acute Leukemia, High-Risk Myelodysplasia, or Aggressive Myeloproliferative Disorders - Full Text View - ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT00588991?term=veliparib%2C+topotecan&rank=2 (accessed on 14 July 2019).
- Study of Folfiri/Cetuximab in FcGammaRIIIa V/V Stage IV Colorectal Cancer Patients - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03874026?term=cetuximab&draw=2&rank=15 (accessed on 14 July 2019).
- Neoadjuvant Chemotherapy Combined With Cetuximab for EGFR Wild Type Locally Advanced Rectal Cancer - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03391843?term=cetuximab&draw=2&rank=4 (accessed on 14 July 2019).
- Necitumumab in the Neoadjuvant Setting With Gemcitabine in Surgically Resectable - 14X-US-I001 - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03574818?term=necitumumab&draw=2&rank=1 (accessed on 14 July 2019).
- A Study of Perioperative Chemotherapy Plus Panitumumab in Patients With Colorectal Cancer Liver Metastases - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01260415?term=panitumumab&draw=3 (accessed on 14 July 2019).
- Yoshino, T.; Uetake, H.; Tsuchihara, K.; Shitara, K.; Yamazaki, K.; Oki, E.; Sato, T.; Naitoh, T.; Komatsu, Y.; Kato, T.; et al. Rationale for and Design of the PARADIGM Study: Randomized Phase III Study of mFOLFOX6 Plus Bevacizumab or Panitumumab in Chemotherapy-naïve Patients With RAS ( KRAS/NRAS ) Wild-type, Metastatic Colorectal Cancer. Clin. Colorectal Cancer 2017, 16, 158–163. [Google Scholar] [CrossRef] [PubMed]
- DAPHNe: Paclitaxel/Trastuzumab/Pertuzumab in HER2-Positive BC - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03716180?term=pertuzumab&rank=7 (accessed on 14 July 2019).
- Copanlisib (BAY 80-6946) in Combination With Gemcitabine and Cisplatin in Advanced Cholangiocarcinoma - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02631590?term=copanlisib&draw=5&rank=32 (accessed on 14 July 2019).
- A Phase 1b/2 Study of IPI-145 Plus FCR in Previously Untreated, Younger Patients With CLL - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02158091?term=duvelisib&draw=4&rank=28 (accessed on 14 July 2019).
- Hauke, R.J.; Infante, J.R.; Rubin, M.S.; Shih, K.C.; Arrowsmith, E.R.; Hainsworth, J.D. Everolimus in combination with paclitaxel and carboplatin in patients with metastatic melanoma: a phase II trial of the Sarah Cannon Research Institute Oncology Research Consortium. Melanoma Res. 2013, 23, 468–473. [Google Scholar] [CrossRef]
- Everolimus With and Without Temozolomide in Adult Low Grade Glioma - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02023905?term=temozolomide%2C+pi3k&rank=1 (accessed on 14 July 2019).
- Zelenetz, A.D.; Barrientos, J.C.; Brown, J.R.; Coiffier, B.; Delgado, J.; Egyed, M.; Ghia, P.; Illés, Á.; Jurczak, W.; Marlton, P.; et al. Idelalisib or placebo in combination with bendamustine and rituximab in patients with relapsed or refractory chronic lymphocytic leukaemia: interim results from a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2017, 18, 297–311. [Google Scholar] [CrossRef] [Green Version]
- Dunn, L.A.; Fury, M.G.; Xiao, H.; Baxi, S.S.; Sherman, E.J.; Korte, S.; Pfister, C.; Haque, S.; Katabi, N.; Ho, A.L.; et al. A phase II study of temsirolimus added to low-dose weekly carboplatin and paclitaxel for patients with recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC). Ann. Oncol. 2017, 28, 2533–2538. [Google Scholar] [CrossRef] [PubMed]
- A Study of Avastin (Bevacizumab) and Xeloda (Capecitabine) in Patients With Advanced or Metastatic Liver Cancer - Full Text View - ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT02013830?term=02013830&rank=1 (accessed on 14 July 2019).
- McWilliams, R.R.; Allred, J.B.; Slostad, J.A.; Katipamula, R.; Dronca, R.S.; Rumilla, K.M.; Erickson, L.A.; Bryce, A.H.; Joseph, R.W.; Kottschade, L.A.; et al. NCCTG N0879 (Alliance): A randomized phase 2 cooperative group trial of carboplatin, paclitaxel, and bevacizumab ± everolimus for metastatic melanoma. Cancer 2018, 124, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Onvansertib in Combination With FOLFIRI and Bevacizumab for Second Line Treatment of Metastatic Colorectal Cancer Patients With a Kras Mutation - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03829410?term=bevacizumab%2C+FOLFIRI&rank=3 (accessed on 14 July 2019).
- Van den Bent, M.J.; Klein, M.; Smits, M.; Reijneveld, J.C.; French, P.J.; Clement, P.; de Vos, F.Y.F.; Wick, A.; Mulholland, P.J.; Taphoorn, M.J.B.; et al. Bevacizumab and temozolomide in patients with first recurrence of WHO grade II and III glioma, without 1p/19q co-deletion (TAVAREC): A randomised controlled phase 2 EORTC trial. Lancet Oncol. 2018, 19, 1170–1179. [Google Scholar] [CrossRef]
- Lenvatinib and Weekly Paclitaxel for Patients With Recurrent Endometrial or Ovarian Cancer - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02788708?term=lenvatinib&rank=10 (accessed on 14 July 2019).
- Ramucirumab Plus FOLFIRI Versus Ramucirumab Plus Paclitaxel in Patients With Advanced or Metastatic Gastric Cancer, Who Failed One Prior Line of Palliative Chemotherapy - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03081143?term=ramucirumab%2C+folfiri&rank=2 (accessed on 14 July 2019).
- Regorafenib Combined With Irinotecan as Second-line in Patients With Metastatic Gastro-oesophageal Adenocarcinomas - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03722108?term=regorafenib&draw=2&rank=6 (accessed on 14 July 2019).
- Treating Relapsed/Recurrent/Refractory Pediatric Solid Tumors With Sorafenib in Combination With Irinotecan - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02747537?term=irinotecan%2C+vegfr&rank=3 (accessed on 14 July 2019).
- Santoro, A.; Su, W.-C.; Navarro, A.; Simonelli, M.; Yang, J.C.-H.; Ardizzoni, A.; Barlesi, F.; Kang, J.H.; Didominick, S.; Abdelhady, A.; et al. 1392PDose-determination results from a phase Ib/II study of ceritinib (CER) + ribociclib (RIB) in ALK-positive (ALK+) non-small cell lung cancer (NSCLC). Ann. Oncol. 2018, 29. [Google Scholar] [CrossRef]
- Wood, A.C.; Krytska, K.; Ryles, H.T.; Infarinato, N.R.; Sano, R.; Hansel, T.D.; Hart, L.S.; King, F.J.; Smith, T.R.; Ainscow, E.; et al. Dual ALK and CDK4/6 Inhibition Demonstrates Synergy against Neuroblastoma. Clin. Cancer Res. 2017, 23, 2856–2868. [Google Scholar] [CrossRef] [PubMed]
- Next Generation Personalized Neuroblastoma Therapy (NEPENTHE). Available online: https://clinicaltrials.gov/ct2/show/NCT02780128?term=ceritinib%2C+ribociclib&rank=2 (accessed on 14 July 2019).
- Crizotinib in Combination With Enzalutamide in Metastatic Castration-resistant Prostate Cancer - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02207504?term=crizotinib&recrs=de&rank=4 (accessed on 14 July 2019).
- Tripathi, A.; Supko, J.G.; Gray, K.P.; Melnick, Z.; Taplin, M.-E.; Choudhury, A.D.; Pomerantz, M.; Bellmunt, J.; Yu, C.; Sun, Z.; et al. 852PPharmacokinetic (PK) analysis of concurrent administration of enzalutamide (enza) and crizotinib (crizo) in patients with metastatic castration resistant prostate cancer (CRPC). Ann. Oncol. 2018, 29. [Google Scholar] [CrossRef]
- Kato, S.; Jardim, D.L.; Johnson, F.M.; Subbiah, V.; Piha-Paul, S.; Tsimberidou, A.M.; Falchook, G.S.; Karp, D.; Zinner, R.; Wheler, J.; et al. Phase I study of the combination of crizotinib (as a MET inhibitor) and dasatinib (as a c-SRC inhibitor) in patients with advanced cancer. Invest. New Drugs 2018, 36, 416–423. [Google Scholar] [CrossRef]
- Dasatinib and Crizotinib in Advanced Cancer - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01744652?term=crizotinib&recrs=de&rank=6 (accessed on 14 July 2019).
- Study of the Combination of Crizotinib and Dasatinib in Pediatric Research Participants With Diffuse Pontine Glioma (DIPG) and High-Grade Glioma (HGG) - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01644773?term=crizotinib&recrs=de&rank=22 (accessed on 14 July 2019).
- Michmerhuizen, N.L.; Leonard, E.; Matovina, C.; Harris, M.; Herbst, G.; Kulkarni, A.; Zhai, J.; Jiang, H.; Carey, T.E.; Brenner, J.C. Rationale for Using Irreversible Epidermal Growth Factor Receptor Inhibitors in Combination with Phosphatidylinositol 3-Kinase Inhibitors for Advanced Head and Neck Squamous Cell Carcinoma. Mol. Pharmacol. 2019, 95, 528–536. [Google Scholar] [CrossRef] [Green Version]
- Copanlisib in Association With Cetuximab in Patients With Recurrent and/or Metastatic Head and Neck Squamous Cell Carcinomas Harboring a PI3KCA Mutation/Amplification and/or a PTEN Loss - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02822482?term=copanlisib&rank=2 (accessed on 14 July 2019).
- Pazopanib and Everolimus in PI3KCA Mutation Positive/PTEN Loss Patients - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01430572 (accessed on 14 July 2019).
- Barr, P.M.; Saylors, G.B.; Spurgeon, S.E.; Cheson, B.D.; Greenwald, D.R.; OBrien, S.M.; Liem, A.K.D.; Mclntyre, R.E.; Joshi, A.; Abella-Dominicis, E.; et al. Phase 2 study of idelalisib and entospletinib: pneumonitis limits combination therapy in relapsed refractory CLL and NHL. Blood 2016, 127, 2411–2415. [Google Scholar] [CrossRef] [Green Version]
- A Study of Abemaciclib (LY2835219) Alone or in Combination With Other Agents in Participants With Previously Treated Pancreatic Ductal Adenocarcinoma - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02981342?term=NCT02981342&rank=1 (accessed on 14 July 2019).
- Keam, B.; Tahara, M.; Lin, J.-C.; Sacco, A.G.; Melichar, B.; Baney, T.; Hoffman, J.; Wang, D.D.; Wang, S.L.; Martini, J.-F.; et al. An international, multicenter, randomized, double-blind, placebo-controlled, parallel-group phase 2 study of palbociclib (an oral CDK4/6 inhibitor) plus cetuximab in patients with recurrent/metastatic (R/M) squamous cell carcinoma of the head and neck (SCCHN). J. Clin. Oncol. 2016, 34, TPS6102. [Google Scholar]
- Sullivan, R.J.; Amaria, R.N.; Lawrence, D.P.; Brennan, J.; Leister, C.; Singh, R.; Legos, J.; Thurm, H.; Yan, L.; Flaherty, K.T.; et al. Abstract PR06: Phase 1b dose-escalation study of trametinib (MEKi) plus palbociclib (CDK4/6i) in patients with advanced solid tumors. In Proceedings of the MAPK Pathways; American Association for Cancer Research: Philadelphia, PA, USA, 2015; p. PR06. [Google Scholar]
- A Study to Investigate the Safety, Pharmacokinetics, Pharmacodynamics, and Anti-Cancer Activity of Trametinib in Combination With Palbociclib in Subjects With Solid Tumors - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02065063 (accessed on 15 July 2019).
- Berghoff, A.S.; Preusser, M. Targeted Therapies for Melanoma Brain Metastases. Curr. Treat. Options Neurol. 2017, 19, 13. [Google Scholar] [CrossRef] [PubMed]
- Vemurafenib and Cobimetinib Combination in BRAF Mutated Melanoma With Brain Metastasis - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02537600?term=vemurafenib&rank=7 (accessed on 14 July 2019).
- A Study of Tarceva (Erlotinib) and Avastin (Bevacizumab) in Patients With Advanced or Metastatic Liver Cancer. - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00605722?term=00605722&rank=1 (accessed on 14 July 2019).
- Nissan, M.H.; Rosen, N.; Solit, D.B. ERK Pathway Inhibitors: How Low Should We Go? Cancer Discov. 2013, 3, 719–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- Pelster, M.S.; Amaria, R.N. Combined targeted therapy and immunotherapy in melanoma: a review of the impact on the tumor microenvironment and outcomes of early clinical trials. Ther. Adv. Med. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, B.S.; Durinck, S.; Stawiski, E.W.; Yin, J.; Wang, W.; Lin, E.; Moffat, J.; Martin, S.E.; Modrusan, Z.; Seshagiri, S. ERK Mutations and amplification confer resistance to ERK-inhibitor therapy. Clin. Cancer Res. 2018, 24, 4044–4055. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Infante, J.R.; Janku, F.; Lee Wong, D.J.; Sosman, J.A.; Keedy, V.; Patel, M.R.; Shapiro, G.I.; Mier, J.W.; Tolcher, A.W.; et al. First-in-class ERK1/2 inhibitor ulixertinib (BVD-523) in patients with MAPK mutant advanced solid tumors: Results of a phase I dose-escalation and expansion study. Cancer Discov. 2018, 8, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Hanker, A.B.; Kaklamani, V.; Arteaga, C.L. Challenges for the clinical development of PI3K inhibitors: Strategies to improve their impact in solid tumors. Cancer Discov. 2019, 9. [Google Scholar] [CrossRef]
- Curigliano, G.; Shah, R.R. Safety and Tolerability of Phosphatidylinositol-3-Kinase (PI3K) Inhibitors in Oncology. Drug Saf. 2019, 42, 247–262. [Google Scholar] [CrossRef]
- Song, M.; Bode, A.M.; Dong, Z.; Lee, M.H. AKt as a therapeutic target for cancer. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Sampath, D.; Malik, A.; Plunkett, W.; Nowak, B.; Williams, B.; Burton, M.; Verstovsek, S.; Faderl, S.; Garcia-Manero, G.; List, A.F.; et al. Phase I clinical, pharmacokinetic, and pharmacodynamic study of the Akt-inhibitor triciribine phosphate monohydrate in patients with advanced hematologic malignancies. Leuk. Res. 2013, 37, 1461–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunyoto, T.; Potet, J.; Boelaert, M. Why miltefosine - A life-saving drug for leishmaniasis-is unavailable to people who need it the most. BMJ Glob. Heal. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ghidini, M.; Petrelli, F.; Ghidini, A.; Tomasello, G.; Hahne, J.C.; Passalacqua, R.; Barni, S. Clinical development of mTor inhibitors for renal cancer. Expert Opin. Investig. Drugs 2017, 26, 1229–1237. [Google Scholar] [CrossRef] [PubMed]
- Teng, Q.-X.; Ashar, Y.V.; Gupta, P.; Gadee, E.; Fan, Y.-F.; Reznik, S.E.; Wurpel, J.N.D.; Chen, Z.-S. Revisiting mTOR inhibitors as anticancer agents. Drug Discov. Today 2019. [Google Scholar] [CrossRef]
- Prasad, V.; De Jesús, K.; Mailankody, S. The high price of anticancer drugs: origins, implications, barriers, solutions. Nat. Rev. Clin. Oncol. 2017, 14, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Turk, A.A.; Wisinski, K.B. PARP inhibitors in breast cancer: Bringing synthetic lethality to the bedside. Cancer 2018, 124, 2498–2506. [Google Scholar] [CrossRef]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Heinzel, A.; Marhold, M.; Mayer, P.; Schwarz, M.; Tomasich, E.; Lukas, A.; Krainer, M.; Perco, P. Synthetic lethality guiding selection of drug combinations in ovarian cancer. PLoS ONE 2019, 14, e0210859. [Google Scholar] [CrossRef]
- Gupta, S.C.; Sung, B.; Prasad, S.; Webb, L.J.; Aggarwal, B.B. Cancer drug discovery by repurposing: Teaching new tricks to old dogs. Trends Pharmacol. Sci. 2013. [Google Scholar] [CrossRef]
- Bertolini, F.; Sukhatme, V.P.; Bouche, G. Drug repurposing in oncology-patient and health systems opportunities. Nat. Rev. Clin. Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shen, C.; Liu, Z.; Peng, F.; Chen, X.; Yang, G.; Zhang, D.; Yin, Z.; Ma, J.; Zheng, Z.; et al. Nitazoxanide, an antiprotozoal drug, inhibits late-stage autophagy and promotes ING1-induced cell cycle arrest in glioblastoma. Cell Death Dis. 2018, 9, 1032. [Google Scholar] [CrossRef] [PubMed]
- Hendouei, N.; Saghafi, F.; Shadfar, F.; Hosseinimehr, S.J. Molecular mechanisms of anti-psychotic drugs for improvement of cancer treatment. Eur. J. Pharmacol. 2019, 856, 172402. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-Y.; Lee, W.-T.; Cheng, C.-H.; Chen, K.-C.; Chou, C.-M.; Chung, C.-H.; Sun, M.-S.; Cheng, H.-W.; Ho, M.-N.; Lin, C.-W. Repositioning antipsychotic chlorpromazine for treating colorectal cancer by inhibiting sirtuin 1. Oncotarget 2015, 6, 27580–27595. [Google Scholar] [CrossRef] [PubMed]
- Soletti, R.C.; Biasoli, D.; Rodrigues, N.A.L.V.; Delou, J.M.A.; Maciel, R.; Chagas, V.L.A.; Martins, R.A.P.; Rehen, S.K.; Borges, H.L. Inhibition of pRB Pathway Differentially Modulates Apoptosis in Esophageal Cancer Cells. Transl. Oncol. 2017, 10, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Laurent, M.; Brahmi, M.; Dufresne, A.; Meeus, P.; Karanian, M.; Ray-Coquard, I.; Blay, J.-Y. Adjuvant therapy with imatinib in gastrointestinal stromal tumors (GISTs)—review and perspectives. Transl. Gastroenterol. Hepatol. 2019, 4, 24. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Patel, C.J. A standard database for drug repositioning. Sci. Data 2017, 4, 170029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aberle, M.R.; Burkhart, R.A.; Tiriac, H.; Olde Damink, S.W.M.; Dejong, C.H.C.; Tuveson, D.A.; van Dam, R.M. Patient-derived organoid models help define personalized management of gastrointestinal cancer. Br. J. Surg. 2018, 105, e48–e60. [Google Scholar] [CrossRef]


{kind=link}
| Top | Cytotoxic Drugs (N = 59) | Drugs Affected | % | Targeted Drugs (N = 117) | Drugs Affected | % |
|---|---|---|---|---|---|---|
| 1 | ABC transporters | 21 | 36 | MAPK family | 34 | 29 |
| 2 | Enzymatic detoxification | 9 | 17 | PI3K-AKT-mTOR | 33 | 28 |
| 3 | Mutation in and/or downregulation of topoisomerases I/II | 7 | 12 | EGF and EGFR | 21 | 18 |
| 4 | Mutation in and/or overexpression of tubulins | 6 | 10 | PTEN | 14 | 12 |
| 5 | Decreased dCK | 6 | 8 | ABC transporters | 14 | 12 |
| 6 | Increased activity of GST | 5 | 8 | IGFs | 14 | 12 |
| 7 | Activation of NF-κB | 4 | 7 | JAK/STAT | 14 | 12 |
| 8 | Increased MGMT | 4 | 7 | BCL-2 family | 13 | 12 |
| 9 | Increased levels of ALDH1 | 3 | 5 | FGFs | 12 | 11 |
| 10 | Silencing or mutations in TP53 | 3 | 5 | ERBB2 (HER2) | 12 | 11 |
| Top | Cytotoxic (N = 59) | Drugs Affected | % | Targeted therapies (N = 117) | Drugs Affected | % |
|---|---|---|---|---|---|---|
| 1 | Metabolism (Detoxification transporters and enzymes, protection from ROS, increased glucose metabolism) | 21 | 36 | Sustaining Proliferative Signaling (GF, Hedgehog, MAPK, PI3K, WNT, autophagy induction) | 65 | 56 |
| 2 | Sustaining Proliferative Signaling (GF, Hedgehog, MAPK, PI3K, WNT, autophagy induction) | 19 | 32 | Cell Death Evasion (BCL2 family, TP53, MDM2, PTEN, NF-KB, autophagy induction) | 41 | 35 |
| 3 | Cell Death Evasion (BCL2 family, TP53, MDM2, PTEN, NF-KB, autophagy induction) | 10 | 17 | Angiogenesis (EGF, IGF, FGF, VEGF, PDGF) | 31 | 26 |
| 4 | Genome instability (TP53, MDM2, NHEJ, RAD51, CHEK1/2, BRCA1/2, HDAC) | 5 | 8 | Metabolism (Detoxification transporters and enzymes, protection from ROS, increased glucose metabolism) | 21 | 18 |
| 5 | Evading growth suppressors (TP53, RB, cyclins, CDKs, p16, p18, p21) | 4 | 7 | Genome instability (TP53, MDM2, NHEJ, RAD51, CHEK1/2, BRCA1/2, HDAC, p21) | 18 | 15 |
| 6 | Inflammation (NF-kB) | 4 | 7 | Immune checkpoint (CD19, CD20, CTLA-4, NT5E, PCLP, PD-1, PD-L1) | 16 | 14 |
| 7 | Angiogenesis (EGF, IGF, FGF, VEGF, PDGF) | 2 | 3 | Evading growth suppressors (TP53, RB, cyclins, CDKs, p16, p18, p21) | 13 | 11 |
| 8 | Immune checkpoint (CD19, CD20, CTLA-4, NT5E, PCLP, PD-1, PD-L1) | 0 | 0 | EMT, invasion, and metastasis (EMT phenotype, integrin) | 13 | 11 |
| 9 | EMT, invasion, and metastasis (EMT phenotype, integrin) | 0 | 0 | Inflammation (NF-kB) | 12 | 10 |
| 10 | Replicative immortality (WNT, Hedgehog, TERT) | 0 | 0 | Replicative immortality (WNT, Hedgehog, TERT) | 8 | 7 |
| Signalling Pathway Affected | Targeted Drug/Inhibitor Drug | Cytotoxic Drugs | Clinical Trial Phase | Indications | Obs | References |
|---|---|---|---|---|---|---|
| BCR-ABL | Bosutinib | Pemetrexed | Ongoing phase I | Bladder, cervical, NSCLC, ovarian | Recruiting | [109] |
| Dasatinib | Carboplatin + Paclitaxel | Phase I completed | Ovarian | Recommended for phase II | [110] | |
| BCL-2 | Venetoclax | Azacitidine | Ongoing phase II | AML | Elderly patients | [111] |
| Cyclophosphamide, etoposide, doxorubicin, methotrexate, 6-mercaptopurine, cytarabine | ALL | Ongoing phase II | Older patients with relapsed or refractory ALL | [112] | ||
| CDK4/6 | Abemaciclib | Pemetrexed, or gemcitabine | Ongoing phase I | NSCLC | For stage IV patients | [113] |
| Temozolomide | Ongoing phase II | Glioblastoma | [114] | |||
| Palbociclib | Carboplatin | Ongoing phase II | Metastatic head and neck squamous cell carcinoma | [115] | ||
| Nab-paclitaxel | Phase I completed | mPDAC | No results posted; last update in May 30, 2019. | [116] | ||
| Temozolomide + irinotecan | Ongoing phase I | Solid tumors, neuroblastoma, medulloblastoma | For children, adolescents and young adults | [117] | ||
| Ribociclib | Docetaxel + Prednisone | Ongoing phase Ib/II | mCRPC | [118] | ||
| Gemcitabine | Ongoing phase I | Malignant brain tumors | [119] | |||
| Paclitaxel + Carboplatin | Ongoing phase I | Ovarian cancer, fallopian tube cancer, peritoneal carcinoma | [120] | |||
| DNA DAMAGE REPAIR | Niraparib | Temozolomide | Ongoing phase Ib/II | SCLC | [121] | |
| Olaparib | Cisplatin | Ongoing phase I | Advanced NSCLC | [122] | ||
| Rucaparib | Irinotecan | Ongoing phase Ib | Solid tumors | For patients with DNA repair defects in solid tumors | [123] | |
| Veliparib | FOLFIRI | Ongoing phase II | Pancreatic (metastatic) | Second line therapy | [124] | |
| Phase I completed | Gastric | Recommended for further investigation | [124] | |||
| Irinotecan | Ongoing phase I | Breast, lung, ovarian, pancreatic, Hodgkin’s lymphoma | For cancer that is metastatic or cannot be removed | [125] | ||
| Temozolomide | Ongoing phase I | ALL | [126] | |||
| Phase II completed | CRC | Recommended for further investigation | [127] | |||
| Topotecan | Ongoing phase I | Acute leukemias | [128] | |||
| EGFR | Cetuximab | FOLFIRI | Ongoing phase II | CRC | For patients with FcγRIIIa polymorphism and wild-type KRAS, NRAS and BRAF | [129] |
| FOLFOXIRI | Ongoing phase II | Locally advanced rectal carcinoma | For EGFR wild type patients | [130] | ||
| Necitumumab | Gemcitabine + Cisplatin | Ongoing phase II | Stage IB, II or IIIA squamous NSCLC | Neoadjuvant therapy | [131] | |
| Panitumumab | FOLFOX/FOLFIRI | Phase II completed | Liver (metastatic) | For patients with wild-type KRAS; no results posted; last update May 14, 2019. | [132] | |
| mFOLFOX6 + bevacizumab/panitumumab | Ongoing phase III | Advanced/recurrent CRC | First-line therapy for patients with KRAS/NRAS wild-type tumors | [133] | ||
| Pertuzumab | Paclitaxel + Trastuzumab | Ongoing phase I | HER2-positive breast cancer | [134] | ||
| PI3K-AKT-MTOR | Copanlisib | Gemcitabine | Phase I completed | Cholangiocarcinoma | No results yet. | [135] |
| Duvelisib | Fludarabine + cyclophosphamide + rituximab (FCR) | Ongoing phase Ib/II | CLL | [136] | ||
| Everolimus | Carboplatin + Paclitaxel | Phase II completed | Melanoma | Everolimus failed to improve efficacy | [137] | |
| Temozolomide | Ongoing phase II | Low-grade glioma | [138] | |||
| Idelalisib | Bendamustine + Rituximab | Phase III completed | Relapsed or refractory CLL | Improved PFS but with serious adverse events and infections | [139] | |
| Temsirolimus | Carboplatin + Paclitaxel | Phase II completed | Recurrent or metastatic head and neck | Recommended further investigation for PI3K/mTOR mutations | [140] | |
| VEGF | Bevacizumab | Capecitabine | Phase II completed | Advanced or Metastatic Liver Cancer | "All patients presented serious adverse events; only 9.1% presented objective response" | [141] |
| Carboplatin + Paclitaxel (CPB) | Phase II completed | Melanoma | Recommended for phase III | [142] | ||
| Carboplatin + Paclitaxel + Everolimus (CPBE) | Phase II completed | Melanoma | Failed to improve PFS compared to CPB | |||
| FOLFIRI (+ Onvansertib) | Ongoing phase Ib/II | mCRC | Second line therapy for patients with KRAS mutation | [143] | ||
| Temozolomide | Phase II completed | Glioma (grade II/III) | Failed to improve 1-year OS | [144] | ||
| Lenvatinib | Paclitaxel | Ongoing phase I | Endometrial, ovarian, fallopian tube, or primary peritoneal cancer | [145] | ||
| Ramucirumab | FOLFIRI | Ongoing phase II | Gastric | For previous failed therapy | [146] | |
| Regorafenib | Irinotecan | Ongoing phase II | Metastatic gastro-esophageal adenocarcinomas | Second line therapy | [147] | |
| Sorafenib | Irinotecan | Ongoing phase II | Pediatric solid tumors | Patients with mutations in Raf, PDGFR, VEGFR, Flt-3, KIT, JAK, STAT, RAS, MEK, or ERK | [148] |
| Signalling Pathways | Targeted Therapy | Clinical Trial Phase | Type of Tumor | Rationale | References |
|---|---|---|---|---|---|
| ALK AND CDK4/6 | Ceritinib + Ribociclib | Phase I completed | NSCLC | ALK+ NSCLC tumors | [149] |
| Ongoing phase I | Neuroblastoma | In vitro synergy (lower phospho-RB1 levels only in ALK+ NB cells) | [150,151] | ||
| MET AND AR | Crizotinib + Enzalutamide | Ongoing phase I | mCRPC | AR inhibition upregulates MET (off-target effect for crizotinib) | [152,153] |
| PDGFR AND C-SRC | Crizotinib + Dasatinib | Ongoing phase I | Solid malignancies | Downstream effects of MET require c-Src, whose inhibition upregulates MET | [154,155] |
| Phase I completed | High-grade glioma | PDGFR upregulated in gliomas (off-target for dasatinib) | [156] | ||
| PI3K AND EGFR | Copanlisib + Cetuximab | Ongoing phase Ib/II | HNSCC | Aberrant PI3K signaling confers resistance to cetuximab and both pathways are upregulated in HNSCC | [157,158] |
| VEGFR AND MTOR | Pazopanib + Everolimus | Ongoing phase I | Solid tumors | mTOR pathway activation confers resistance to anti-VEGF therapy | [159] |
| BCR | Idelalisib + Entospletinib | Phase II completed | Lymphoid malignancies | Synergistic effect with simultaneous inhibition of multiple kinases in the BCR pathway (PI3K and Syk), but limited by severe life-threatening adverse effects. | [160] |
| CDK4/6 AND PI3K/MTOR | Abemaciclib + LY3023414 | Ongoing phase II | PDAC | Enhanced PI3K/mTOR activity confers resistance to CDKi therapy | [161] |
| Ribociclib + Everolimus | Ongoing phase II | mPDAC | [105] | ||
| CDK4/6 AND EGFR | Palbociclib + Cetuximab | Ongoing phase II | HNSCC | EGFR overexpression is an oncogenic driver and there is either a frequent loss of CDKN2A or amplification of CCND1 | [162] |
| CDK4/6 AND MEK | Palbociclib + Trametinib | Phase Ib completed | Advanced solid malignancies | CDK activity confers resistance to MEKi | [163,164] |
| BRAF AND MEK | Dabrafenib + Trametinib | Ongoing phase II | Melanoma and brain metastases | MAPK pathway reactivation confers resistance to BRAFi. Melanoma brain metastasis seem to lack ABCB1 expression. Combination can cross blood-brain barrier. | [165] |
| Vemurafenib + Cobimetinib | Ongoing phase II | [166] | |||
| VEGFR AND EGFR | Bevacizumab + Erlotinib | Phase II completed | Advanced or Metastatic Liver Cancer | Dual blockade of tumor neovascularization and proliferative signaling. | [167] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delou, J.M.A.; Souza, A.S.O.; Souza, L.C.M.; Borges, H.L. Highlights in Resistance Mechanism Pathways for Combination Therapy. Cells 2019, 8, 1013. https://doi.org/10.3390/cells8091013
Delou JMA, Souza ASO, Souza LCM, Borges HL. Highlights in Resistance Mechanism Pathways for Combination Therapy. Cells. 2019; 8(9):1013. https://doi.org/10.3390/cells8091013
Chicago/Turabian StyleDelou, João M. A., Alana S. O. Souza, Leonel C. M. Souza, and Helena L. Borges. 2019. "Highlights in Resistance Mechanism Pathways for Combination Therapy" Cells 8, no. 9: 1013. https://doi.org/10.3390/cells8091013
APA StyleDelou, J. M. A., Souza, A. S. O., Souza, L. C. M., & Borges, H. L. (2019). Highlights in Resistance Mechanism Pathways for Combination Therapy. Cells, 8(9), 1013. https://doi.org/10.3390/cells8091013