Regulation of Ketogenic Enzyme HMGCS2 by Wnt/β-catenin/PPARγ Pathway in Intestinal Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Treatment, and Transfection
2.2. Western Blot Analysis
2.3. Intestinal Organoid Culture
2.4. RNA Isolation and Real Time Reverse Transcription-PCR (RT-PCR) Analysis
2.5. Chromatin Immunoprecipitation (ChIP) Analysis
2.6. βHB Assay
2.7. PPARγ and PPARα Transcription Factor Assays
2.8. Measurement of Glycolysis
2.9. Intestinal Alkaline Phosphatase Activity
2.10. Statistical Analysis
3. Results
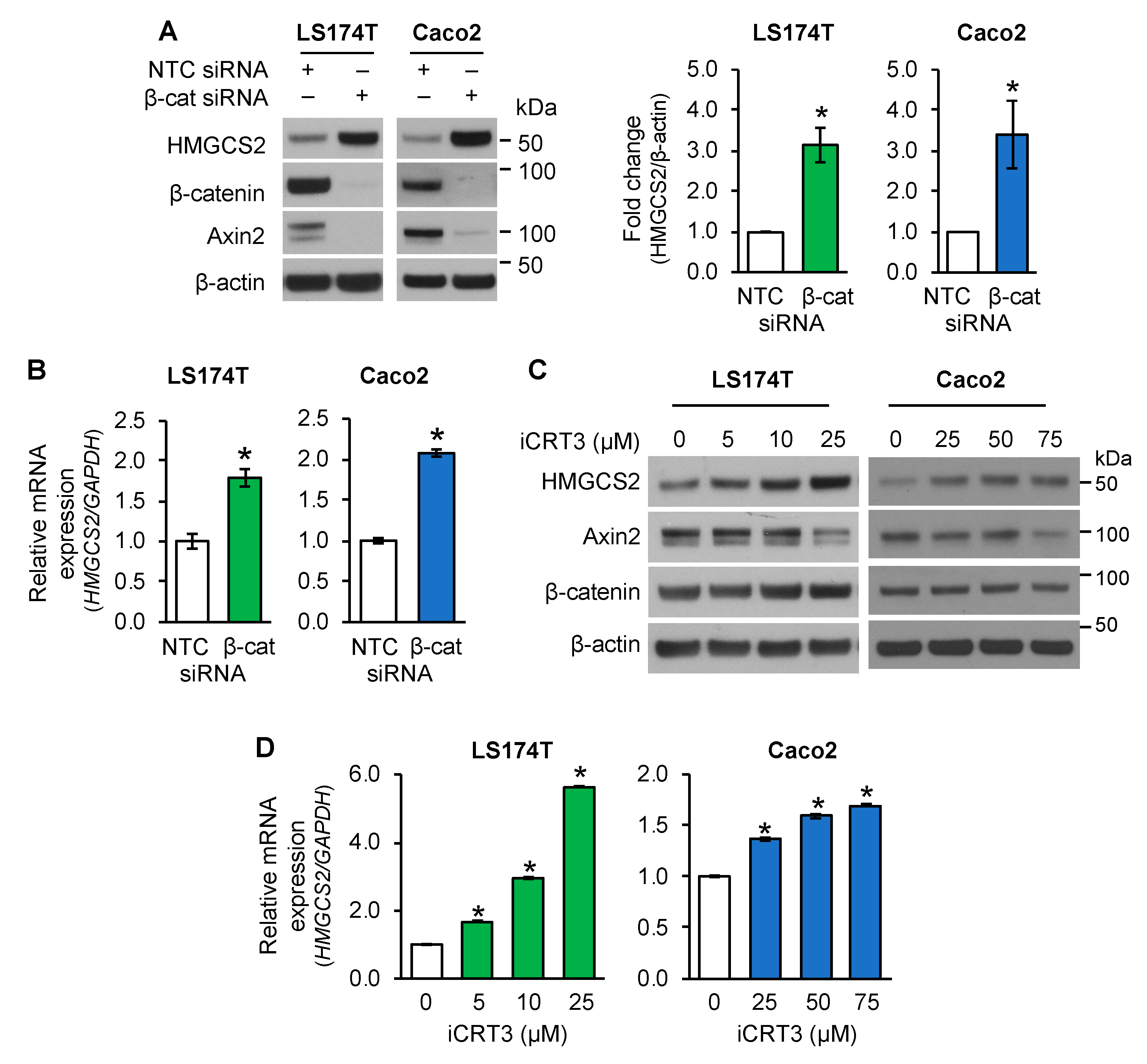
3.1. Inhibition of Wnt/β-Catenin Pathway Increased HMGCS2 Expression in Human Intestinal Cancer Cell Lines
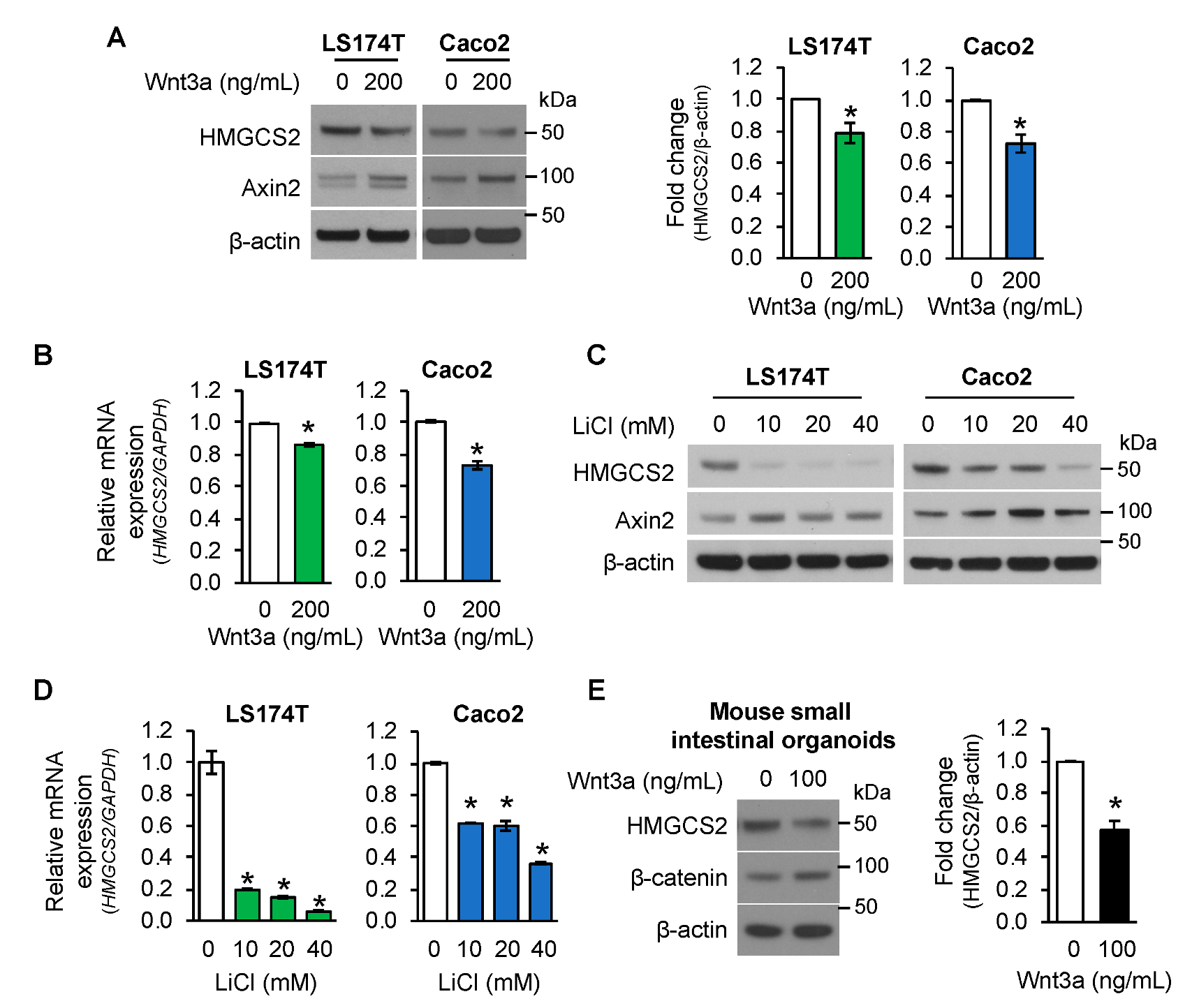
3.2. Activation of Wnt/β-Catenin Signaling Suppressed HMGCS2 Expression in Human Intestinal Cancer Cells and Mouse Small Intestinal Organoids
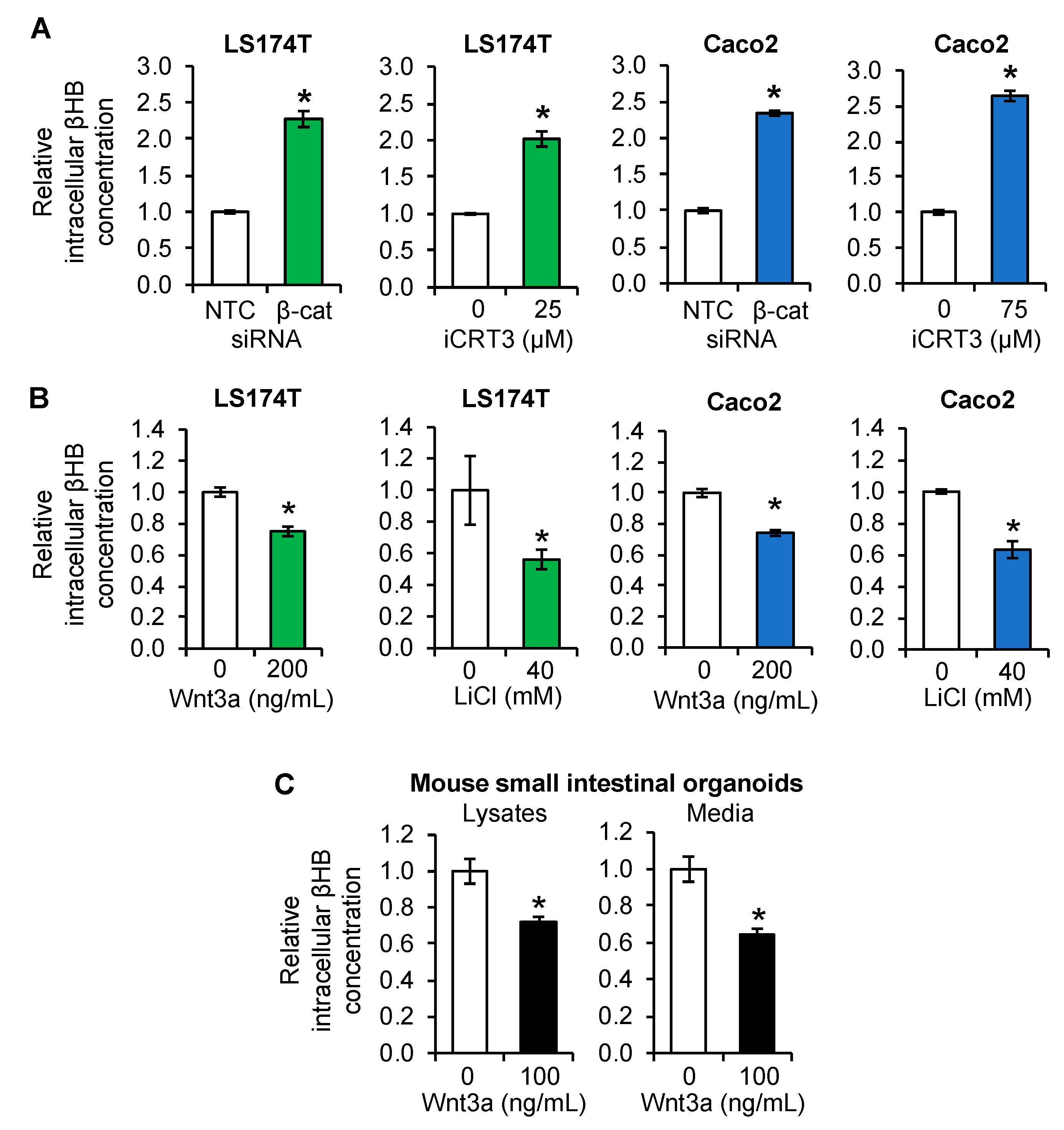
3.3. Regulation of Ketogenesis by Wnt/β-Catenin Signaling in Intestinal Cancer Cell Lines and Organoids
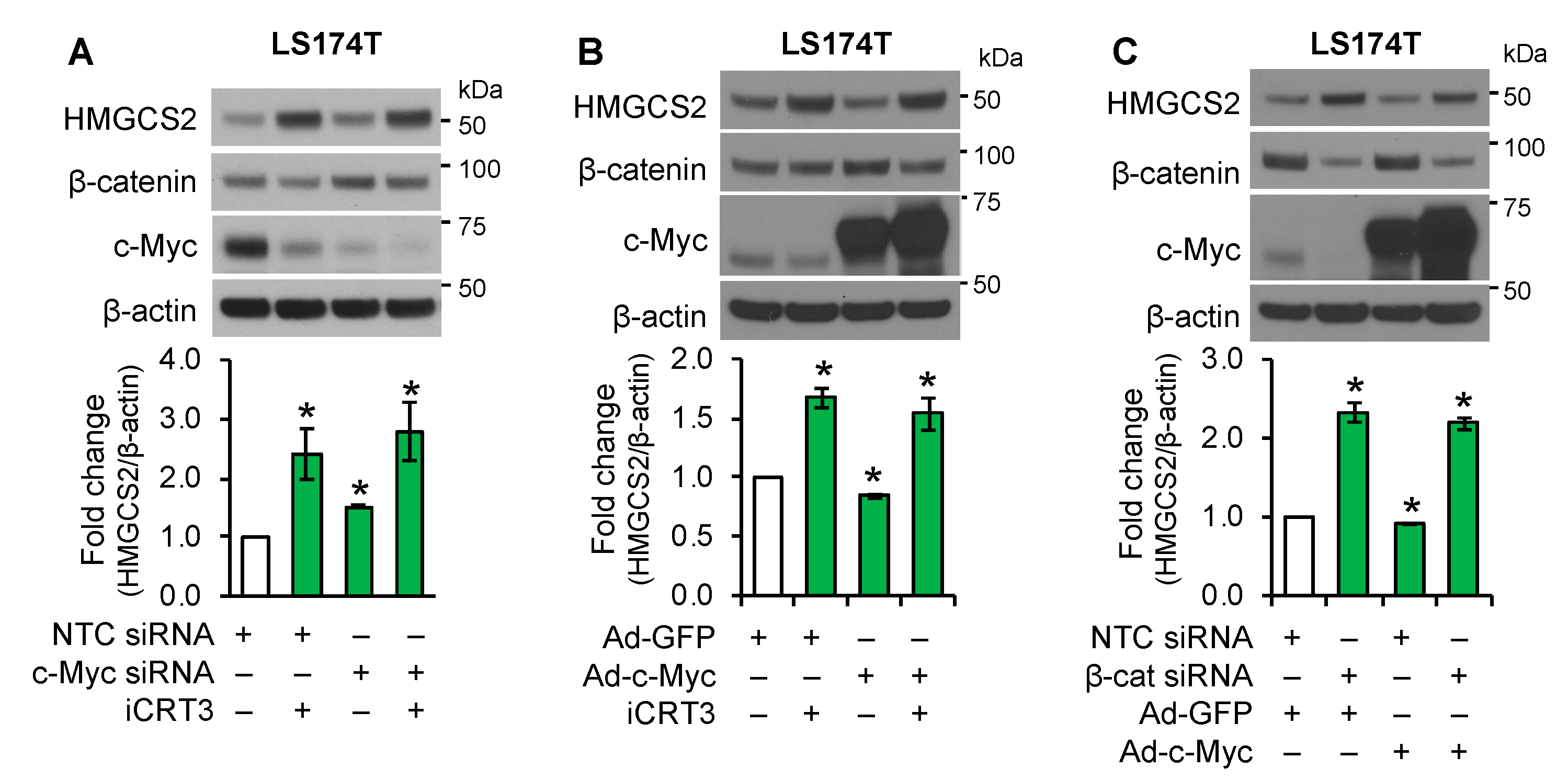
3.4. Wnt/β-Catenin Signaling Regulated Ketogenesis Independent of c-Myc
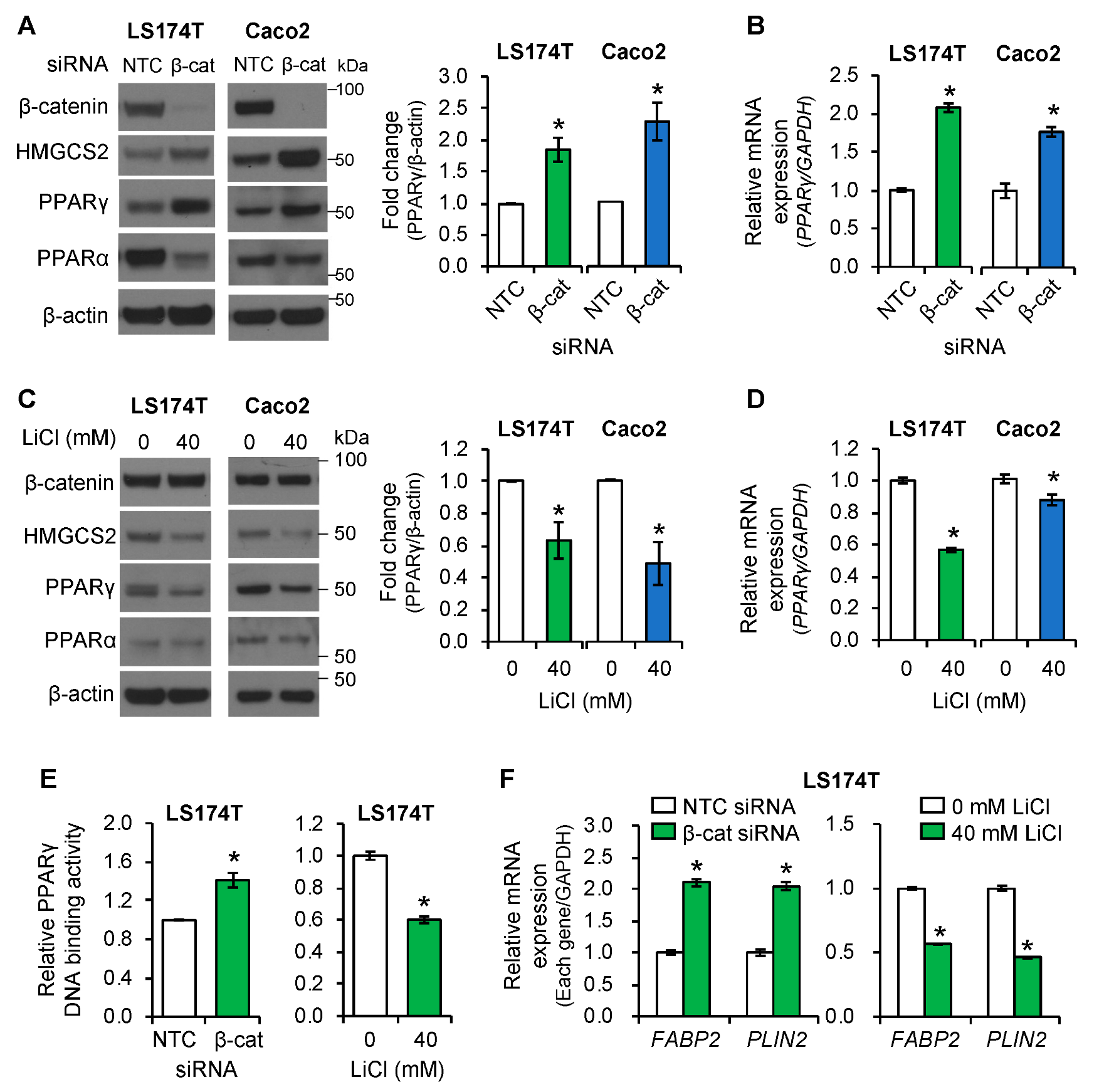
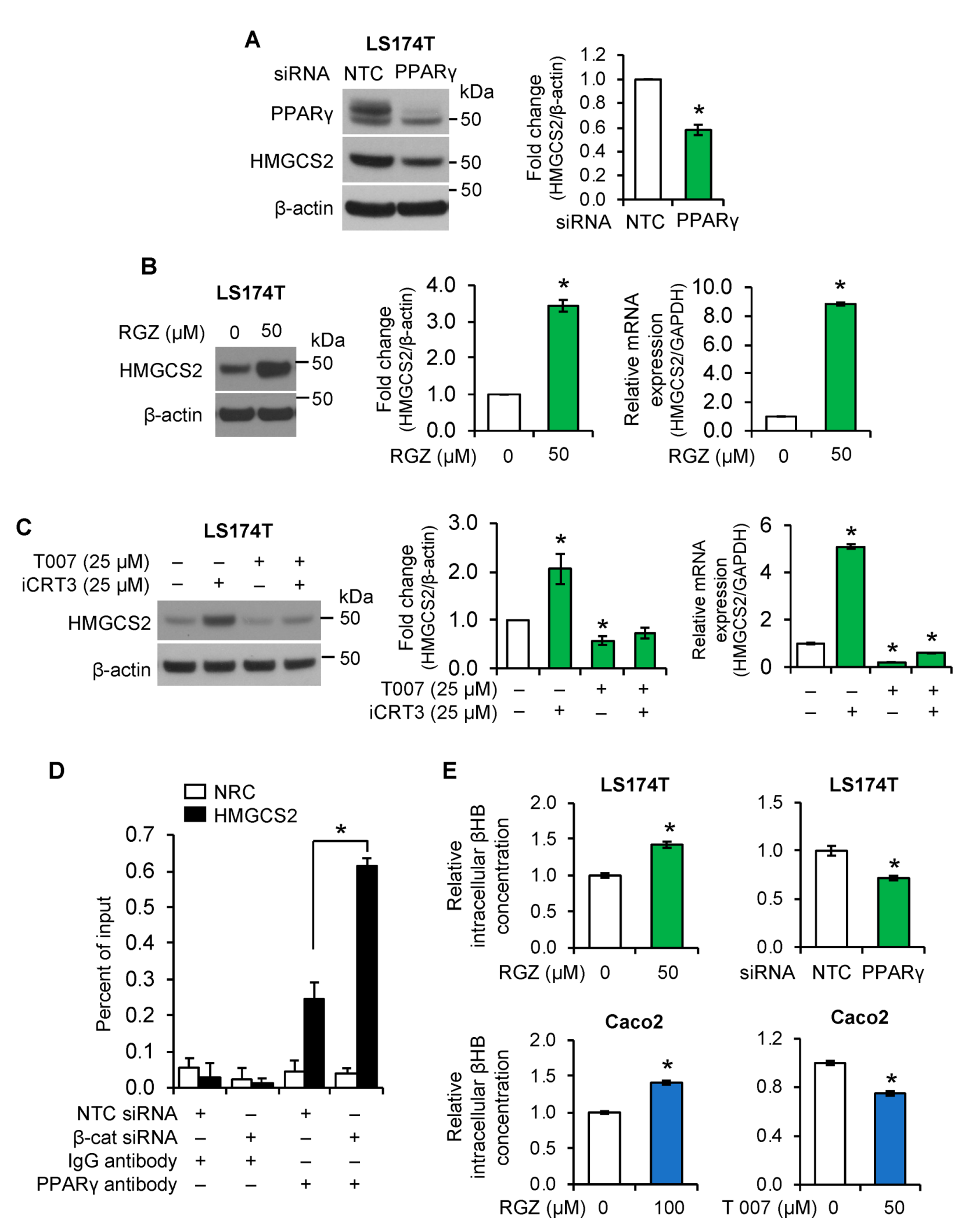
3.5. PPARγ Mediated the Role of Wnt/β-Catenin Signaling in the Regulation of Ketogenesis
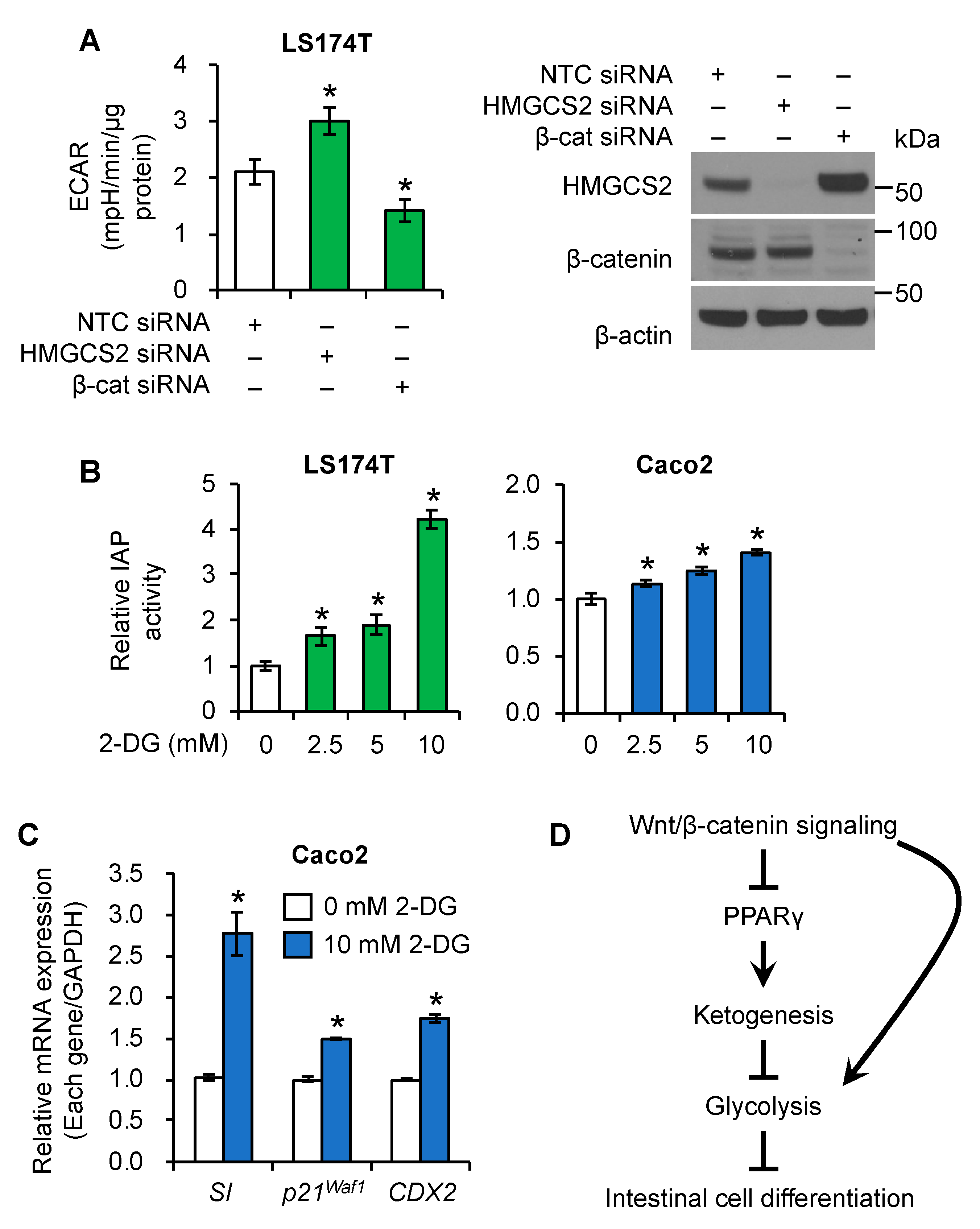
3.6. Regulation of Glycolysis by Wnt/β-Catenin/HMGCS2 Pathway
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Noah, T.K.; Donahue, B.; Shroyer, N.F. Intestinal development and differentiation. Exp. Cell Res. 2011, 317, 2702–2710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, A.M.; Watson, A.J.M. Deciphering the Complex Signaling Systems That Regulate Intestinal Epithelial Cell Death Processes and Shedding. Front. Immunol. 2017, 8, 841. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Fletcher, R.B.; Ngai, J. Cellular mechanisms of epithelial stem cell self-renewal and differentiation during homeostasis and repair. Wiley Interdiscip. Rev. Dev. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Scoville, D.H.; Sato, T.; He, X.C.; Li, L. Current view: Intestinal stem cells and signaling. Gastroenterology 2008, 134, 849–864. [Google Scholar] [CrossRef] [PubMed]
- Negroni, A.; Cucchiara, S.; Stronati, L. Apoptosis, Necrosis, and Necroptosis in the Gut and Intestinal Homeostasis. Mediat. Inflamm. 2015, 2015, 250762. [Google Scholar] [CrossRef] [PubMed]
- Van der Flier, L.G.; Clevers, H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu. Rev. Physiol. 2009, 71, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Van der Heijden, M.; Vermeulen, L. Stem cells in homeostasis and cancer of the gut. Mol. Cancer 2019, 18, 66. [Google Scholar] [CrossRef] [PubMed]
- Klaus, A.; Birchmeier, W. Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer 2008, 8, 387–398. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- Koch, S. Extrinsic control of Wnt signaling in the intestine. Differentiation 2017, 97, 1–8. [Google Scholar] [CrossRef]
- Krausova, M.; Korinek, V. Wnt signaling in adult intestinal stem cells and cancer. Cell Signal. 2014, 26, 570–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fearon, E.R. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.H.; Barker, N. Wnt Signaling in Adult Epithelial Stem Cells and Cancer. Prog. Mol. Biol. Transl. Sci. 2018, 153, 21–79. [Google Scholar] [CrossRef] [PubMed]
- Varga, T.; Czimmerer, Z.; Nagy, L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim. Biophys. Acta 2011, 1812, 1007–1022. [Google Scholar] [CrossRef] [PubMed]
- Fucci, A.; Colangelo, T.; Votino, C.; Pancione, M.; Sabatino, L.; Colantuoni, V. The role of peroxisome proliferator-activated receptors in the esophageal, gastric, and colorectal cancer. PPAR Res. 2012, 2012, 242498. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.C.; Gil-Gomez, G.; Hegardt, F.G.; Haro, D. Peroxisome proliferator-activated receptor mediates induction of the mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase gene by fatty acids. J. Biol. Chem. 1994, 269, 18767–18772. [Google Scholar]
- Grabacka, M.; Pierzchalska, M.; Dean, M.; Reiss, K. Regulation of Ketone Body Metabolism and the Role of PPARalpha. Int. J. Mol. Sci. 2016, 17, 2093. [Google Scholar] [CrossRef] [PubMed]
- Sikder, K.; Shukla, S.K.; Patel, N.; Singh, H.; Rafiq, K. High Fat Diet Upregulates Fatty Acid Oxidation and Ketogenesis via Intervention of PPAR-gamma. Cell Physiol. Biochem. 2018, 48, 1317–1331. [Google Scholar] [CrossRef]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARgamma signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Lecarpentier, Y.; Claes, V.; Vallee, A.; Hebert, J.L. Thermodynamics in cancers: Opposing interactions between PPAR gamma and the canonical WNT/beta-catenin pathway. Clin. Transl. Med. 2017, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Vallee, A.; Lecarpentier, Y.; Guillevin, R.; Vallee, J.N. Opposite Interplay Between the Canonical WNT/beta-Catenin Pathway and PPAR Gamma: A Potential Therapeutic Target in Gliomas. Neurosci. Bull. 2018, 34, 573–588. [Google Scholar] [CrossRef] [PubMed]
- Hegardt, F.G. Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase: A control enzyme in ketogenesis. Biochem. J. 1999, 338, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.C.; Verdin, E. Ketone bodies as signaling metabolites. Trends Endocrinol. Metab. 2014, 25, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Camarero, N.; Mascaro, C.; Mayordomo, C.; Vilardell, F.; Haro, D.; Marrero, P.F. Ketogenic HMGCS2 is a c-Myc target gene expressed in differentiated cells of human colonic epithelium and down-regulated in colon cancer. Mol. Cancer Res. 2006, 4, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Saraon, P.; Cretu, D.; Musrap, N.; Karagiannis, G.S.; Batruch, I.; Drabovich, A.P.; van der Kwast, T.; Mizokami, A.; Morrissey, C.; Jarvi, K.; et al. Quantitative proteomics reveals that enzymes of the ketogenic pathway are associated with prostate cancer progression. Mol. Cell Proteom. 2013, 12, 1589–1601. [Google Scholar] [CrossRef] [PubMed]
- Gromov, P.; Espinoza, J.A.; Talman, M.L.; Honma, N.; Kroman, N.; Timmermans Wielenga, V.; Moreira, J.M.; Gromova, I. FABP7 and HMGCS2 are novel protein markers for apocrine differentiation categorizing apocrine carcinoma of the breast. PLoS ONE 2014, 9, e112024. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Wu, Y.; Qin, Y.; Wang, H.; Jia, Y.; Yang, S.; Luo, S.; Wang, Q. Predictive significance of HMGCS2 for prognosis in resected Chinese esophageal squamous cell carcinoma patients. Onco Targets Ther. 2017, 10, 2553–2560. [Google Scholar] [CrossRef]
- Su, S.G.; Yang, M.; Zhang, M.F.; Peng, Q.Z.; Li, M.Y.; Liu, L.P.; Bao, S.Y. miR-107-mediated decrease of HMGCS2 indicates poor outcomes and promotes cell migration in hepatocellular carcinoma. Int. J. Biochem. Cell Biol. 2017, 91, 53–59. [Google Scholar] [CrossRef]
- Wang, Q.; Zhou, Y.; Rychahou, P.; Fan, T.W.; Lane, A.N.; Weiss, H.L.; Evers, B.M. Ketogenesis contributes to intestinal cell differentiation. Cell Death Differ. 2017, 24, 458–468. [Google Scholar] [CrossRef]
- Spit, M.; Koo, B.K.; Maurice, M.M. Tales from the crypt: Intestinal niche signals in tissue renewal, plasticity and cancer. Open Biol. 2018, 8. [Google Scholar] [CrossRef]
- Kim, J.T.; Liu, C.; Zaytseva, Y.Y.; Weiss, H.L.; Townsend, C.M., Jr.; Evers, B.M. Neurotensin, a novel target of Wnt/beta-catenin pathway, promotes growth of neuroendocrine tumor cells. Int. J. Cancer 2015, 136, 1475–1481. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.T.; Li, J.; Song, J.; Lee, E.Y.; Weiss, H.L.; Townsend, C.M., Jr.; Evers, B.M. Differential expression and tumorigenic function of neurotensin receptor 1 in neuroendocrine tumor cells. Oncotarget 2015, 6, 26960–26970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Colman, M.J.; Schewe, M.; Meerlo, M.; Stigter, E.; Gerrits, J.; Pras-Raves, M.; Sacchetti, A.; Hornsveld, M.; Oost, K.C.; Snippert, H.J.; et al. Interplay between metabolic identities in the intestinal crypt supports stem cell function. Nature 2017, 543, 424–427. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.J.P.; Goh, F.G.; Banks, H.; Krausgruber, T.; Kotenko, S.V.; Foxwell, B.M.J.; Udalova, I.A. The role of transposable elements in the regulation of IFN-lambda1 gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11564–11569. [Google Scholar] [CrossRef] [PubMed]
- Lacazette, E. A laboratory practical illustrating the use of the ChIP-qPCR method in a robust model: Estrogen receptor alpha immunoprecipitation using Mcf-7 culture cells. Biochem. Mol. Biol. Educ 2017, 45, 152–160. [Google Scholar] [CrossRef]
- Lin, X.; Tirichine, L.; Bowler, C. Protocol: Chromatin immunoprecipitation (ChIP) methodology to investigate histone modifications in two model diatom species. Plant Methods 2012, 8, 48. [Google Scholar] [CrossRef]
- Jho, E.H.; Zhang, T.; Domon, C.; Joo, C.K.; Freund, J.N.; Costantini, F. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol. Cell Biol. 2002, 22, 1172–1183. [Google Scholar] [CrossRef]
- Gonsalves, F.C.; Klein, K.; Carson, B.B.; Katz, S.; Ekas, L.A.; Evans, S.; Nagourney, R.; Cardozo, T.; Brown, A.M.; DasGupta, R. An RNAi-based chemical genetic screen identifies three small-molecule inhibitors of the Wnt/wingless signaling pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 5954–5963. [Google Scholar] [CrossRef]
- Caspi, M.; Zilberberg, A.; Eldar-Finkelman, H.; Rosin-Arbesfeld, R. Nuclear GSK-3beta inhibits the canonical Wnt signalling pathway in a beta-catenin phosphorylation-independent manner. Oncogene 2008, 27, 3546–3555. [Google Scholar] [CrossRef]
- Vidal, F.; de Araujo, W.M.; Cruz, A.L.; Tanaka, M.N.; Viola, J.P.; Morgado-Diaz, J.A. Lithium reduces tumorigenic potential in response to EGF signaling in human colorectal cancer cells. Int. J. Oncol. 2011, 38, 1365–1373. [Google Scholar] [CrossRef]
- Gupta, R.A.; Brockman, J.A.; Sarraf, P.; Willson, T.M.; DuBois, R.N. Target genes of peroxisome proliferator-activated receptor gamma in colorectal cancer cells. J. Biol. Chem. 2001, 276, 29681–29687. [Google Scholar] [CrossRef] [PubMed]
- Jansson, E.A.; Are, A.; Greicius, G.; Kuo, I.C.; Kelly, D.; Arulampalam, V.; Pettersson, S. The Wnt/beta-catenin signaling pathway targets PPARgamma activity in colon cancer cells. Proc. Natl. Acad. Sci. USA 2005, 102, 1460–1465. [Google Scholar] [CrossRef] [PubMed]
- Dang, Y.F.; Jiang, X.N.; Gong, F.L.; Guo, X.L. New insights into molecular mechanisms of rosiglitazone in monotherapy or combination therapy against cancers. Chem. Biol. Interact. 2018, 296, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Burton, J.D.; Goldenberg, D.M.; Blumenthal, R.D. Potential of peroxisome proliferator-activated receptor gamma antagonist compounds as therapeutic agents for a wide range of cancer types. PPAR Res. 2008, 2008, 494161. [Google Scholar] [CrossRef] [PubMed]
- Pate, K.T.; Stringari, C.; Sprowl-Tanio, S.; Wang, K.; TeSlaa, T.; Hoverter, N.P.; McQuade, M.M.; Garner, C.; Digman, M.A.; Teitell, M.A.; et al. Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J. 2014, 33, 1454–1473. [Google Scholar] [CrossRef]
- Thompson, C.B. Wnt meets Warburg: Another piece in the puzzle? EMBO J. 2014, 33, 1420–1422. [Google Scholar] [CrossRef]
- Sherwood, V. WNT signaling: An emerging mediator of cancer cell metabolism? Mol. Cell Biol. 2015, 35, 2–10. [Google Scholar] [CrossRef]
- Stringari, C.; Edwards, R.A.; Pate, K.T.; Waterman, M.L.; Donovan, P.J.; Gratton, E. Metabolic trajectory of cellular differentiation in small intestine by Phasor Fluorescence Lifetime Microscopy of NADH. Sci. Rep. 2012, 2, 568. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Li, J.; Wang, F.; Hu, J.; Wang, S.; Sun, Y. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014, 355, 176–183. [Google Scholar] [CrossRef]
- Evers, B.M. Intestinal cell differentiation: Cellular mechanisms and the search for the perfect model focus on “involvement of p21(WAF1/Cip1) and p27(Kip1) in intestinal epithelial cell differentiation”. Am. J. Physiol. 1999, 276, C1243–C1244. [Google Scholar] [CrossRef]
- Saad, R.S.; Ghorab, Z.; Khalifa, M.A.; Xu, M. CDX2 as a marker for intestinal differentiation: Its utility and limitations. World J. Gastrointest. Surg. 2011, 3, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.T.; Li, J.; Jang, E.R.; Gulhati, P.; Rychahou, P.G.; Napier, D.L.; Wang, C.; Weiss, H.L.; Lee, E.Y.; Anthony, L.; et al. Deregulation of Wnt/beta-catenin signaling through genetic or epigenetic alterations in human neuroendocrine tumors. Carcinogenesis 2013, 34, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Henderson, J.T.; Beghtel, H.; van den Born, M.M.; Sancho, E.; Huls, G.; Meeldijk, J.; Robertson, J.; van de Wetering, M.; Pawson, T.; et al. Beta-catenin and TCF mediate cell positioning in the intestinal epithelium by controlling the expression of EphB/ephrinB. Cell 2002, 111, 251–263. [Google Scholar] [CrossRef]
- Blache, P.; van de Wetering, M.; Duluc, I.; Domon, C.; Berta, P.; Freund, J.N.; Clevers, H.; Jay, P. SOX9 is an intestine crypt transcription factor, is regulated by the Wnt pathway, and represses the CDX2 and MUC2 genes. J. Cell Biol. 2004, 166, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Fevr, T.; Robine, S.; Louvard, D.; Huelsken, J. Wnt/beta-catenin is essential for intestinal homeostasis and maintenance of intestinal stem cells. Mol. Cell Biol. 2007, 27, 7551–7559. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhou, Y.; Rychahou, P.; Harris, J.W.; Zaytseva, Y.Y.; Liu, J.; Wang, C.; Weiss, H.L.; Liu, C.; Lee, E.Y.; et al. Deptor is a novel target of Wnt/beta-catenin/c-Myc and contributes to colorectal cancer cell growth. Cancer Res. 2018. [Google Scholar] [CrossRef]
- Hao, G.W.; Chen, Y.S.; He, D.M.; Wang, H.Y.; Wu, G.H.; Zhang, B. Growth of human colon cancer cells in nude mice is delayed by ketogenic diet with or without omega-3 fatty acids and medium-chain triglycerides. Asian Pac. J. Cancer Prev. 2015, 16, 2061–2068. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; O’Donnell, K.A.; Zeller, K.I.; Nguyen, T.; Osthus, R.C.; Li, F. The c-Myc target gene network. Semin. Cancer Biol. 2006, 16, 253–264. [Google Scholar] [CrossRef]
- Sipos, F.; Firneisz, G.; Muzes, G. Therapeutic aspects of c-MYC signaling in inflammatory and cancerous colonic diseases. World J. Gastroenterol. 2016, 22, 7938–7950. [Google Scholar] [CrossRef]
- Poole, C.J.; van Riggelen, J. MYC-Master Regulator of the Cancer Epigenome and Transcriptome. Genes 2017, 8, 142. [Google Scholar] [CrossRef]
- Rennoll, S.; Yochum, G. Regulation of MYC gene expression by aberrant Wnt/beta-catenin signaling in colorectal cancer. World J. Biol. Chem. 2015, 6, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, L.; Pancione, M.; Votino, C.; Colangelo, T.; Lupo, A.; Novellino, E.; Lavecchia, A.; Colantuoni, V. Emerging role of the beta-catenin-PPARgamma axis in the pathogenesis of colorectal cancer. World J. Gastroenterol. 2014, 20, 7137–7151. [Google Scholar] [CrossRef] [PubMed]
- Michalik, L.; Desvergne, B.; Wahli, W. Peroxisome-proliferator-activated receptors and cancers: Complex stories. Nat. Rev. Cancer 2004, 4, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Bajwa, P.J.; Lee, J.W.; Straus, D.S.; Lytle, C. Activation of PPARgamma by rosiglitazone attenuates intestinal Cl- secretion. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G82–G89. [Google Scholar] [CrossRef] [PubMed]
- Mansen, A.; Guardiola-Diaz, H.; Rafter, J.; Branting, C.; Gustafsson, J.A. Expression of the peroxisome proliferator-activated receptor (PPAR) in the mouse colonic mucosa. Biochem. Biophys. Res. Commun. 1996, 222, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Lecarpentier, Y.; Claes, V.; Vallee, A.; Hebert, J.L. Interactions between PPAR Gamma and the Canonical Wnt/Beta-Catenin Pathway in Type 2 Diabetes and Colon Cancer. PPAR Res. 2017, 2017, 5879090. [Google Scholar] [CrossRef] [PubMed]
- Vallee, A.; Lecarpentier, Y. Crosstalk Between Peroxisome Proliferator-Activated Receptor Gamma and the Canonical WNT/beta-Catenin Pathway in Chronic Inflammation and Oxidative Stress During Carcinogenesis. Front. Immunol. 2018, 9, 745. [Google Scholar] [CrossRef]
- Lund, T.M.; Ploug, K.B.; Iversen, A.; Jensen, A.A.; Jansen-Olesen, I. The metabolic impact of beta-hydroxybutyrate on neurotransmission: Reduced glycolysis mediates changes in calcium responses and KATP channel receptor sensitivity. J. Neurochem. 2015, 132, 520–531. [Google Scholar] [CrossRef]
- Yellen, G. Ketone bodies, glycolysis, and KATP channels in the mechanism of the ketogenic diet. Epilepsia 2008, 49 (Suppl. 8), 80–82. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.T.; Li, C.; Weiss, H.L.; Zhou, Y.; Liu, C.; Wang, Q.; Evers, B.M. Regulation of Ketogenic Enzyme HMGCS2 by Wnt/β-catenin/PPARγ Pathway in Intestinal Cells. Cells 2019, 8, 1106. https://doi.org/10.3390/cells8091106
Kim JT, Li C, Weiss HL, Zhou Y, Liu C, Wang Q, Evers BM. Regulation of Ketogenic Enzyme HMGCS2 by Wnt/β-catenin/PPARγ Pathway in Intestinal Cells. Cells. 2019; 8(9):1106. https://doi.org/10.3390/cells8091106
Chicago/Turabian StyleKim, Ji Tae, Chang Li, Heidi L. Weiss, Yuning Zhou, Chunming Liu, Qingding Wang, and B. Mark Evers. 2019. "Regulation of Ketogenic Enzyme HMGCS2 by Wnt/β-catenin/PPARγ Pathway in Intestinal Cells" Cells 8, no. 9: 1106. https://doi.org/10.3390/cells8091106
APA StyleKim, J. T., Li, C., Weiss, H. L., Zhou, Y., Liu, C., Wang, Q., & Evers, B. M. (2019). Regulation of Ketogenic Enzyme HMGCS2 by Wnt/β-catenin/PPARγ Pathway in Intestinal Cells. Cells, 8(9), 1106. https://doi.org/10.3390/cells8091106