FOXO3a from the Nucleus to the Mitochondria: A Round Trip in Cellular Stress Response

 , ,
, ,
Abstract
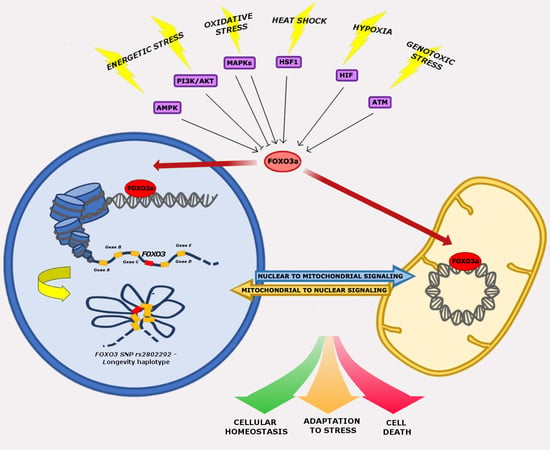
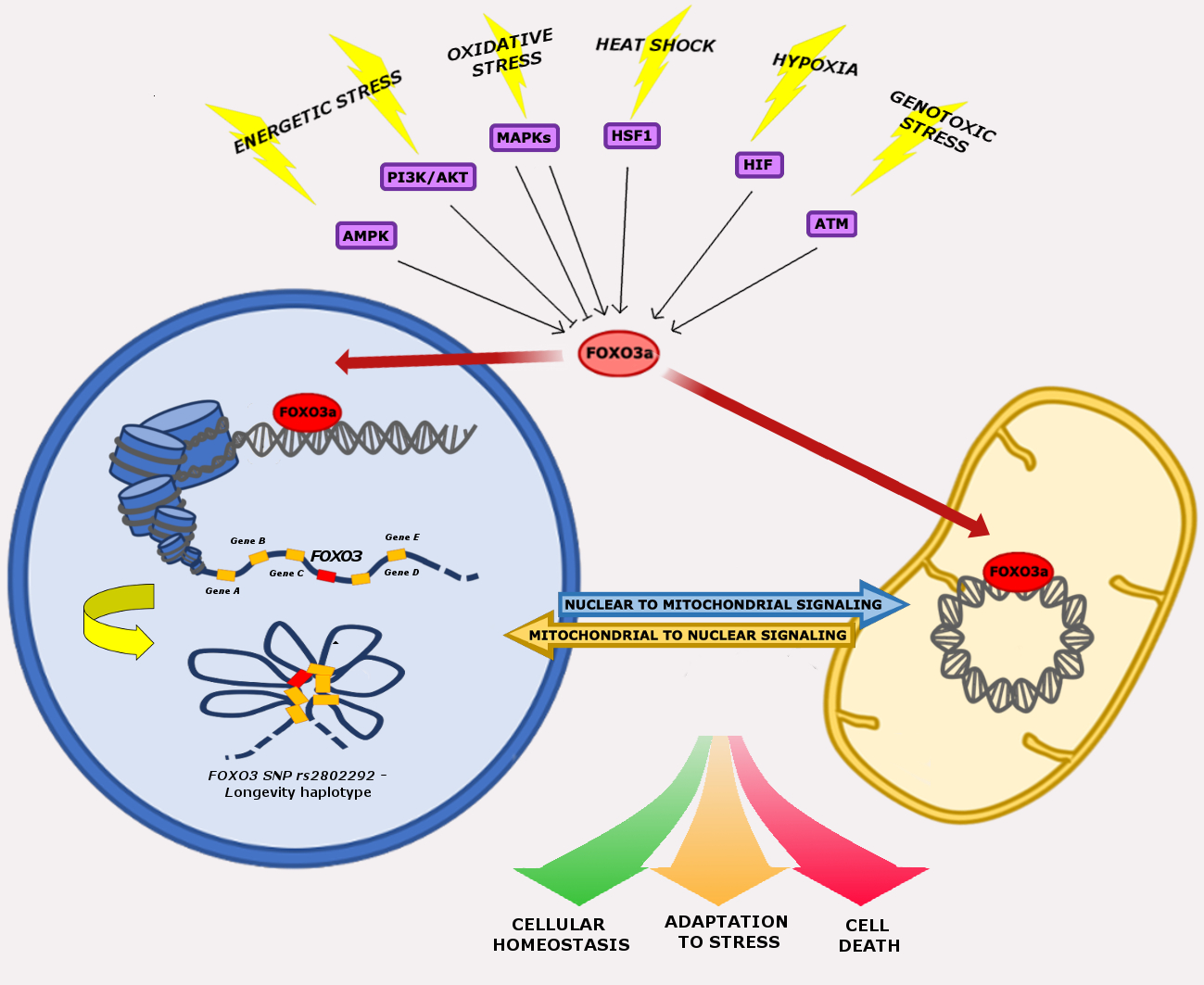
:
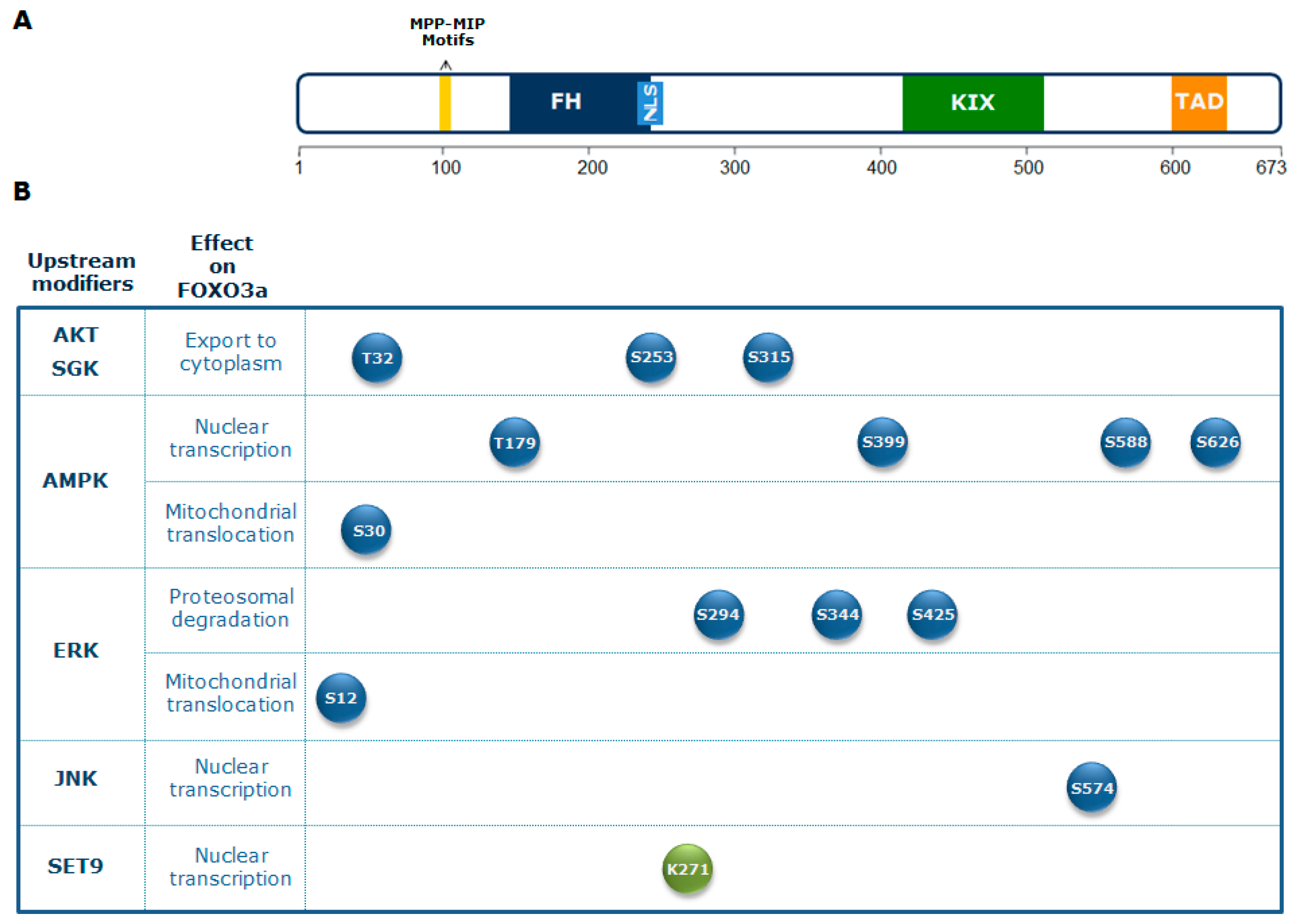
{kind=link}
{kind=link}
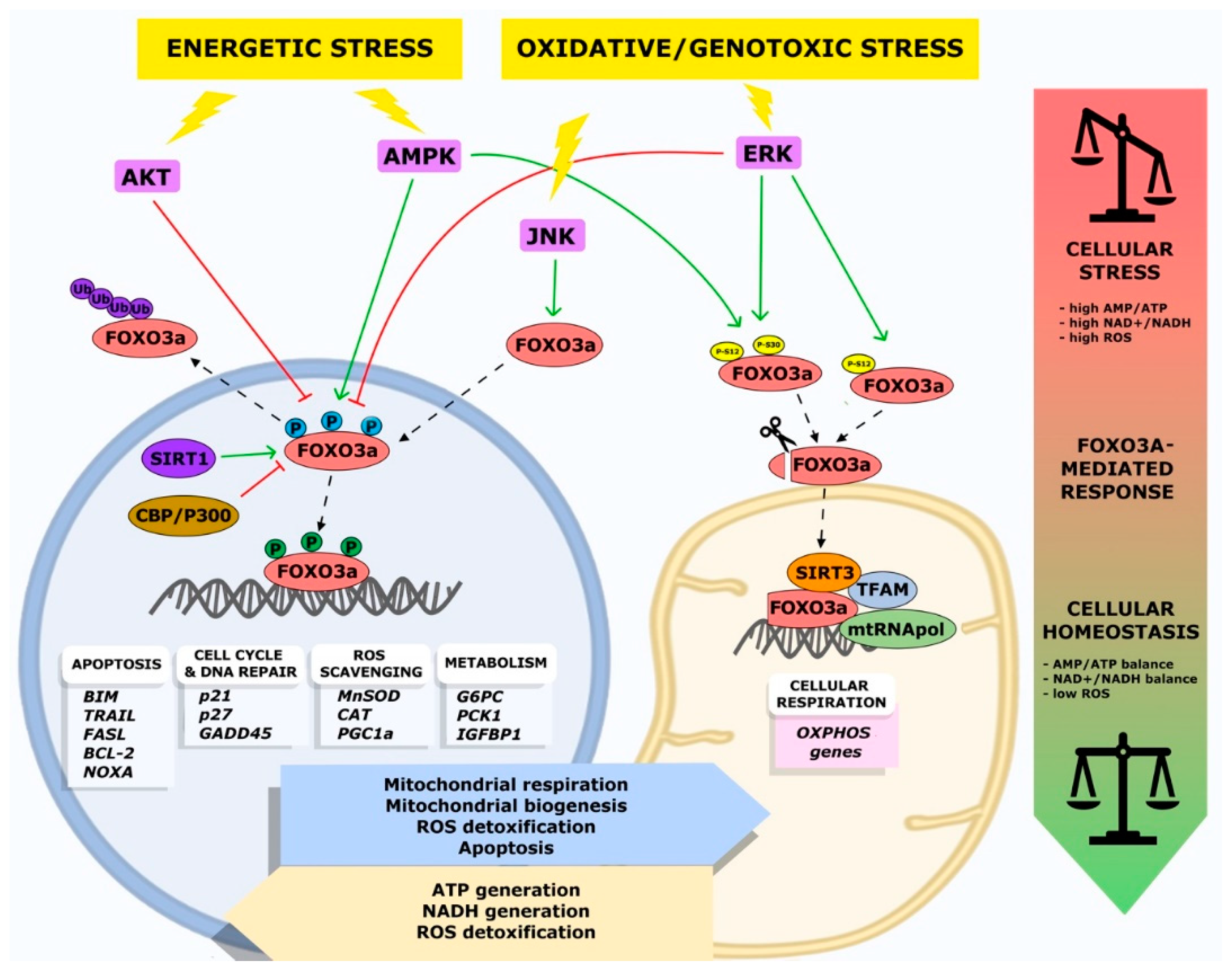{kind=link}
1. Introduction
2. Upstream Regulation of FOXO3a
2.1. The PI3K-AKT-FOXO3a and AMPK-FOXO3a Axes in Nutrient Signaling and Energetic Stress
2.2. The MAPK-FOXO3a Axis in Oxidative Stress
2.3. The HIF1α-FOXO3a Axis in Hypoxic Stress
2.4. Upstream Regulation of FOXO3a in Heat Shock Stress
2.5. Upstream Regulation of FOXO3a in Genotoxic Stress
3. Architecture of FOXO3a Domains and its Nuclear Functions
4. Roles of Nuclear FOXO3a in Cellular Stress Response
4.1. Role of FOXO3a in the Regulation of Genes Involved in Metabolic/Energetic Stress
4.2. Role of FOXO3a in the Regulation of Genes Involved in Oxidative Stress
4.3. Roles of FOXO3a in the Regulation of Hypoxic Stress Response
4.4. Roles of FOXO3a in the Regulation of Genotoxic Stress
4.5. FOXO3a: A Regulator of Cell Fate in Stress Conditions
5. Role of Mitochondrial FOXO3a in Cellular Stress Response
5.1. Mitochondrial FOXO3a in Metabolic Stress Response
5.2. Mitochondrial FOXO3a-Mediated Chemoresistance in Chemotherapy-Dependent Genotoxic Stress
6. FOXO3a-Dependent Nuclear and Mitochondrial Crosstalk in Cellular Stress Response
6.1. Nuclear FOXO3a-Dependent Effects in Stressed Mitochondria
6.2. Mitochondrial FOXO3a-Dependent Effects in Stressed Cells
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Galluzzi, L.; Yamazaki, T.; Kroemer, G. Linking cellular stress responses to systemic homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Kourtis, N.; Tavernarakis, N. Cellular stress response pathways and ageing: Intricate molecular relationships. EMBO J. 2011, 30, 2520–2531. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Kehrer, J.P.; Lund, L.G. Cellular reducing equivalents and oxidative stress. Free. Radic. Biol. Med. 1994, 17, 65–75. [Google Scholar] [CrossRef]
- Slimen, I.B.; Najar, T.; Ghram, A.; Dabbebi, H.; Ben Mrad, M.; Abdrabbah, M. Reactive oxygen species, heat stress and oxidative-induced mitochondrial damage. Rev. Int. J. Hyperth. 2014, 30, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef]
- Richter, K.; Haslbeck, M.; Buchner, J. The heat shock response: Life on the verge of death. Mol. Cell 2010, 40, 253–266. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Davis, R.J. Cell Signaling and Stress Responses. Cold Spring Harb. Perspect. Biol. 2016, 8, a006072. [Google Scholar] [CrossRef] [Green Version]
- De Nadal, E.; Ammerer, G.; Posas, F. Controlling gene expression in response to stress. Nat. Rev. Genet. 2011, 12, 833–845. [Google Scholar] [CrossRef]
- Golson, M.L.; Kaestner, K.H. Fox transcription factors: From development to disease. Development 2016, 143, 4558–4570. [Google Scholar] [CrossRef] [PubMed]
- Eijkelenboom, A.; Burgering, B.M. FOXOs: Signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 2013, 14, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Van der Horst, A.; Burgering, B.M. Stressing the role of FoxO proteins in lifespan and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Riddle, D.L.; Swanson, M.M.; Albert, P.S. Interacting genes in nematode dauer larva formation. Nature 1981, 290, 668–671. [Google Scholar] [CrossRef] [PubMed]
- Donovan, M.R.; Marr, M.T. dFOXO Activates Large and Small Heat Shock Protein Genes in Response to Oxidative Stress to Maintain Proteostasis in Drosophila. J. Biol. Chem. 2016, 291, 19042–19050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, A.K.; Webb, A.E. Regulation of FOXO Factors in Mammalian Cells. Curr. Top. Dev. Biol. 2018, 127, 165–192. [Google Scholar] [PubMed]
- Hosaka, T.; Biggs, W.H.; Tieu, D.; Boyer, A.D.; Varki, N.M.; Cavenee, W.K.; Arden, K.C. Disruption of forkhead transcription factor (FOXO) family members in mice reveals their functional diversification. Proc. Natl. Acad. Sci. USA 2004, 101, 2975–2980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Z.; Ren, L.; Wu, D.; Yang, X.; Zhou, Z.; Nie, Q.; Jiang, G.; Xue, S.; Weng, W.; Qiu, Y. Overexpression of FoxO3a is associated with glioblastoma progression and predicts poor patient prognosis. Int. J. Cancer 2017, 140, 2792–2804. [Google Scholar] [CrossRef] [Green Version]
- Santo, E.E.; Stroeken, P.; Sluis, P.V.; Koster, J.; Versteeg, R.; Westerhout, E.M. FOXO3a is a major target of inactivation by PI3K/AKT signaling in aggressive neuroblastoma. Cancer Res. 2013, 73, 2189–2198. [Google Scholar] [CrossRef]
- Su, B.; Liu, H.; Wang, X.; Chen, S.G.; Siedlak, S.L.; Kondo, E.; Choi, R.; Takeda, A.; Castellani, R.J.; Perry, G.; et al. Ectopic localization of FOXO3a protein in Lewy bodies in Lewy body dementia and Parkinson’s disease. Mol. Neurodegener. 2009, 4, 32. [Google Scholar] [CrossRef]
- Wong, H.K.; Veremeyko, T.; Patel, N.; Lemere, C.A.; Walsh, D.M.; Esau, C.; Vanderburg, C.; Krichevsky, A.M. De-repression of FOXO3a death axis by microRNA-132 and -212 causes neuronal apoptosis in Alzheimer’s disease. Hum. Mol. Genet. 2013, 22, 3077–3092. [Google Scholar] [CrossRef] [PubMed]
- Martins, R.; Lithgow, G.J.; Link, W. Long live FOXO: Unraveling the role of FOXO proteins in aging and longevity. Aging Cell 2016, 15, 196–207. [Google Scholar] [CrossRef]
- Willcox, B.J.; Donlon, T.A.; He, Q.; Chen, R.; Grove, J.S.; Yano, K.; Masaki, K.H.; Willcox, D.C.; Rodriguez, B.; Curb, J.D. FOXO3A genotype is strongly associated with human longevity. Proc. Natl. Acad. Sci. USA 2008, 105, 13987–13992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medema, R.H.; Kops, G.J.; Bos, J.L.; Burgering, B.M. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 2000, 404, 782–787. [Google Scholar] [CrossRef]
- Le, H.V.; Shen, L.; Anderson, S.A.; Massagué, J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell 2004, 117, 211–223. [Google Scholar]
- Tran, H.; Brunet, A.; Grenier, J.M.; Datta, S.R.; Fornace, A.J.; DiStefano, P.S.; Chiang, L.W.; Greenberg, M.E. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science 2002, 296, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Lim, S.W.; Jin, L.; Luo, K.; Jin, J.; Shin, Y.J.; Hong, S.Y.; Yang, C.W. Klotho enhances FoxO3-mediated manganese superoxide dismutase expression by negatively regulating PI3K/AKT pathway during tacrolimus-induced oxidative stress. Cell Death Dis. 2017, 8, e2972. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yu, T.; Huang, P. Post-translational modifications of FOXO family proteins (Review). Mol. Med. Rep. 2016, 14, 4931–4941. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hu, S.; Liu, L. Phosphorylation and acetylation modifications of FOXO3a: Independently or synergistically? Oncol. Lett. 2017, 13, 2867–2872. [Google Scholar] [CrossRef]
- Calnan, D.R.; Brunet, A. The FoxO code. Oncogene 2008, 27, 2276–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef] [PubMed]
- Scott, P.H.; Brunn, G.J.; Kohn, A.D.; Roth, R.A.; Lawrence, J.C. Evidence of insulin-stimulated phosphorylation and activation of the mammalian target of rapamycin mediated by a protein kinase B signaling pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 7772–7777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgering, B.M.; Kops, G.J. Cell cycle and death control: Long live Forkheads. Trends Biochem. Sci. 2002, 27, 352–360. [Google Scholar] [CrossRef]
- Vanfleteren, J.R.; Braeckman, B.P. Mechanisms of life span determination in Caenorhabditis elegans. Neurobiol. Aging 1999, 20, 487–502. [Google Scholar] [CrossRef]
- Brunet, A.; Park, J.; Tran, H.; Hu, L.S.; Hemmings, B.A.; Greenberg, M.E. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol. Cell Biol. 2001, 21, 952–965. [Google Scholar] [CrossRef] [PubMed]
- Ono, H.; Katagiri, H.; Funaki, M.; Anai, M.; Inukai, K.; Fukushima, Y.; Sakoda, H.; Ogihara, T.; Onishi, Y.; Fujishiro, M.; et al. Regulation of phosphoinositide metabolism, Akt phosphorylation, and glucose transport by PTEN (phosphatase and tensin homolog deleted on chromosome 10) in 3T3-L1 adipocytes. Mol. Endocrinol. 2001, 15, 1411–1422. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Oskoui, P.R.; Banko, M.R.; Maniar, J.M.; Gygi, M.P.; Gygi, S.P.; Brunet, A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J. Biol. Chem. 2007, 282, 30107–30119. [Google Scholar] [CrossRef]
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 2016, 26, 190–201. [Google Scholar] [CrossRef]
- Stapleton, D.; Mitchelhill, K.I.; Gao, G.; Widmer, J.; Michell, B.J.; Teh, T.; House, C.M.; Fernandez, C.S.; Cox, T.; Witters, L.A.; et al. Mammalian AMP-activated protein kinase subfamily. J. Biol. Chem. 1996, 271, 611–614. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Sengupta, S.; Peterson, T.R.; Sabatini, D.M. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol. Cell 2010, 40, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Chiacchiera, F.; Simone, C. The AMPK-FoxO3A axis as a target for cancer treatment. Cell Cycle 2010, 9, 1091–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Marshall, C.B.; Yamamoto, K.; Li, G.Y.; Gasmi-Seabrook, G.M.; Okada, H.; Mak, T.W.; Ikura, M. Structures of KIX domain of CBP in complex with two FOXO3a transactivation domains reveal promiscuity and plasticity in coactivator recruitment. Proc. Natl. Acad. Sci. USA 2012, 109, 6078–6083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiota, M.; Yokomizo, A.; Kashiwagi, E.; Tada, Y.; Inokuchi, J.; Tatsugami, K.; Kuroiwa, K.; Uchiumi, T.; Seki, N.; Naito, S. Foxo3a expression and acetylation regulate cancer cell growth and sensitivity to cisplatin. Cancer Sci. 2010, 101, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Celestini, V.; Tezil, T.; Russo, L.; Fasano, C.; Sanese, P.; Forte, G.; Peserico, A.; Lepore Signorile, M.; Longo, G.; De Rasmo, D.; et al. Uncoupling FoxO3A mitochondrial and nuclear functions in cancer cells undergoing metabolic stress and chemotherapy. Cell Death Dis. 2018, 9, 231. [Google Scholar] [CrossRef]
- Bowman, C.J.; Ayer, D.E.; Dynlacht, B.D. Foxk proteins repress the initiation of starvation-induced atrophy and autophagy programs. Nat. Cell Biol. 2014, 16, 1202–1214. [Google Scholar] [CrossRef]
- Ma, D.; Li, S.; Molusky, M.M.; Lin, J.D. Circadian autophagy rhythm: A link between clock and metabolism? Trends Endocrinol. Metab. 2012, 23, 319–325. [Google Scholar] [CrossRef]
- Chaves, I.; van der Horst, G.T.; Schellevis, R.; Nijman, R.M.; Koerkamp, M.G.; Holstege, F.C.; Smidt, M.P.; Hoekman, M.F. Insulin-FOXO3 signaling modulates circadian rhythms via regulation of clock transcription. Curr. Biol. 2014, 24, 1248–1255. [Google Scholar] [CrossRef]
- Olsen, R.K.; Cornelius, N.; Gregersen, N. Redox signalling and mitochondrial stress responses; lessons from inborn errors of metabolism. J. Inherit. Metab. Dis. 2015, 38, 703–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Lassègue, B.; Martín, A.S.; Griendling, K.K. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res. 2012, 110, 1364–1390. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Näpänkangas, J.P.; Liimatta, E.V.; Joensuu, P.; Bergmann, U.; Ylitalo, K.; Hassinen, I.E. Superoxide production during ischemia-reperfusion in the perfused rat heart: A comparison of two methods of measurement. J. Mol. Cell Cardiol. 2012, 53, 906–915. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef]
- Murata, H.; Ihara, Y.; Nakamura, H.; Yodoi, J.; Sumikawa, K.; Kondo, T. Glutaredoxin exerts an antiapoptotic effect by regulating the redox state of Akt. J. Biol. Chem. 2003, 278, 50226–50233. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [PubMed]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar] [CrossRef] [PubMed]
- Keshet, Y.; Seger, R. The MAP kinase signaling cascades: A system of hundreds of components regulates a diverse array of physiological functions. Methods Mol. Biol. 2010, 661, 3–38. [Google Scholar] [PubMed]
- Yang, J.Y.; Zong, C.S.; Xia, W.; Yamaguchi, H.; Ding, Q.; Xie, X.; Lang, J.Y.; Lai, C.C.; Chang, C.J.; Huang, W.C.; et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell Biol. 2008, 10, 138–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunters, A.; Madureira, P.A.; Pomeranz, K.M.; Aubert, M.; Brosens, J.J.; Cook, S.J.; Burgering, B.M.; Coombes, R.C.; Lam, E.W. Paclitaxel-induced nuclear translocation of FOXO3a in breast cancer cells is mediated by c-Jun NH2-terminal kinase and Akt. Cancer Res. 2006, 66, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.M.; Park, S.H.; Tsai, W.B.; Wang, S.Y.; Ikeda, M.A.; Berek, J.S.; Chen, D.J.; Hu, M.C. FOXO3 signalling links ATM to the p53 apoptotic pathway following DNA damage. Nat. Commun. 2012, 3, 1000. [Google Scholar] [CrossRef] [PubMed]
- Ferber, E.C.; Peck, B.; Delpuech, O.; Bell, G.P.; East, P.; Schulze, A. FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression. Cell Death Differ. 2012, 19, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.K.; McGuire, V.A.; Koo, C.Y.; Muir, K.W.; de Olano, N.; Maifoshie, E.; Kelly, D.J.; McGovern, U.B.; Monteiro, L.J.; Gomes, A.R.; et al. Phosphorylation of FOXO3a on Ser-7 by p38 promotes its nuclear localization in response to doxorubicin. J. Biol. Chem. 2012, 287, 1545–1555. [Google Scholar] [CrossRef] [PubMed]
- Choudhry, H.; Harris, A.L. Advances in Hypoxia-Inducible Factor Biology. Cell Metab. 2018, 27, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Ratcliffe, P.J.; O’Rourke, J.F.; Maxwell, P.H.; Pugh, C.W. Oxygen sensing, hypoxia-inducible factor-1 and the regulation of mammalian gene expression. J. Exp. Biol. 1998, 201, 1153–1162. [Google Scholar]
- Fong, G.H.; Takeda, K. Role and regulation of prolyl hydroxylase domain proteins. Cell Death Differ. 2008, 15, 635–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.T.; Nurse, C.A. Induction of HIF-2alpha is dependent on mitochondrial O2 consumption in an O2-sensitive adrenomedullary chromaffin cell line. Am. J. Physiol. Cell Physiol. 2008, 294, C1305–C1312. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; Mack, N.J.; Ahmad, N. The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox Signal. 2018, 28, 643–661. [Google Scholar] [CrossRef] [PubMed]
- Samarin, J.; Wessel, J.; Cicha, I.; Kroening, S.; Warnecke, C.; Goppelt-Struebe, M. FoxO proteins mediate hypoxic induction of connective tissue growth factor in endothelial cells. J. Biol. Chem. 2010, 285, 4328–4336. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhao, Y.; Xu, M.; Yu, L.; Chen, J.; Yuan, Y.; Zheng, Q.; Niu, X. FoxO3a modulates hypoxia stress induced oxidative stress and apoptosis in cardiac microvascular endothelial cells. PLoS ONE 2013, 8, e80342. [Google Scholar]
- Jensen, K.S.; Binderup, T.; Jensen, K.T.; Therkelsen, I.; Borup, R.; Nilsson, E.; Multhaupt, H.; Bouchard, C.; Quistorff, B.; Kjaer, A.; et al. FoxO3A promotes metabolic adaptation to hypoxia by antagonizing Myc function. EMBO J. 2011, 30, 4554–4570. [Google Scholar] [CrossRef] [PubMed]
- Bakker, W.J.; IHarris, S.; Mak, T.W. FOXO3a is activated in response to hypoxic stress and inhibits HIF1-induced apoptosis via regulation of CITED2. Mol. Cell 2007, 28, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhai, B.; Koivunen, P.; Shin, S.J.; Lu, G.; Liu, J.; Geisen, C.; Chakraborty, A.A.; Moslehi, J.J.; Smalley, D.M.; et al. Prolyl hydroxylation by EglN2 destabilizes FOXO3a by blocking its interaction with the USP9x deubiquitinase. Genes Dev. 2014, 28, 1429–1444. [Google Scholar] [CrossRef] [Green Version]
- Emerling, B.M.; Weinberg, F.; Liu, J.L.; Mak, T.W.; Chandel, N.S. PTEN regulates p300-dependent hypoxia-inducible factor 1 transcriptional activity through Forkhead transcription factor 3a (FOXO3a). Proc. Natl. Acad. Sci. USA 2008, 105, 2622–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funk, R.H.; Nagel, F.; Wonka, F.; Krinke, H.E.; Gölfert, F.; Hofer, A. Effects of heat shock on the functional morphology of cell organelles observed by video-enhanced microscopy. Anat. Rec. 1999, 255, 458–464. [Google Scholar] [CrossRef]
- Gavrilova, L.P.; Korpacheva, I.I.; Semushina, S.G.; Iashin, V.A. [Heat shock induces simultaneous rearrangements of all known cytoskeletal filaments in normal interphase fibroblasts]. Tsitologiia 2012, 54, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.T.; Morange, M.; Bensaude, O. Protein denaturation during heat shock and related stress. Escherichia coli beta-galactosidase and Photinus pyralis luciferase inactivation in mouse cells. J. Biol. Chem. 1989, 264, 10487–10492. [Google Scholar] [PubMed]
- Su, Z.; Tang, Y.; Ritchey, L.E.; Tack, D.C.; Zhu, M.; Bevilacqua, P.C.; Assmann, S.M. Genome-wide RNA structurome reprogramming by acute heat shock globally regulates mRNA abundance. Proc. Natl. Acad. Sci. USA 2018, 115, 12170–12175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velichko, A.K.; Petrova, N.V.; Kantidze, O.L.; Razin, S.V. Dual effect of heat shock on DNA replication and genome integrity. Mol. Biol. Cell 2012, 23, 3450–3460. [Google Scholar] [CrossRef]
- Li, J.; Labbadia, J.; Morimoto, R.I. Rethinking HSF1 in Stress, Development, and Organismal Health. Trends Cell Biol. 2017, 27, 895–905. [Google Scholar] [CrossRef]
- Jaeger, A.M.; Makley, L.N.; Gestwicki, J.E.; Thiele, D.J. Genomic heat shock element sequences drive cooperative human heat shock factor 1 DNA binding and selectivity. J. Biol. Chem. 2014, 289, 30459–30469. [Google Scholar] [CrossRef]
- Grossi, V.; Forte, G.; Sanese, P.; Peserico, A.; Tezil, T.; Lepore Signorile, M.; Fasano, C.; Lovaglio, R.; Bagnulo, R.; Loconte, D.C.; et al. The longevity SNP rs2802292 uncovered: HSF1 activates stress-dependent expression of FOXO3 through an intronic enhancer. Nucleic Acids Res. 2018, 46, 5587–5600. [Google Scholar] [CrossRef] [Green Version]
- Dayalan Naidu, S.; Sutherland, C.; Zhang, Y.; Risco, A.; de la Vega, L.; Caunt, C.J.; Hastie, C.J.; Lamont, D.J.; Torrente, L.; Chowdhry, S.; et al. Heat Shock Factor 1 Is a Substrate for p38 Mitogen-Activated Protein Kinases. Mol. Cell Biol. 2016, 36, 2403–2417. [Google Scholar] [CrossRef] [Green Version]
- Murphy, C.T.; McCarroll, S.A.; Bargmann, C.I.; Fraser, A.; Kamath, R.S.; Ahringer, J.; Li, H.; Kenyon, C. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 2003, 424, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Hsu, A.L.; Murphy, C.T.; Kenyon, C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 2003, 300, 1142–1145. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, T.; Kobayashi, H.; Kakigi, R.; Sugiura, T.; Naito, H. Heat stress-induced phosphorylation of FoxO3a signalling in rat skeletal muscle. Acta Physiol. 2016, 218, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Ma, J.; Li, T.; Yan, L. Up-regulation of miR-122 protects against neuronal cell death in ischemic stroke through the heat shock protein 70-dependent NF-κB pathway by targeting FOXO3. Exp. Cell Res. 2018, 369, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Skurk, C.; Maatz, H.; Shiojima, I.; Ivashchenko, Y.; Yoon, S.W.; Park, Y.B.; Walsh, K. Akt/FOXO3a signaling modulates the endothelial stress response through regulation of heat shock protein 70 expression. FASEB J. 2005, 19, 1042–1044. [Google Scholar] [CrossRef] [PubMed]
- Houtgraaf, J.H.; Versmissen, J.; van der Giessen, W.J. A concise review of DNA damage checkpoints and repair in mammalian cells. Cardiovasc. Revasc. Med. 2006, 7, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Nestal de Moraes, G.; Bella, L.; Zona, S.; Burton, M.J.; Lam, E.W. Insights into a Critical Role of the FOXO3a-FOXM1 Axis in DNA Damage Response and Genotoxic Drug Resistance. Curr. Drug Targets 2016, 17, 164–177. [Google Scholar] [CrossRef] [Green Version]
- Tsai, W.B.; Chung, Y.M.; Takahashi, Y.; Xu, Z.; Hu, M.C. Functional interaction between FOXO3a and ATM regulates DNA damage response. Nat. Cell Biol. 2008, 10, 460–467. [Google Scholar] [CrossRef] [Green Version]
- Calnan, D.R.; Webb, A.E.; White, J.L.; Stowe, T.R.; Goswami, T.; Shi, X.; Espejo, A.; Bedford, M.T.; Gozani, O.; Gygi, S.P.; et al. Methylation by Set9 modulates FoxO3 stability and transcriptional activity. Aging 2012, 4, 462–479. [Google Scholar] [CrossRef] [Green Version]
- Clark, K.L.; Halay, E.D.; Lai, E.; Burley, S.K. Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nature 1993, 364, 412–420. [Google Scholar] [CrossRef]
- Pierrou, S.; Hellqvist, M.; Samuelsson, L.; Enerbäck, S.; Carlsson, P. Cloning and characterization of seven human forkhead proteins: Binding site specificity and DNA bending. EMBO J. 1994, 13, 5002–5012. [Google Scholar] [CrossRef] [PubMed]
- So, C.W.; Cleary, M.L. MLL-AFX requires the transcriptional effector domains of AFX to transform myeloid progenitors and transdominantly interfere with forkhead protein function. Mol. Cell Biol. 2002, 22, 6542–6552. [Google Scholar] [CrossRef] [PubMed]
- So, C.W.; Cleary, M.L. Common mechanism for oncogenic activation of MLL by forkhead family proteins. Blood 2003, 101, 633–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuyama, T.; Nakazawa, T.; Nakano, I.; Mori, N. Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-16 homologues. Biochem. J. 2000, 349, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Xuan, Z.; Zhang, M.Q. From worm to human: Bioinformatics approaches to identify FOXO target genes. Mech. Ageing Dev. 2005, 126, 209–215. [Google Scholar] [CrossRef]
- Eijkelenboom, A.; Mokry, M.; de Wit, E.; Smits, L.M.; Polderman, P.E.; van Triest, M.H.; van Boxtel, R.; Schulze, A.; de Laat, W.; Cuppen, E.; et al. Genome-wide analysis of FOXO3 mediated transcription regulation through RNA polymerase II profiling. Mol. Syst. Biol. 2013, 9, 638. [Google Scholar] [CrossRef]
- Eijkelenboom, A.; Mokry, M.; Smits, L.M.; Nieuwenhuis, E.E.; Burgering, B.M. FOXO3 selectively amplifies enhancer activity to establish target gene regulation. Cell Rep. 2013, 5, 1664–1678. [Google Scholar] [CrossRef]
- Donlon, T.A.; Curb, J.D.; He, Q.; Grove, J.S.; Masaki, K.H.; Rodriguez, B.; Elliott, A.; Willcox, D.C.; Willcox, B.J. FOXO3 gene variants and human aging: Coding variants may not be key players. J. Gerontol. A Biol. Sci. Med. Sci. 2012, 67, 1132–1139. [Google Scholar] [CrossRef]
- Forte, G.; Grossi, V.; Celestini, V.; Lucisano, G.; Scardapane, M.; Varvara, D.; Patruno, M.; Bagnulo, R.; Loconte, D.; Giunti, L.; et al. Characterization of the rs2802292 SNP identifies FOXO3A as a modifier locus predicting cancer risk in patients with PJS and PHTS hamartomatous polyposis syndromes. BMC Cancer 2014, 14, 661. [Google Scholar] [CrossRef]
- Banasik, K.; Ribel-Madsen, R.; Gjesing, A.P.; Wegner, L.; Andersson, A.; Poulsen, P.; Borglykke, A.; Witte, D.R.; Pedersen, O.; Hansen, T.; et al. The FOXO3A rs2802292 G-allele associates with improved peripheral and hepatic insulin sensitivity and increased skeletal muscle-FOXO3A mRNA expression in twins. J. Clin. Endocrinol. Metab. 2011, 96, E119–E124. [Google Scholar] [CrossRef]
- Donlon, T.A.; Morris, B.J.; Chen, R.; Masaki, K.H.; Allsopp, R.C.; Willcox, D.C.; Elliott, A.; Willcox, B.J. FOXO3 longevity interactome on chromosome 6. Aging Cell 2017, 16, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Hall, R.K.; Yamasaki, T.; Kucera, T.; Waltner-Law, M.; O’Brien, R.; Granner, D.K. Regulation of phosphoenolpyruvate carboxykinase and insulin-like growth factor-binding protein-1 gene expression by insulin. The role of winged helix/forkhead proteins. J. Biol. Chem. 2000, 275, 30169–30175. [Google Scholar] [CrossRef] [PubMed]
- Onuma, H.; Vander Kooi, B.T.; Boustead, J.N.; Oeser, J.K.; O’Brien, R.M. Correlation between FOXO1a (FKHR) and FOXO3a (FKHRL1) binding and the inhibition of basal glucose-6-phosphatase catalytic subunit gene transcription by insulin. Mol. Endocrinol. 2006, 20, 2831–2847. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Li, L.; Qi, Y.; Zhu, X.; Gan, B.; DePinho, R.A.; Averitt, T.; Guo, S. Hepatic suppression of Foxo1 and Foxo3 causes hypoglycemia and hyperlipidemia in mice. Endocrinology 2012, 153, 631–646. [Google Scholar] [CrossRef] [PubMed]
- Bertaggia, E.; Coletto, L.; Sandri, M. Posttranslational modifications control FoxO3 activity during denervation. Am. J. Physiol. Cell Physiol. 2012, 302, C587–C596. [Google Scholar] [CrossRef] [PubMed]
- Gai, J.; Ji, M.; Shi, C.; Li, W.; Chen, S.; Wang, Y.; Li, H. FoxO regulates expression of ABCA6, an intracellular ATP-binding-cassette transporter responsive to cholesterol. Int. J. Biochem. Cell Biol. 2013, 45, 2651–2659. [Google Scholar] [CrossRef]
- Bollinger, L.M.; Witczak, C.A.; Houmard, J.A.; Brault, J.J. SMAD3 augments FoxO3-induced MuRF-1 promoter activity in a DNA-binding-dependent manner. Am. J. Physiol. Cell Physiol. 2014, 307, C278–C287. [Google Scholar] [CrossRef]
- Kang, S.H.; Lee, H.A.; Kim, M.; Lee, E.; Sohn, U.D.; Kim, I. Forkhead box O3 plays a role in skeletal muscle atrophy through expression of E3 ubiquitin ligases MuRF-1 and atrogin-1 in Cushing’s syndrome. Am. J. Physiol. Endocrinol. Metab. 2017, 312, E495–E507. [Google Scholar] [CrossRef]
- Waddell, D.S.; Baehr, L.M.; van den Brandt, J.; Johnsen, S.A.; Reichardt, H.M.; Furlow, J.D.; Bodine, S.C. The glucocorticoid receptor and FOXO1 synergistically activate the skeletal muscle atrophy-associated MuRF1 gene. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E785–E797. [Google Scholar] [CrossRef]
- Kops, G.J.; Dansen, T.B.; Polderman, P.E.; Saarloos, I.; Wirtz, K.W.; Coffer, P.J.; Huang, T.T.; Bos, J.L.; Medema, R.H.; Burgering, B.M. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 2002, 419, 316–321. [Google Scholar] [CrossRef]
- Nemoto, S.; Finkel, T. Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science 2002, 295, 2450–2452. [Google Scholar] [CrossRef] [PubMed]
- Olmos, Y.; Valle, I.; Borniquel, S.; Tierrez, A.; Soria, E.; Lamas, S.; Monsalve, M. Mutual dependence of Foxo3a and PGC-1alpha in the induction of oxidative stress genes. J. Biol. Chem. 2009, 284, 14476–14484. [Google Scholar] [CrossRef]
- Brunelle, J.K.; Santore, M.T.; Budinger, G.R.; Tang, Y.; Barrett, T.A.; Zong, W.X.; Kandel, E.; Keith, B.; Simon, M.C.; Thompson, C.B.; et al. c-Myc sensitization to oxygen deprivation-induced cell death is dependent on Bax/Bak, but is independent of p53 and hypoxia-inducible factor-1. J. Biol. Chem. 2004, 279, 4305–4312. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Tindall, D.J. FOXOs, cancer and regulation of apoptosis. Oncogene 2008, 27, 2312–2319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.W.; Chen, P.M.; Hsieh, Y.H.; Weng, C.C.; Chang, C.W.; Yao, C.C.; Hu, L.Y.; Wu, P.E.; Shen, C.Y. Foxo3a-mediated overexpression of microRNA-622 suppresses tumor metastasis by repressing hypoxia-inducible factor-1α in ERK-responsive lung cancer. Oncotarget 2015, 6, 44222–44238. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, S.; Zhang, X.; Luciano, J.P.; Mungamuri, S.K.; Marinkovic, D.; Vercherat, C.; Sarkar, A.; Grisotto, M.; Taneja, R.; Ghaffari, S. Foxo3 is essential for the regulation of ataxia telangiectasia mutated and oxidative stress-mediated homeostasis of hematopoietic stem cells. J. Biol. Chem. 2008, 283, 25692–25705. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Marshall, C.B.; Yamamoto, K.; Li, G.Y.; Plevin, M.J.; You, H.; Mak, T.W.; Ikura, M. Biochemical and structural characterization of an intramolecular interaction in FOXO3a and its binding with p53. J. Mol. Biol. 2008, 384, 590–603. [Google Scholar] [CrossRef]
- Chandramohan, V.; Jeay, S.; Pianetti, S.; Sonenshein, G.E. Reciprocal control of Forkhead box O 3a and c-Myc via the phosphatidylinositol 3-kinase pathway coordinately regulates p27Kip1 levels. J. Immunol. 2004, 172, 5522–5527. [Google Scholar] [CrossRef]
- Lettieri Barbato, D.; Aquilano, K.; Baldelli, S.; Cannata, S.M.; Bernardini, S.; Rotilio, G.; Ciriolo, M.R. Proline oxidase-adipose triglyceride lipase pathway restrains adipose cell death and tissue inflammation. Cell Death Differ. 2014, 21, 113–123. [Google Scholar] [CrossRef]
- Obexer, P.; Geiger, K.; Ambros, P.F.; Meister, B.; Ausserlechner, M.J. FKHRL1-mediated expression of Noxa and Bim induces apoptosis via the mitochondria in neuroblastoma cells. Cell Death Differ. 2007, 14, 534–547. [Google Scholar] [CrossRef]
- Chapuis, N.; Park, S.; Leotoing, L.; Tamburini, J.; Verdier, F.; Bardet, V.; Green, A.S.; Willems, L.; Agou, F.; Ifrah, N.; et al. IκB kinase overcomes PI3K/Akt and ERK/MAPK to control FOXO3a activity in acute myeloid leukemia. Blood 2010, 116, 4240–4250. [Google Scholar] [CrossRef] [PubMed]
- Modur, V.; Nagarajan, R.; Evers, B.M.; Milbrandt, J. FOXO proteins regulate tumor necrosis factor-related apoptosis inducing ligand expression. Implications for PTEN mutation in prostate cancer. J. Biol. Chem. 2002, 277, 47928–47937. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Pellegrini, M.; Tsuchihara, K.; Yamamoto, K.; Hacker, G.; Erlacher, M.; Villunger, A.; Mak, T.W. FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J. Exp. Med. 2006, 203, 1657–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greer, E.L.; Dowlatshahi, D.; Banko, M.R.; Villen, J.; Hoang, K.; Blanchard, D.; Gygi, S.P.; Brunet, A. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr. Biol. 2007, 17, 1646–1656. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, K.M.; Pennington, J.D.; Bisht, K.S.; Aykin-Burns, N.; Kim, H.S.; Mishra, M.; Sun, L.; Nguyen, P.; Ahn, B.H.; Leclerc, J.; et al. SIRT3 interacts with the daf-16 homolog FOXO3a in the mitochondria, as well as increases FOXO3a dependent gene expression. Int. J. Biol. Sci. 2008, 4, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Lombard, D.B.; Alt, F.W.; Cheng, H.L.; Bunkenborg, J.; Streeper, R.S.; Mostoslavsky, R.; Kim, J.; Yancopoulos, G.; Valenzuela, D.; Murphy, A.; et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell Biol. 2007, 27, 8807–8814. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Patel, K.; Muldoon-Jacobs, K.; Bisht, K.S.; Aykin-Burns, N.; Pennington, J.D.; van der Meer, R.; Nguyen, P.; Savage, J.; Owens, K.M.; et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 2010, 17, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Peserico, A.; Chiacchiera, F.; Grossi, V.; Matrone, A.; Latorre, D.; Simonatto, M.; Fusella, A.; Ryall, J.G.; Finley, L.W.; Haigis, M.C.; et al. A novel AMPK-dependent FoxO3A-SIRT3 intramitochondrial complex sensing glucose levels. Cell Mol. Life Sci. 2013, 70, 2015–2029. [Google Scholar] [CrossRef] [PubMed]
- Oakhill, J.S.; Steel, R.; Chen, Z.P.; Scott, J.W.; Ling, N.; Tam, S.; Kemp, B.E. AMPK is a direct adenylate charge-regulated protein kinase. Science 2011, 332, 1433–1435. [Google Scholar] [CrossRef] [PubMed]
- Grossi, V.; Fasano, C.; Celestini, V.; Lepore Signorile, M.; Sanese, P.; Simone, C. Chasing the FOXO3: Insights into Its New Mitochondrial Lair in Colorectal Cancer Landscape. Cancers 2019, 11, 414. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Ke, R.; Xu, Q.; Li, C.; Luo, L.; Huang, D. Mechanisms of AMPK in the maintenance of ATP balance during energy metabolism. Cell Biol. Int. 2018, 42, 384–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gakh, O.; Cavadini, P.; Isaya, G. Mitochondrial processing peptidases. Biochim. Biophys. Acta 2002, 1592, 63–77. [Google Scholar] [CrossRef] [Green Version]
- Klammer, M.; Kaminski, M.; Zedler, A.; Oppermann, F.; Blencke, S.; Marx, S.; Müller, S.; Tebbe, A.; Godl, K.; Schaab, C. Phosphosignature predicts dasatinib response in non-small cell lung cancer. Mol. Cell Proteom. 2012, 11, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertins, P.; Yang, F.; Liu, T.; Mani, D.R.; Petyuk, V.A.; Gillette, M.A.; Clauser, K.R.; Qiao, J.W.; Gritsenko, M.A.; Moore, R.J.; et al. Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol. Cell Proteom. 2014, 13, 1690–1704. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.V.; Vermeulen, M.; Santamaria, A.; Kumar, C.; Miller, M.L.; Jensen, L.J.; Gnad, F.; Cox, J.; Jensen, T.S.; Nigg, E.A.; et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 2010, 3, ra3. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Olsen, J.V.; Daub, H.; Mann, M. Global effects of kinase inhibitors on signaling networks revealed by quantitative phosphoproteomics. Mol. Cell Proteom. 2009, 8, 2796–2808. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; D’Souza, R.C.; Tyanova, S.; Schaab, C.; Wiśniewski, J.R.; Cox, J.; Mann, M. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 2014, 8, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Gurkar, A.U.; Robinson, A.R.; Cui, Y.; Li, X.; Allani, S.K.; Webster, A.; Muravia, M.; Fallahi, M.; Weissbach, H.; Robbins, P.D.; et al. Dysregulation of DAF-16/FOXO3A-mediated stress responses accelerates oxidative DNA damage induced aging. Redox Biol. 2018, 18, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Roncato, R.; Cecchin, E.; Montico, M.; De Mattia, E.; Giodini, L.; Buonadonna, A.; Solfrini, V.; Innocenti, F.; Toffoli, G. Cost Evaluation of Irinotecan-Related Toxicities Associated with the UGT1A1*28 Patient Genotype. Clin. Pharmacol. Ther. 2017, 102, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Rancoule, C.; Guy, J.B.; Vallard, A.; Ben Mrad, M.; Rehailia, A.; Magné, N. 50th anniversary of cisplatin. Bull. Cancer 2017, 104, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Tamemasa, O.; Tezuka, M. Antitumor effect of 5-fluorouracil in combination with some additives which stimulate the formation of antineoplastic 5-fluorouracil nucleotides. Gan 1983, 74, 285–290. [Google Scholar] [PubMed]
- McClendon, A.K.; Osheroff, N. DNA topoisomerase II, genotoxicity, and cancer. Mutat. Res. 2007, 623, 83–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, F.; Dai, C.; Xie, J.; Hu, X. Biochemical issues in estimation of cytosolic free NAD/NADH ratio. PLoS ONE 2012, 7, e34525. [Google Scholar] [CrossRef] [PubMed]
- Golinska, M.; Troy, H.; Chung, Y.L.; McSheehy, P.M.; Mayr, M.; Yin, X.; Ly, L.; Williams, K.J.; Airley, R.E.; Harris, A.L.; et al. Adaptation to HIF-1 deficiency by upregulation of the AMP/ATP ratio and phosphofructokinase activation in hepatomas. BMC Cancer 2011, 11, 198. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, A.; Loh, K.; Galic, S.; Fam, B.; Shields, B.; Wiede, F.; Tremblay, M.L.; Watt, M.J.; Andrikopoulos, S.; Tiganis, T. T-cell protein tyrosine phosphatase attenuates STAT3 and insulin signaling in the liver to regulate gluconeogenesis. Diabetes 2010, 59, 1906–1914. [Google Scholar] [CrossRef] [PubMed]
- Kume, S.; Uzu, T.; Horiike, K.; Chin-Kanasaki, M.; Isshiki, K.; Araki, S.; Sugimoto, T.; Haneda, M.; Kashiwagi, A.; Koya, D. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J. Clin. Investig. 2010, 120, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Ney, P.A. Mitochondrial autophagy: Origins, significance, and role of BNIP3 and NIX. Biochim. Biophys. Acta 2015, 1853, 2775–2783. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Long, B.; Jiao, J.Q.; Wang, J.X.; Liu, J.P.; Li, Q.; Li, P.F. miR-484 regulates mitochondrial network through targeting Fis1. Nat. Commun. 2012, 3, 781. [Google Scholar] [CrossRef] [PubMed]
- Colombo, S.L.; Moncada, S. AMPKalpha1 regulates the antioxidant status of vascular endothelial cells. Biochem. J. 2009, 421, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Furukawa-Hibi, Y.; Chen, C.; Horio, Y.; Isobe, K.; Ikeda, K.; Motoyama, N. SIRT1 is critical regulator of FOXO-mediated transcription in response to oxidative stress. Int. J. Mol. Med. 2005, 16, 237–243. [Google Scholar] [CrossRef]
- Cullen, J.J.; Weydert, C.; Hinkhouse, M.M.; Ritchie, J.; Domann, F.E.; Spitz, D.; Oberley, L.W. The role of manganese superoxide dismutase in the growth of pancreatic adenocarcinoma. Cancer Res. 2003, 63, 1297–1303. [Google Scholar]
- Oberley, L.W. Mechanism of the tumor suppressive effect of MnSOD overexpression. Biomed. Pharmacother. 2005, 59, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Smith, B.J.; Oberley, L.W. Enzymatic activity is necessary for the tumor-suppressive effects of MnSOD. Antioxid. Redox Signal. 2006, 8, 1283–1293. [Google Scholar] [CrossRef]
- Delpuech, O.; Griffiths, B.; East, P.; Essafi, A.; Lam, E.W.; Burgering, B.; Downward, J.; Schulze, A. Induction of Mxi1-SR alpha by FOXO3a contributes to repression of Myc-dependent gene expression. Mol. Cell Biol. 2007, 27, 4917–4930. [Google Scholar] [CrossRef]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef]
- Anderson, R.; Prolla, T. PGC-1alpha in aging and anti-aging interventions. Biochim. Biophys. Acta 2009, 1790, 1059–1066. [Google Scholar] [CrossRef]
- Ura, S.; Masuyama, N.; Graves, J.D.; Gotoh, Y. MST1-JNK promotes apoptosis via caspase-dependent and independent pathways. Genes Cells 2001, 6, 519–530. [Google Scholar] [CrossRef]
- Lehtinen, M.K.; Yuan, Z.; Boag, P.R.; Yang, Y.; Villén, J.; Becker, E.B.; DiBacco, S.; de la Iglesia, N.; Gygi, S.; Blackwell, T.K.; et al. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell 2006, 125, 987–1001. [Google Scholar] [CrossRef] [PubMed]
- Finley, L.W.; Haigis, M.C. The coordination of nuclear and mitochondrial communication during aging and calorie restriction. Ageing Res. Rev. 2009, 8, 173–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quirós, P.M.; Mottis, A.; Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 2016, 17, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.T.; Hoogenraad, N.J. Mitochondrial-nuclear communications. Annu. Rev. Biochem. 2007, 76, 701–722. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Wang, X.L.; Tong, M.M.; Gan, L.; Chen, H.; Wu, S.S.; Chen, J.X.; Li, R.L.; Wu, Y.; Zhang, H.Y.; et al. SIRT6 protects cardiomyocytes against ischemia/reperfusion injury by augmenting FoxO3α-dependent antioxidant defense mechanisms. Basic Res. Cardiol. 2016, 111, 13. [Google Scholar] [CrossRef] [PubMed]
- Hagenbuchner, J.; Ausserlechner, M.J. Mitochondria and FOXO3: Breath or die. Front. Physiol. 2013, 4, 147. [Google Scholar] [CrossRef] [PubMed]
- Obexer, P.; Hagenbuchner, J.; Unterkircher, T.; Sachsenmaier, N.; Seifarth, C.; Böck, G.; Porto, V.; Geiger, K.; Ausserlechner, M. Repression of BIRC5/survivin by FOXO3/FKHRL1 sensitizes human neuroblastoma cells to DNA damage-induced apoptosis. Mol. Biol. Cell 2009, 20, 2041–2048. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Gao, B.; Li, N.; Wang, J.; Qiu, C.; Zhang, G.; Liu, M.; Zhang, R.; Li, C.; Ji, G.; et al. Sirt3 deficiency exacerbates diabetic cardiac dysfunction: Role of Foxo3A-Parkin-mediated mitophagy. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1973–1983. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.J.; Li, Y.F.; Liu, F.L.; Deng, J.; Wu, Y.P.; Yuan, W.L.; Tsoi, B.; Chen, J.L.; Wang, Q.; Cai, S.H.; et al. A SIRT3/AMPK/autophagy network orchestrates the protective effects of trans-resveratrol in stressed peritoneal macrophages and RAW 264.7 macrophages. Free Radic. Biol. Med. 2016, 95, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Qiao, A.; Wang, K.; Yuan, Y.; Guan, Y.; Ren, X.; Li, L.; Chen, X.; Li, F.; Chen, A.F.; Zhou, J.; et al. Correction: Sirt3-mediated mitophagy protects tumor cells against apoptosis under hypoxia. Oncotarget 2018, 9, 27318. [Google Scholar] [CrossRef]
- Pi, H.; Xu, S.; Reiter, R.J.; Guo, P.; Zhang, L.; Li, Y.; Li, M.; Cao, Z.; Tian, L.; Xie, J.; et al. SIRT3-SOD2-mROS-dependent autophagy in cadmium-induced hepatotoxicity and salvage by melatonin. Autophagy 2015, 11, 1037–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, A.H.; Shieh, S.S.; Wang, D.L. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic. Biol. Med. 2013, 63, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.Q.; Wang, K.; Lv, D.Y.; Li, P.F. Foxo3a inhibits cardiomyocyte hypertrophy through transactivating catalase. J. Biol. Chem. 2008, 283, 29730–29739. [Google Scholar] [CrossRef] [PubMed]
- Chiribau, C.B.; Cheng, L.; Cucoranu, I.C.; Yu, Y.S.; Clempus, R.E.; Sorescu, D. FOXO3A regulates peroxiredoxin III expression in human cardiac fibroblasts. J. Biol. Chem. 2008, 283, 8211–8217. [Google Scholar] [CrossRef]
- Morris, B.J.; Willcox, D.C.; Donlon, T.A.; Willcox, B.J. FOXO3: A Major Gene for Human Longevity—A Mini-Review. Gerontology 2015, 61, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Jeon, S.M.; Bhaskar, P.T.; Nogueira, V.; Sundararajan, D.; Tonic, I.; Park, Y.; Hay, N. FoxOs inhibit mTORC1 and activate Akt by inducing the expression of Sestrin3 and Rictor. Dev. Cell 2010, 18, 592–604. [Google Scholar] [CrossRef]
- Hagenbuchner, J.; Kuznetsov, A.; Hermann, M.; Hausott, B.; Obexer, P.; Ausserlechner, M.J. FOXO3-induced reactive oxygen species are regulated by BCL2L11 (Bim) and SESN3. J. Cell Sci. 2012, 125, 1191–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundaresan, N.R.; Samant, S.A.; Pillai, V.B.; Rajamohan, S.B.; Gupta, M.P. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol. Cell Biol. 2008, 28, 6384–6401. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fasano, C.; Disciglio, V.; Bertora, S.; Lepore Signorile, M.; Simone, C. FOXO3a from the Nucleus to the Mitochondria: A Round Trip in Cellular Stress Response. Cells 2019, 8, 1110. https://doi.org/10.3390/cells8091110
Fasano C, Disciglio V, Bertora S, Lepore Signorile M, Simone C. FOXO3a from the Nucleus to the Mitochondria: A Round Trip in Cellular Stress Response. Cells. 2019; 8(9):1110. https://doi.org/10.3390/cells8091110
Chicago/Turabian StyleFasano, Candida, Vittoria Disciglio, Stefania Bertora, Martina Lepore Signorile, and Cristiano Simone. 2019. "FOXO3a from the Nucleus to the Mitochondria: A Round Trip in Cellular Stress Response" Cells 8, no. 9: 1110. https://doi.org/10.3390/cells8091110
APA StyleFasano, C., Disciglio, V., Bertora, S., Lepore Signorile, M., & Simone, C. (2019). FOXO3a from the Nucleus to the Mitochondria: A Round Trip in Cellular Stress Response. Cells, 8(9), 1110. https://doi.org/10.3390/cells8091110