Hyperglycemia-Induced Aberrant Cell Proliferation; A Metabolic Challenge Mediated by Protein O-GlcNAc Modification

 , ,
, ,
Abstract
:1. Introduction
2. Diabetes and Its Effect on Cell Cycle Regulation
2.1. Metabolic Effects
2.2. Oxidative Stress and Intracellular Consequences
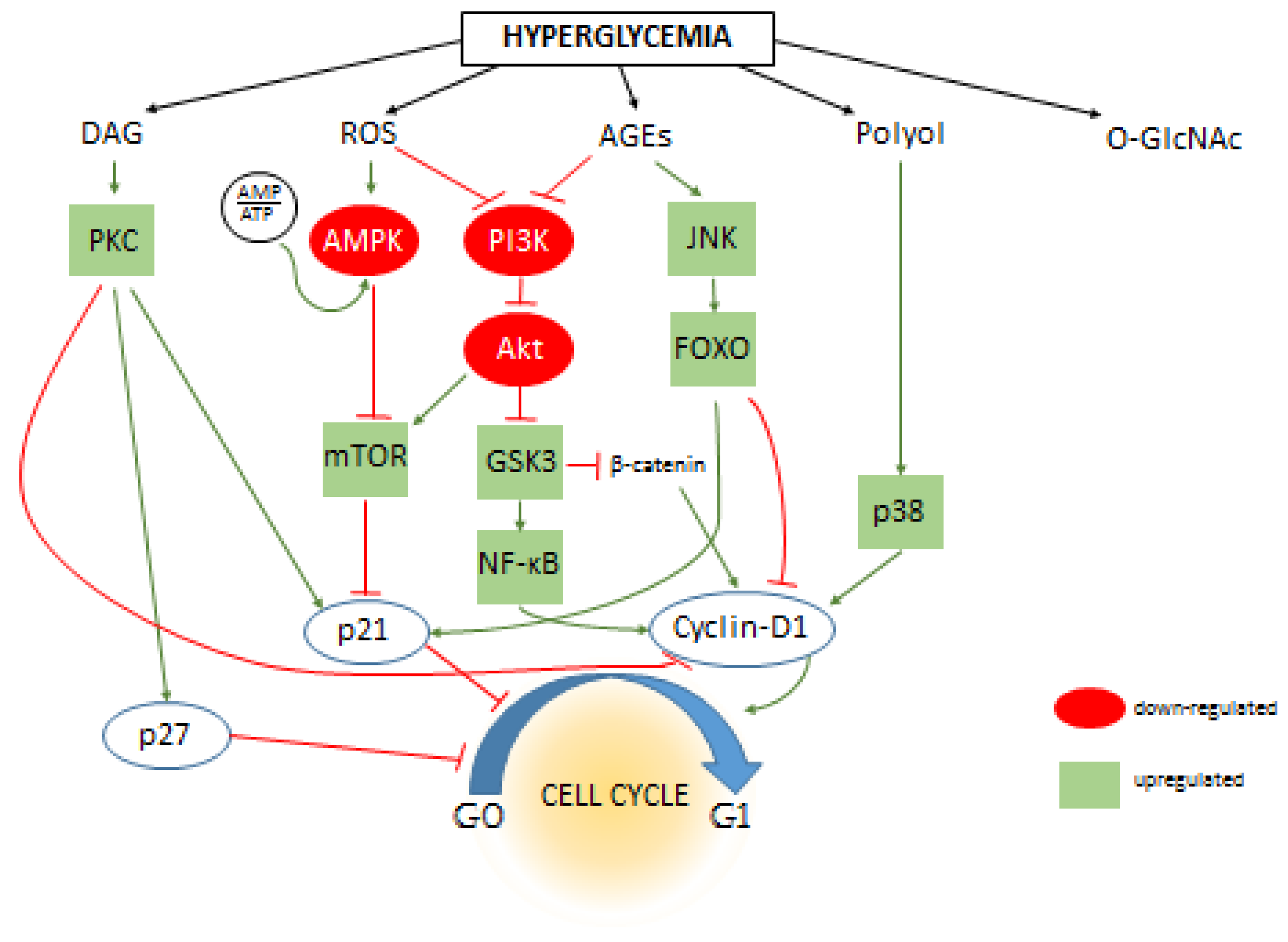
2.3. Downstream Signaling Pathways Regulating Cell Cycle During Hyperglycemia
3. Cell Proliferative Disorders Related to Diabetes
3.1. Cancer Development
3.2. Skeletal Growth and Bone Remodeling
3.3. Insufficient Tissue Regeneration
3.4. Renal Mesangial Cell Growth
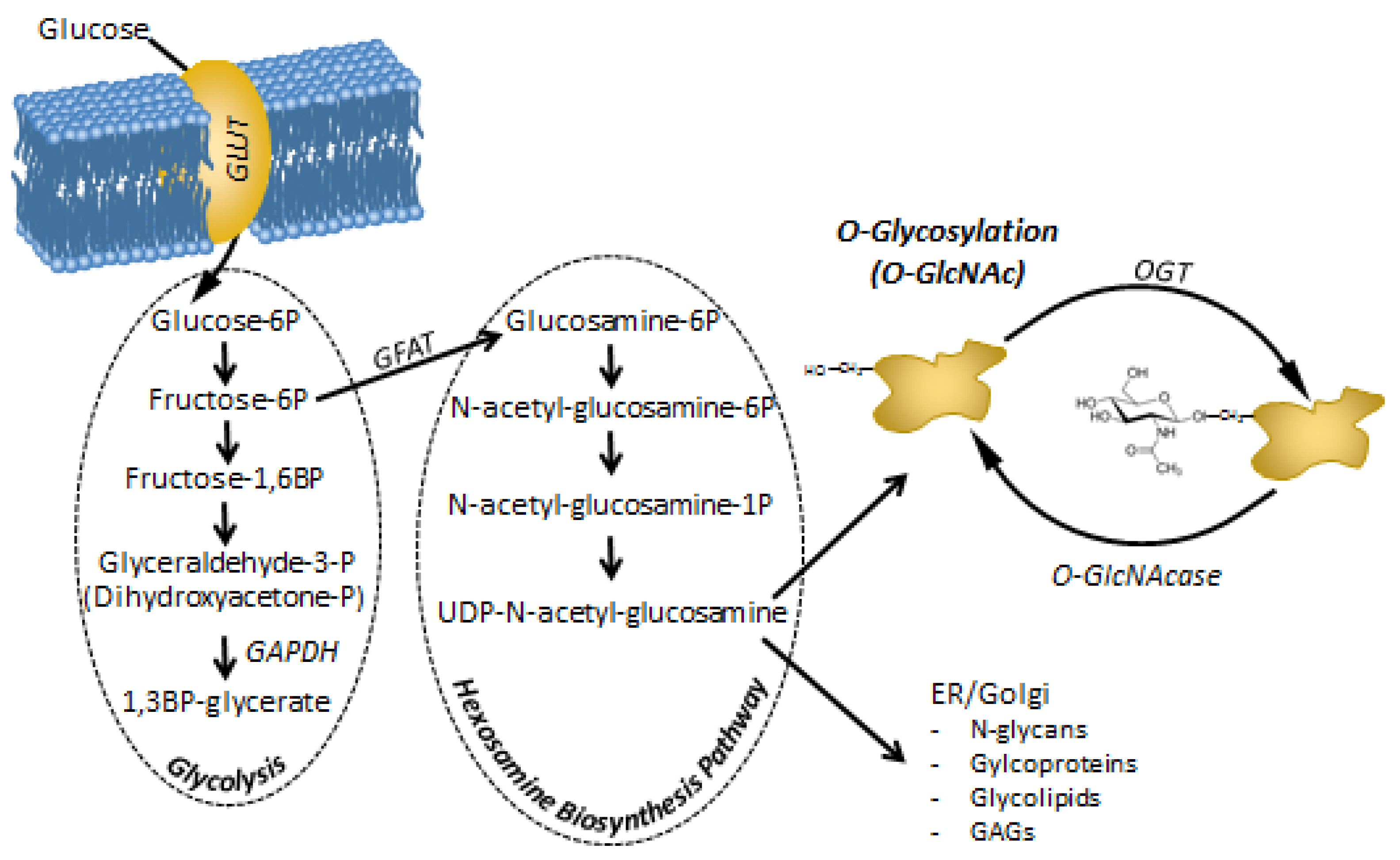
4. O-GlcNAc and Its Effect on Cell Cycle Regulation
5. Cell Proliferative Disorders as a Consequence of O-GlcNAc Disturbances
5.1. Pancreatic Beta Cell Regulation
5.2. Cancer Development
5.3. Embryonal Development
5.4. Endothelial Cell Proliferation, Wound Healing
5.5. Immune Cell Proliferation
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jacob, F.; Monod, J. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 1961, 3, 318–356. [Google Scholar] [CrossRef]
- Loomis, W.F.; Magasanik, B. Glucose-lactose diauxie in Escherichia coli. J. Bacteriol. 1967, 93, 1397–1401. [Google Scholar] [PubMed]
- Stoebel, D.M.; Dean, A.M.; Dykhuizen, D.E. The cost of expression of Escherichia coli lac operon proteins is in the process, not in the products. Genetics 2008, 178, 1653–1660. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Report on Diabetes. Available online: https://www.who.int/diabetes/global-report/en/ (accessed on 11 July 2019).
- Forouhi, N.G.; Wareham, N.J. Epidemiology of diabetes. Medicine (Abingdon) 2014, 42, 698–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshpande, A.D.; Harris-Hayes, M.; Schootman, M. Epidemiology of diabetes and diabetes-related complications. Phys. Ther. 2008, 88, 1254–1264. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, E.; Harlan, D.M.; Archer, M.C.; Bergenstal, R.M.; Gapstur, S.M.; Habel, L.A.; Pollak, M.; Regensteiner, J.G.; Yee, D. Diabetes and cancer: A consensus report. Diabetes Care 2010, 33, 1674–1685. [Google Scholar] [CrossRef]
- Duan, W.; Shen, X.; Lei, J.; Xu, Q.; Yu, Y.; Li, R.; Wu, E.; Ma, Q. Hyperglycemia, a Neglected Factor during Cancer Progression. Biomed Res. Int. 2014, 2014, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varma, S.; Lal, B.K.; Zheng, R.; Breslin, J.W.; Saito, S.; Pappas, P.J.; Hobson, R.W.; Durán, W.N. Hyperglycemia alters PI3k and Akt signaling and leads to endothelial cell proliferative dysfunction. Am. J. Physiol. Circ. Physiol. 2005, 289, H1744–H1751. [Google Scholar] [CrossRef] [PubMed]
- Brem, H.; Tomic-Canic, M. Cellular and molecular basis of wound healing in diabetes. J. Clin. Investig. 2007, 117, 1219–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohkuma, T.; Peters, S.A.E.; Woodward, M. Sex differences in the association between diabetes and cancer: A systematic review and meta-analysis of 121 cohorts including 20 million individuals and one million events. Diabetologia 2018, 61, 2140–2154. [Google Scholar] [CrossRef] [PubMed]
- Berlanga-Acosta, J.; Schultz, G.S.; López-Mola, E.; Guillen-Nieto, G.; García-Siverio, M.; Herrera-Martínez, L. Glucose Toxic Effects on Granulation Tissue Productive Cells: The Diabetics’ Impaired Healing. Biomed Res. Int. 2013, 2013, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Greenhalgh, D.G. Tissue Repair in Models of Diabetes Mellitus: A Review. In Wound Healing; Humana Press: Totowa, NJ, USA, 2003; pp. 181–190. [Google Scholar]
- Linderkamp, O.; Ruef, P.; Zilow, E.P.; Hoffmann, G.F. Impaired deformability of erythrocytes and neutrophils in children with newly diagnosed insulin-dependent diabetes mellitus. Diabetologia 1999, 42, 865–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, J.F.; Mora, C. Role of inflammation in diabetic complications. Nephrol. Dial. Transplant. 2005, 20, 2601–2604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellen, K.E.; Hotamisligil, G.S. Inflammation, stress, and diabetes. J. Clin. Investig. 2005, 115, 1111–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alba-Loureiro, T.C.; Munhoz, C.D.; Martins, J.O.; Cerchiaro, G.A.; Scavone, C.; Curi, R.; Sannomiya, P. Neutrophil function and metabolism in individuals with diabetes mellitus. Braz. J. Med. Biol. Res. 2007, 40, 1037–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touch, S.; Clément, K.; André, S. T Cell Populations and Functions Are Altered in Human Obesity and Type 2 Diabetes. Curr. Diab. Rep. 2017, 17, 81. [Google Scholar] [CrossRef]
- Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 2014, 37, S81–S90. [CrossRef]
- Tokarz, V.L.; MacDonald, P.E.; Klip, A. The cell biology of systemic insulin function. J. Cell Biol. 2018, 217, 2273–2289. [Google Scholar] [CrossRef] [Green Version]
- Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes—2018. Diabetes Care 2018, 41, S13–S27. [CrossRef]
- Wilcox, N.S.; Rui, J.; Hebrok, M.; Herold, K.C. Life and death of β cells in Type 1 diabetes: A comprehensive review. J. Autoimmun. 2016, 71, 51–58. [Google Scholar] [CrossRef]
- Taylor, R. Insulin Resistance and Type 2 Diabetes. Diabetes 2012, 61, 778–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahn, S.E. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia 2003, 46, 3–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandimalla, R.; Thirumala, V.; Reddy, P.H. Is Alzheimer’s disease a Type 3 Diabetes? A critical appraisal. Biochim. Biophys. Acta. Mol. Basis Dis. 2017, 1863, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Forbes, J.M.; Cooper, M.E. Mechanisms of Diabetic Complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef]
- Lopez, R.; Arumugam, A.; Joseph, R.; Monga, K.; Boopalan, T.; Agullo, P.; Gutierrez, C.; Nandy, S.; Subramani, R.; de la Rosa, J.M.; et al. Hyperglycemia Enhances the Proliferation of Non-Tumorigenic and Malignant Mammary Epithelial Cells through Increased leptin/IGF1R Signaling and Activation of AKT/mTOR. PLoS ONE 2013, 8, e79708. [Google Scholar] [CrossRef]
- Wolf, G. Cell cycle regulation in diabetic nephropathy. Kidney Int. 2000, 58, S59–S66. [Google Scholar] [CrossRef] [Green Version]
- Masson, E.; Lagarde, M.; Wiernsperger, N.; El Bawab, S. Hyperglycemia and glucosamine-induced mesangial cell cycle arrest and hypertrophy: Common or independent mechanisms? IUBMB Life (Int. Union Biochem. Mol. Biol. Life) 2006, 58, 381–388. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Choudhury, R.G.; Winters, A.; Prah, J.; Lin, W.; Liu, R.; Yang, S.H. Hyperglycemia Alters Astrocyte Metabolism and Inhibits Astrocyte Proliferation. Aging Dis. 2018, 9, 674. [Google Scholar] [CrossRef] [PubMed]
- McClelland, D.L.; Satoorian, T.S.; Walker, L.M.; Sparks, N.R.L.L.; Pulyanina, P.Y.; Nieden, N.I. Glucose-Induced Oxidative Stress Reduces Proliferation in Embryonic Stem Cells via FOXO3A/β-Catenin-Dependent Transcription of p21cip1. Stem Cell Reports 2016, 7, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Weiss, U.; Cervar, M.; Puerstner, P.; Schmut, O.; Haas, J.; Mauschitz, R.; Arikan, G.; Desoye, G. Hyperglycaemia in vitro alters the proliferation and mitochondrial activity of the choriocarcinoma cell lines BeWo, JAR and JEG-3 as models for human first-trimester trophoblast. Diabetologia 2001, 44, 209–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasconcelos-dos-Santos, A.; de Queiroz, R.M.; da Costa Rodrigues, B.; Todeschini, A.R.; Dias, W.B. Hyperglycemia and aberrant O-GlcNAcylation: Contributions to tumor progression. J. Bioenerg. Biomembr. 2018, 50, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, J.; Cai, Y.; Peng, S.; Wang, J.; Xiao, Z.; Wang, Y.; Tao, Y.; Li, J.; Leng, Q.; et al. Hyperglycaemia-induced miR-301a promotes cell proliferation by repressing p21 and Smad4 in prostate cancer. Cancer Lett. 2018, 418, 211–220. [Google Scholar] [CrossRef]
- Kim, D.; Ahn, B.N.; Kim, Y.; Hur, D.Y.; Yang, J.W.; Park, G.B.; Jang, J.E.; Lee, E.J.; Kwon, M.J.; Kim, T.N.; et al. High Glucose with Insulin Induces Cell Cycle Progression and Activation of Oncogenic Signaling of Bladder Epithelial Cells Cotreated with Metformin and Pioglitazone. J. Diabetes Res. 2019, 2019, 2376512. [Google Scholar] [CrossRef] [PubMed]
- Fried, R.C.; Mullen, J.; Stein, T.P.; Yuhas, J.; Miller, E.; Buzby, G.P. The effects of glucose and amino acids on tumor and host DNA synthesis. J. Surg. Res. 1985, 39, 461–469. [Google Scholar] [CrossRef]
- Watanabe, A.; Tanaka, R.; Takeda, N.; Washiyama, K. DNA synthesis, blood flow, and glucose utilization in experimental rat brain tumors. J. Neurosurg. 1989, 70, 86–91. [Google Scholar] [CrossRef] [Green Version]
- Jannière, L.; Canceill, D.; Suski, C.; Kanga, S.; Dalmais, B.; Lestini, R.; Monnier, A.F.; Chapuis, J.; Bolotin, A.; Titok, M.; et al. Genetic Evidence for a Link Between Glycolysis and DNA Replication. PLoS ONE 2007, 2, e447. [Google Scholar] [CrossRef]
- Takahashi, S.; Izawa, Y.; Suzuki, N. Astroglial Pentose Phosphate Pathway Rates in Response to High-Glucose Environments. ASN Neuro 2012, 4, AN20120002. [Google Scholar] [CrossRef] [Green Version]
- Peiró, C.; Romacho, T.; Azcutia, V.; Villalobos, L.; Fernández, E.; Bolaños, J.P.; Moncada, S.; Sánchez-Ferrer, C.F. Inflammation, glucose, and vascular cell damage: The role of the pentose phosphate pathway. Cardiovasc. Diabetol. 2016, 15, 82. [Google Scholar] [CrossRef] [PubMed]
- Vizán, P.; Alcarraz-Vizán, G.; Díaz-Moralli, S.; Solovjeva, O.N.; Frederiks, W.M.; Cascante, M. Modulation of pentose phosphate pathway during cell cycle progression in human colon adenocarcinoma cell line HT29. Int. J. Cancer 2009, 124, 2789–2796. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Wang, L.; Huang, D.; Li, Y.; Yang, D.; Li, T.; Li, F.; Sun, L.; Wei, H.; He, K.; et al. Polo-like kinase 1 coordinates biosynthesis during cell cycle progression by directly activating pentose phosphate pathway. Nat. Commun. 2017, 8, 1506. [Google Scholar] [CrossRef] [PubMed]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.C.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.; Krüger, A.; Alam, T.M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. 2015, 90, 927–963. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.; Manda, G. Metabolic Pathways of the Warburg Effect in Health and Disease: Perspectives of Choice, Chain or Chance. Int. J. Mol. Sci. 2017, 18, 2755. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Da Veiga Moreira, J.; Peres, S.; Steyaert, J.M.; Bigan, E.; Paulevé, L.; Nogueira, M.L.; Schwartz, L. Cell cycle progression is regulated by intertwined redox oscillators. Theor. Biol. Med. Model. 2015, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Rietman, E.A.; Friesen, D.E.; Hahnfeldt, P.; Gatenby, R.; Hlatky, L.; Tuszynski, J.A. An integrated multidisciplinary model describing initiation of cancer and the Warburg hypothesis. Theor. Biol. Med. Model. 2013, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Jones, W.; Bianchi, K. Aerobic glycolysis: Beyond proliferation. Front. Immunol. 2015, 6, 227. [Google Scholar] [CrossRef]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Du, X.L.; Edelstein, D.; Dimmeler, S.; Ju, Q.; Sui, C.; Brownlee, M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J. Clin. Investig. 2001, 108, 1341–1348. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Cuezva, J.M. The H+-ATP synthase: A gate to ROS-mediated cell death or cell survival. Biochim. Biophys. Acta Bioenerg. 2014, 1837, 1099–1112. [Google Scholar] [CrossRef] [Green Version]
- Szendroedi, J.; Schmid, A.I.; Chmelik, M.; Toth, C.; Brehm, A.; Krssak, M.; Nowotny, P.; Wolzt, M.; Waldhausl, W.; Roden, M. Muscle mitochondrial ATP synthesis and glucose transport/phosphorylation in type 2 diabetes. PLoS Med. 2007, 4, e154. [Google Scholar] [CrossRef]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Garcia, R.; Shulman, G.I. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med. 2004, 350, 664–671. [Google Scholar] [CrossRef]
- Kwak, S.H.; Park, K.S.; Lee, K.U.; Lee, H.K. Mitochondrial metabolism and diabetes. J. Diabetes Investig. 2010, 1, 161–169. [Google Scholar] [CrossRef]
- Rocha, L.A.; Oliveira, K.S.; Migliolo, L.; Franco, O.L. Effect of Moderate Exercise on Mitochondrial Proteome in Heart Tissue of Spontaneous Hypertensive Rats. Am. J. Hypertens. 2016, 29, 696–704. [Google Scholar] [CrossRef]
- Farhat, F.; Dupas, J.; Amérand, A.; Goanvec, C.; Feray, A.; Simon, B.; Guegueniat, N.; Moisan, C. Effect of exercise training on oxidative stress and mitochondrial function in rat heart and gastrocnemius muscle. Redox Rep. 2015, 20, 60–68. [Google Scholar] [CrossRef]
- Havens, C.G.; Ho, A.; Yoshioka, N.; Dowdy, S.F. Regulation of late G1/S phase transition and APC Cdh1 by reactive oxygen species. Mol. Cell. Biol. 2006, 26, 4701–4711. [Google Scholar] [CrossRef]
- Barni, S.; Sciola, L.; Spano, A.; Pippia, P. Static cytofluorometry and fluorescence morphology of mitochondria and DNA in proliferating fibroblasts. Biotech. Histochem. 1996, 71, 66–70. [Google Scholar] [CrossRef]
- Buranasin, P.; Mizutani, K.; Iwasaki, K.; Pawaputanon Na Mahasarakham, C.; Kido, D.; Takeda, K.; Izumi, Y. High glucose-induced oxidative stress impairs proliferation and migration of human gingival fibroblasts. PLoS ONE 2018, 13, e0201855. [Google Scholar] [CrossRef]
- Boonstra, J.; Post, J.A. Molecular events associated with reactive oxygen species and cell cycle progression in mammalian cells. Gene 2004, 337, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Nowotny, K.; Jung, T.; Höhn, A.; Weber, D.; Grune, T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef]
- Black, A.R.; Black, J.D. Protein kinase C signaling and cell cycle regulation. Front. Immunol. 2012, 3, 423. [Google Scholar] [CrossRef]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced Glycation End Products. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef] [Green Version]
- Wautier, M.P.; Chappey, O.; Corda, S.; Stern, D.M.; Schmidt, A.M.; Wautier, J.L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E685–E694. [Google Scholar] [CrossRef]
- SCHMIDT, A.; STERN, D. RAGE: A New Target for the Prevention and Treatment of the Vascular and Inflammatory Complications of Diabetes. Trends Endocrinol. Metab. 2000, 11, 368–375. [Google Scholar] [CrossRef]
- Ledoux, A.C.; Perkins, N.D. NF-κB and the cell cycle. Biochem. Soc. Trans. 2014, 42, 76–81. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef]
- Schmidt, A.; Kuhla, B.; Bigl, K.; Münch, G.; Arendt, T. Cell cycle related signaling in Neuro2a cells proceeds via the receptor for advanced glycation end products. J. Neural Transm. 2007, 114, 1413–1424. [Google Scholar] [CrossRef]
- Xu, X.C.; Abuduhadeer, X.; Zhang, W.B.; Li, T.; Gao, H.; Wang, Y.H. Knockdown of RAGE inhibits growth and invasion of gastric cancer cells. Eur. J. Histochem. 2013, 57, e36. [Google Scholar] [CrossRef]
- Franke, S.; Sommer, M.; Rüster, C.; Bondeva, T.; Marticke, J.; Hofmann, G.; Hein, G.; Wolf, G. Advanced glycation end products induce cell cycle arrest and proinflammatory changes in osteoarthritic fibroblast-like synovial cells. Arthritis Res. Ther. 2009, 11, R136. [Google Scholar] [CrossRef]
- Schwab, A.; Siddiqui, A.; Vazakidou, M.E.; Napoli, F.; Böttcher, M.; Menchicchi, B.; Raza, U.; Saatci, Ö.; Krebs, A.M.; Ferrazzi, F.; et al. Polyol Pathway Links Glucose Metabolism to the Aggressiveness of Cancer Cells. Cancer Res. 2018, 78, 1604–1618. [Google Scholar] [CrossRef] [Green Version]
- Criollo, A.; Galluzzi, L.; Maiuri, M.C.; Tasdemir, E.; Lavandero, S.; Kroemer, G. Mitochondrial control of cell death induced by hyperosmotic stress. Apoptosis 2007, 12, 3–18. [Google Scholar] [CrossRef]
- Lu, X.; Li, C.; Wang, Y.K.; Jiang, K.; Gai, X.D. Sorbitol induces apoptosis of human colorectal cancer cells via p38 MAPK signal transduction. Oncol. Lett. 2014, 7, 1992–1996. [Google Scholar] [CrossRef]
- Faust, D.; Schmitt, C.; Oesch, F.; Oesch-Bartlomowicz, B.; Schreck, I.; Weiss, C.; Dietrich, C. Differential p38-dependent signalling in response to cellular stress and mitogenic stimulation in fibroblasts. Cell Commun. Signal. 2012, 10, 6. [Google Scholar] [CrossRef]
- Thornton, T.M.; Rincon, M. Non-Classical P38 Map Kinase Functions: Cell Cycle Checkpoints and Survival. Int. J. Biol. Sci. 2009, 5, 44–52. [Google Scholar] [CrossRef]
- Hart, G.W.; Housley, M.P.; Slawson, C. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature 2007, 446, 1017–1022. [Google Scholar] [CrossRef]
- Nagy, T.; Miseta, A.; Kovacs, L. Protein-associated O-GlcNAc, a multifunctional mechanism in cell signaling and its role in the pathogenesis of diabetes, stress and malignant diseases. Biochem. Med. 2007, 17, 162–177. [Google Scholar] [CrossRef]
- Hart, G.W.; Slawson, C.; Ramirez-Correa, G.; Lagerlof, O. Cross talk between O-GlcNAcylation and phosphorylation: Roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 2011, 80, 825–858. [Google Scholar] [CrossRef]
- Li, H.; Peng, W.; Zhuang, J.; Lu, Y.; Jian, W.; Wei, Y.; Li, W.; Xu, Y. Vaspin attenuates high glucose-induced vascular smooth muscle cells proliferation and chemokinesis by inhibiting the MAPK, PI3K/Akt, and NF-κB signaling pathways. Atherosclerosis 2013, 228, 61–68. [Google Scholar] [CrossRef]
- Scott-Drechsel, D.E.; Rugonyi, S.; Marks, D.L.; Thornburg, K.L.; Hinds, M.T. Hyperglycemia slows embryonic growth and suppresses cell cycle via cyclin D1 and p21. Diabetes 2013, 62, 234–242. [Google Scholar] [CrossRef]
- Loughlin, D.T.; Artlett, C.M. Modification of Collagen by 3-Deoxyglucosone Alters Wound Healing through Differential Regulation of p38 MAP Kinase. PLoS ONE 2011, 6, e18676. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Cuyàs, E.; Corominas-Faja, B.; Joven, J.; Menendez, J.A. Cell cycle regulation by the nutrient-sensing mammalian target of rapamycin (mTOR) pathway. Methods Mol. Biol. 2014, 1170, 113–144. [Google Scholar] [CrossRef]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef]
- Xie, J.; Wang, X.; Proud, C.G. mTOR inhibitors in cancer therapy. F1000Research 2016, 5. [Google Scholar] [CrossRef]
- Llanos, S.; García-Pedrero, J.M.; Morgado-Palacin, L.; Rodrigo, J.P.; Serrano, M. Stabilization of p21 by mTORC1/4E-BP1 predicts clinical outcome of head and neck cancers. Nat. Commun. 2016, 7, 10438. [Google Scholar] [CrossRef]
- Hidalgo, M.; Rowinsky, E.K. The rapamycin-sensitive signal transduction pathway as a target for cancer therapy. Oncogene 2000, 19, 6680–6686. [Google Scholar] [CrossRef] [Green Version]
- Umezawa, S.; Higurashi, T.; Nakajima, A. AMPK: Therapeutic Target for Diabetes and Cancer Prevention. Curr. Pharm. Des. 2017, 23, 3629–3644. [Google Scholar] [CrossRef]
- Zi, F.; Zi, H.; Li, Y.; He, J.; Shi, Q.; Cai, Z. Metformin and cancer: An existing drug for cancer prevention and therapy. Oncol. Lett. 2018, 15, 683–690. [Google Scholar] [CrossRef]
- Huang, H.; Tindall, D.J. Dynamic FoxO transcription factors. J. Cell Sci. 2007, 120, 2479–2487. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Meng, X.; Sun, X.; Liu, M.; Gao, S.; Zhao, J.; Pei, F.; Yu, H. Wnt/β-catenin signaling pathway may regulate cell cycle and expression of cyclin A and cyclin E protein in hepatocellular carcinoma cells. Cell Cycle 2009, 8, 1567–1570. [Google Scholar] [CrossRef]
- Rowlands, T.M.; Pechenkina, I.V.; Hatsell, S.; Cowin, P. Beta-catenin and cyclin D1: Connecting development to breast cancer. Cell Cycle 2004, 3, 145–148. [Google Scholar] [CrossRef]
- Korswagen, H.C. Regulation of the Wnt/β-catenin pathway by redox signaling. Dev. Cell 2006, 10, 687–688. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y. Crosstalk Between Peroxisome Proliferator-Activated Receptor Gamma and the Canonical WNT/β-Catenin Pathway in Chronic Inflammation and Oxidative Stress During Carcinogenesis. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Liu, X.; Yao, Z. Chronic over-nutrition and dysregulation of GSK3 in diseases. Nutr. Metab. (Lond.) 2016, 13, 49. [Google Scholar] [CrossRef]
- Huang, X.; Liu, G.; Guo, J.; Su, Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int. J. Biol. Sci. 2018, 14, 1483–1496. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.; Hu, Z.; Xu, H.; Xu, J.; Zhang, S.; Zhong, Y.; He, X.; Wang, N. Advanced glycation endproducts trigger autophagy in cadiomyocyte via RAGE/PI3K/AKT/mTOR pathway. Cardiovasc. Diabetol. 2014, 13, 78. [Google Scholar] [CrossRef]
- Fakhruddin, S.; Alanazi, W.; Jackson, K.E. Diabetes-Induced Reactive Oxygen Species: Mechanism of Their Generation and Role in Renal Injury. J. Diabetes Res. 2017, 2017, 1–30. [Google Scholar] [CrossRef]
- Loh, K.; Deng, H.; Fukushima, A.; Cai, X.; Boivin, B.; Galic, S.; Bruce, C.; Shields, B.J.; Skiba, B.; Ooms, L.M.; et al. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009, 10, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Takahashi-Yanaga, F.; Sasaguri, T. GSK-3beta regulates cyclin D1 expression: A new target for chemotherapy. Cell. Signal. 2008, 20, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Jope, R.S.; Yuskaitis, C.J.; Beurel, E. Glycogen synthase kinase-3 (GSK3): Inflammation, diseases, and therapeutics. Neurochem. Res. 2007, 32, 577–595. [Google Scholar] [CrossRef] [PubMed]
- Walz, A.; Ugolkov, A.; Chandra, S.; Kozikowski, A.; Carneiro, B.A.; O’Halloran, T.V.; Giles, F.J.; Billadeau, D.D.; Mazar, A.P. Molecular Pathways: Revisiting Glycogen Synthase Kinase-3β as a Target for the Treatment of Cancer. Clin. Cancer Res. 2017, 23, 1891–1897. [Google Scholar] [CrossRef] [PubMed]
- Saengboonmee, C.; Seubwai, W.; Pairojkul, C.; Wongkham, S. High glucose enhances progression of cholangiocarcinoma cells via STAT3 activation. Sci. Rep. 2016, 6, 18995. [Google Scholar] [CrossRef]
- Masur, K.; Vetter, C.; Hinz, A.; Tomas, N.; Henrich, H.; Niggemann, B.; Zänker, K.S. Diabetogenic glucose and insulin concentrations modulate transcriptome and protein levels involved in tumour cell migration, adhesion and proliferation. Br. J. Cancer 2011, 104, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Onodera, Y.; Nam, J.M.; Bissell, M.J. Increased sugar uptake promotes oncogenesis via EPAC/RAP1 and O-GlcNAc pathways. J. Clin. Investig. 2014, 124, 367–384. [Google Scholar] [CrossRef]
- Vigneri, P.; Frasca, F.; Sciacca, L.; Pandini, G.; Vigneri, R. Diabetes and cancer. Endocr. Relat. Cancer 2009, 16, 1103–1123. [Google Scholar] [CrossRef] [Green Version]
- Shikata, K.; Ninomiya, T.; Kiyohara, Y. Diabetes mellitus and cancer risk: Review of the epidemiological evidence. Cancer Sci. 2013, 104, 9–14. [Google Scholar] [CrossRef]
- Wang, C.; Wang, X.; Gong, G.; Ben, Q.; Qiu, W.; Chen, Y.; Li, G.; Wang, L. Increased risk of hepatocellular carcinoma in patients with diabetes mellitus: A systematic review and meta-analysis of cohort studies. Int. J. Cancer 2012, 130, 1639–1648. [Google Scholar] [CrossRef]
- Ben, Q.; Cai, Q.; Li, Z.; Yuan, Y.; Ning, X.; Deng, S.; Wang, K. The relationship between new-onset diabetes mellitus and pancreatic cancer risk: A case-control study. Eur. J. Cancer 2011, 47, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Ben, Q.; Xu, M.; Ning, X.; Liu, J.; Hong, S.; Huang, W.; Zhang, H.; Li, Z. Diabetes mellitus and risk of pancreatic cancer: A meta-analysis of cohort studies. Eur. J. Cancer 2011, 47, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Mantzoros, C.S.; Wolk, A. Diabetes mellitus and risk of breast cancer: A meta-analysis. Int. J. Cancer 2007, 121, 856–862. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Wolk, A. Diabetes mellitus and incidence of kidney cancer: A meta-analysis of cohort studies. Diabetologia 2011, 54, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Ben, Q.; Shen, H.; Lu, W.; Zhang, Y.; Zhu, J. Diabetes mellitus and incidence and mortality of colorectal cancer: A systematic review and meta-analysis of cohort studies. Eur. J. Epidemiol. 2011, 26, 863–876. [Google Scholar] [CrossRef] [PubMed]
- Mitri, J.; Castillo, J.; Pittas, A.G. Diabetes and risk of Non-Hodgkin’s lymphoma: A meta-analysis of observational studies. Diabetes Care 2008, 31, 2391–2397. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.B.; Ren, G.S. Diabetes mellitus and prognosis in women with breast cancer: A systematic review and meta-analysis. Medicine (Baltimore) 2016, 95, e5602. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos-Dos-Santos, A.; Loponte, H.F.B.R.; Mantuano, N.R.; Oliveira, I.A.; de Paula, I.F.; Teixeira, L.K.; De-Freitas-Junior, J.C.M.; Gondim, K.C.; Heise, N.; Mohana-Borges, R.; et al. Hyperglycemia exacerbates colon cancer malignancy through hexosamine biosynthetic pathway. Oncogenesis 2017, 6, e306. [Google Scholar] [CrossRef] [PubMed]
- Lucena, M.C.; Carvalho-Cruz, P.; Donadio, J.L.; Oliveira, I.A.; de Queiroz, R.M.; Marinho-Carvalho, M.M.; Sola-Penna, M.; de Paula, I.F.; Gondim, K.C.; McComb, M.E.; et al. Epithelial Mesenchymal Transition Induces Aberrant Glycosylation through Hexosamine Biosynthetic Pathway Activation. J. Biol. Chem. 2016, 291, 12917–12929. [Google Scholar] [CrossRef] [Green Version]
- Aurer, I.; Lauc, G.; Dumić, J.; Rendić, D.; Matisić, D.; Milos, M.; Heffer-Lauc, M.; Flogel, M.; Labar, B. Aberrant glycosylation of Igg heavy chain in multiple myeloma. Coll. Antropol. 2007, 31, 247–251. [Google Scholar]
- Holst, S.; Stavenhagen, K.; Balog, C.I.A.; Koeleman, C.A.M.; McDonnell, L.M.; Mayboroda, O.A.; Verhoeven, A.; Mesker, W.E.; Tollenaar, R.A.E.M.; Deelder, A.M.; et al. Investigations on aberrant glycosylation of glycosphingolipids in colorectal cancer tissues using liquid chromatography and matrix-assisted laser desorption time-of-flight mass spectrometry (MALDI-TOF-MS). Mol. Cell. Proteomics 2013, 12, 3081–3093. [Google Scholar] [CrossRef] [PubMed]
- Vavasseur, F.; Dole, K.; Yang, J.; Matta, K.L.; Myerscough, N.; Corfield, A.; Paraskeva, C.; Brockhausen, I. O-glycan biosynthesis in human colorectal adenoma cells during progression to cancer. Eur. J. Biochem. 1994, 222, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Patwa, T.H.; Xu, L.; Shedden, K.; Misek, D.E.; Tuck, M.; Jin, G.; Ruffin, M.T.; Turgeon, D.K.; Synal, S.; et al. Plasma glycoprotein profiling for colorectal cancer biomarker identification by lectin glycoarray and lectin blot. J. Proteome Res. 2008, 7, 1693–1703. [Google Scholar] [CrossRef]
- Holst, S.; Wuhrer, M.; Rombouts, Y. Glycosylation characteristics of colorectal cancer. Adv. Cancer Res. 2015, 126, 203–256. [Google Scholar] [CrossRef] [PubMed]
- Laskar, J.; Bhattacharjee, K.; Sengupta, M.; Choudhury, Y. Anti-Diabetic Drugs: Cure or Risk Factors for Cancer? Pathol. Oncol. Res. 2018, 24, 745–755. [Google Scholar] [CrossRef]
- Zhang, Z.-J.; Zheng, Z.-J.; Kan, H.; Song, Y.; Cui, W.; Zhao, G.; Kip, K.E. Reduced risk of colorectal cancer with metformin therapy in patients with type 2 diabetes: A meta-analysis. Diabetes Care 2011, 34, 2323–2328. [Google Scholar] [CrossRef]
- Algire, C.; Amrein, L.; Zakikhani, M.; Panasci, L.; Pollak, M. Metformin blocks the stimulative effect of a high-energy diet on colon carcinoma growth in vivo and is associated with reduced expression of fatty acid synthase. Endocr. Relat. Cancer 2010, 17, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Buzzai, M.; Jones, R.G.; Amaravadi, R.K.; Lum, J.J.; DeBerardinis, R.J.; Zhao, F.; Viollet, B.; Thompson, C.B. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007, 67, 6745–6752. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Mok, M.T.; Sun, H.; Chan, A.W.; Huang, Y.; Cheng, A.S.; Xu, G. The anti-diabetic drug exenatide, a glucagon-like peptide-1 receptor agonist, counteracts hepatocarcinogenesis through cAMP-PKA-EGFR-STAT3 axis. Oncogene 2017, 36, 4135–4149. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, S.E.; Hoepelman, A.I. Immune dysfunction in patients with diabetes mellitus (DM). FEMS Immunol. Med. Microbiol. 1999, 26, 259–265. [Google Scholar] [CrossRef]
- Moutschen, M.P.; Scheen, A.J.; Lefebvre, P.J. Impaired immune responses in diabetes mellitus: Analysis of the factors and mechanisms involved. Relevance to the increased susceptibility of diabetic patients to specific infections. Diabete Metab. 1992, 18, 187–201. [Google Scholar] [PubMed]
- Zhou, T.; Hu, Z.; Yang, S.; Sun, L.; Yu, Z.; Wang, G. Role of Adaptive and Innate Immunity in Type 2 Diabetes Mellitus. J. Diabetes Res. 2018, 2018, 1–9. [Google Scholar] [CrossRef]
- Nagareddy, P.R.; Murphy, A.J.; Stirzaker, R.A.; Hu, Y.; Yu, S.; Miller, R.G.; Ramkhelawon, B.; Distel, E.; Westerterp, M.; Huang, L.S.; et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 2013, 17, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Tencerová, M.; Kračmerová, J.; Krauzová, E.; Mališová, L.; Kováčová, Z.; Wedellová, Z.; Šiklová, M.; Štich, V.; Rossmeislová, L. Experimental Hyperglycemia Induces an Increase of Monocyte and T-Lymphocyte Content in Adipose Tissue of Healthy Obese Women. PLoS ONE 2015, 10, e0122872. [Google Scholar] [CrossRef] [PubMed]
- Swamy, M.; Pathak, S.; Grzes, K.M.; Damerow, S.; Sinclair, L.V.; van Aalten, D.M.F.; Cantrell, D.A. Glucose and glutamine fuel protein O-GlcNAcylation to control T cell self-renewal and malignancy. Nat. Immunol. 2016, 17, 712–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Press Release: The Nobel Prize in Physiology or Medicine 2018. Available online: https://www.nobelprize.org/prizes/medicine/2018/press-release/ (accessed on 31 July 2019).
- Curran, M.A.; Montalvo, W.; Yagita, H.; Allison, J.P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 4275–4280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fainsod-Levi, T.; Gershkovitz, M.; Völs, S.; Kumar, S.; Khawaled, S.; Sagiv, J.Y.; Sionov, R.V.; Grunewald, M.; Keshet, E.; Granot, Z. Hyperglycemia Impairs Neutrophil Mobilization Leading to Enhanced Metastatic Seeding. Cell Rep. 2017, 21, 2384–2392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conroy, M.J.; Dunne, M.R.; Donohoe, C.L.; Reynolds, J. V Obesity-associated cancer: An immunological perspective. Proc. Nutr. Soc. 2016, 75, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Tsentidis, C.; Gourgiotis, D.; Kossiva, L.; Doulgeraki, A.; Marmarinos, A.; Galli-Tsinopoulou, A.; Karavanaki, K. Higher levels of s-RANKL and osteoprotegerin in children and adolescents with type 1 diabetes mellitus may indicate increased osteoclast signaling and predisposition to lower bone mass: A multivariate cross-sectional analysis. Osteoporos. Int. 2016, 27, 1631–1643. [Google Scholar] [CrossRef]
- El-Bialy, T.; Aboul-Azm, S.F.; El-Sakhawy, M. Study of craniofacial morphology and skeletal maturation in juvenile diabetics (Type I). Am. J. Orthod. Dentofac. Orthop. 2000, 118, 189–195. [Google Scholar] [CrossRef]
- Sterky, G. Growth pattern in juvenile diabetes. Acta Paediatr. Scand. 1967, 177, 80–82. [Google Scholar] [CrossRef] [PubMed]
- Sundararaghavan, V.; Mazur, M.M.; Evans, B.; Liu, J.; Ebraheim, N.A. Diabetes and bone health: Latest evidence and clinical implications. Ther. Adv. Musculoskelet. Dis. 2017, 9, 67–74. [Google Scholar] [CrossRef] [PubMed]
- DeShields, S.C.; Cunningham, T.D. Comparison of osteoporosis in US adults with type 1 and type 2 diabetes mellitus. J. Endocrinol. Investig. 2018, 41, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.R.; Haynes, K.; Leonard, M.B.; Willi, S.M.; Denburg, M.R. Type 1 diabetes is associated with an increased risk of fracture across the life span: A population-based cohort study using The Health Improvement Network (THIN). Diabetes Care 2015, 38, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Rathinavelu, S.; Guidry-Elizondo, C.; Banu, J. Molecular Modulation of Osteoblasts and Osteoclasts in Type 2 Diabetes. J. Diabetes Res. 2018, 2018, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, M.F.; Flowers, S.; Bhattacharya, R.; Faibish, D.; Behl, Y.; Kotton, D.N.; Gerstenfeld, L.; Moran, E.; Graves, D.T. FOXO1 modulates osteoblast differentiation. Bone 2011, 48, 1043–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preshaw, P.M.; Alba, A.L.; Herrera, D.; Jepsen, S.; Konstantinidis, A.; Makrilakis, K.; Taylor, R. Periodontitis and diabetes: A two-way relationship. Diabetologia 2012, 55, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Löe, H. Periodontal disease. The sixth complication of diabetes mellitus. Diabetes Care 1993, 16, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G.W.; Burt, B.A.; Becker, M.P.; Genco, R.J.; Shlossman, M. Glycemic control and alveolar bone loss progression in type 2 diabetes. Ann. Periodontol. 1998, 3, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, L.; Wang, N.; Feng, X.; Bi, L. Periodontal ligament remodeling and alveolar bone resorption during orthodontic tooth movement in rats with diabetes. Diabetes Technol. Ther. 2010, 12, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Arita, K.; Hotokezaka, H.; Hashimoto, M.; Nakano-Tajima, T.; Kurohama, T.; Kondo, T.; Darendeliler, M.A.; Yoshida, N. Effects of diabetes on tooth movement and root resorption after orthodontic force application in rats. Orthod. Craniofac. Res. 2016, 19, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Braga, S.M.G.; Taddei, S.R.d.A.; Andrade, I.; Queiroz-Junior, C.M.; Garlet, G.P.; Repeke, C.E.; Teixeira, M.M.; da Silva, T.A. Effect of diabetes on orthodontic tooth movement in a mouse model. Eur. J. Oral Sci. 2011, 119, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Ohgi, S.; Johnson, P.W. Glucose modulates growth of gingival fibroblasts and periodontal ligament cells: Correlation with expression of basic fibroblast growth factor. J. Periodontal Res. 1996, 31, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, C.Z. High glucose improves healing of periodontal wound by inhibiting proliferation and osteogenetic differentiation of human PDL cells. Int. Wound J. 2016, 13, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Taguchi, Y.; Tominaga, K.; Kimura, D.; Yamawaki, I.; Noguchi, M.; Yamauchi, N.; Tamura, I.; Tanaka, A.; Umeda, M. High Glucose Concentrations Suppress the Proliferation of Human Periodontal Ligament Stem Cells and Their Differentiation Into Osteoblasts. J. Periodontol. 2016, 87, e44–e51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Liang, Q.; Kang, W.; Ge, S. Metformin facilitates the proliferation, migration and osteogenic differentiation of periodontal ligament stem cells in vitro. Cell Biol. Int. 2019. [Google Scholar] [CrossRef] [PubMed]
- Alavi, A.; Sibbald, R.G.; Mayer, D.; Goodman, L.; Botros, M.; Armstrong, D.G.; Woo, K.; Boeni, T.; Ayello, E.A.; Kirsner, R.S. Diabetic foot ulcers: Part I. Pathophysiology and prevention. J. Am. Acad. Dermatol. 2014, 70, e1–e18. [Google Scholar] [CrossRef] [PubMed]
- Okonkwo, U.A.; DiPietro, L.A. Diabetes and Wound Angiogenesis. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]
- Dinh, T.; Tecilazich, F.; Kafanas, A.; Doupis, J.; Gnardellis, C.; Leal, E.; Tellechea, A.; Pradhan, L.; Lyons, T.E.; Giurini, J.M.; et al. Mechanisms involved in the development and healing of diabetic foot ulceration. Diabetes 2012, 61, 2937–2947. [Google Scholar] [CrossRef]
- Den Dekker, A.; Davis, F.M.; Kunkel, S.L.; Gallagher, K.A. Targeting epigenetic mechanisms in diabetic wound healing. Transl. Res. 2019, 204, 39–50. [Google Scholar] [CrossRef]
- Yevdokimova, N.Y. High glucose-induced alterations of extracellular matrix of human skin fibroblasts are not dependent on TSP-1-TGFbeta1 pathway. J. Diabetes Complicat. 2003, 17, 355–364. [Google Scholar] [CrossRef]
- Hehenberger, K.; Hansson, A. High glucose-induced growth factor resistance in human fibroblasts can be reversed by antioxidants and protein kinase C-inhibitors. Cell Biochem. Funct. 1997, 15, 197–201. [Google Scholar] [CrossRef]
- Goldstein, S.; Moerman, E.J.; Soeldner, J.S.; Gleason, R.E.; Barnett, D.M. Diabetes mellitus and genetic prediabetes. Decreased replicative capacity of cultured skin fibroblasts. J. Clin. Investig. 1979, 63, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Grazul-Bilska, A.T.; Luthra, G.; Reynolds, L.P.; Bilski, J.J.; Johnson, M.L.; Adbullah, S.A.; Redmer, D.A.; Abdullah, K.M. Effects of basic fibroblast growth factor (FGF-2) on proliferation of human skin fibroblasts in type II diabetes mellitus. Exp. Clin. Endocrinol. Diabetes 2002, 110, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Rowe, D.W.; Starman, B.J.; Fujimoto, W.Y.; Williams, R.H. Abnormalities in proliferation and protein synthesis in skin fibroblast cultures from patients with diabetes mellitus. Diabetes 1977, 26, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Lerman, O.Z.; Galiano, R.D.; Armour, M.; Levine, J.P.; Gurtner, G.C. Cellular dysfunction in the diabetic fibroblast: Impairment in migration, vascular endothelial growth factor production, and response to hypoxia. Am. J. Pathol. 2003, 162, 303–312. [Google Scholar] [CrossRef]
- Pirola, L.; Balcerczyk, A.; Okabe, J.; El-Osta, A. Epigenetic phenomena linked to diabetic complications. Nat. Rev. Endocrinol. 2010, 6, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Yamamoto, Y.; Yamamoto, H. Diabetes alters subsets of endothelial progenitor cells that reside in blood, bone marrow, and spleen. Am. J. Physiol. Cell Physiol. 2012, 302, C892–C901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loomans, C.J.M.; de Koning, E.J.P.; Staal, F.J.T.; Rookmaaker, M.B.; Verseyden, C.; de Boer, H.C.; Verhaar, M.C.; Braam, B.; Rabelink, T.J.; van Zonneveld, A.J. Endothelial progenitor cell dysfunction: A novel concept in the pathogenesis of vascular complications of type 1 diabetes. Diabetes 2004, 53, 195–199. [Google Scholar] [CrossRef]
- Tamarat, R.; Silvestre, J.-S.; Le Ricousse-Roussanne, S.; Barateau, V.; Lecomte-Raclet, L.; Clergue, M.; Duriez, M.; Tobelem, G.; Lévy, B.I. Impairment in ischemia-induced neovascularization in diabetes: Bone marrow mononuclear cell dysfunction and therapeutic potential of placenta growth factor treatment. Am. J. Pathol. 2004, 164, 457–466. [Google Scholar] [CrossRef]
- Shen, C.; Li, Q.; Zhang, Y.C.; Ma, G.; Feng, Y.; Zhu, Q.; Dai, Q.; Chen, Z.; Yao, Y.; Chen, L.; et al. Advanced glycation endproducts increase EPC apoptosis and decrease nitric oxide release via MAPK pathways. Biomed. Pharmacother. 2010, 64, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Hink, U.; Li, H.; Mollnau, H.; Oelze, M.; Matheis, E.; Hartmann, M.; Skatchkov, M.; Thaiss, F.; Stahl, R.A.; Warnholtz, A.; et al. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ. Res. 2001, 88, E14–E22. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; LoGerfo, F.W.; Guthrie, P.; Pradhan, L. Effect of hyperglycemia and neuropeptides on interleukin-8 expression and angiogenesis in dermal microvascular endothelial cells. J. Vasc. Surg. 2011, 53, 1654–1660.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, A.; Djurdjev, O.; Beaulieu, M.; Er, L. Variability and risk factors for kidney disease progression and death following attainment of stage 4 CKD in a referred cohort. Am. J. Kidney Dis. 2008, 52, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.S.; Hsu, Y.H. The role of IL-20 in chronic kidney disease and diabetic nephropathy: Pathogenic and therapeutic implications. J. Leukoc. Biol. 2018, 104, 919–923. [Google Scholar] [CrossRef] [PubMed]
- Masson, E.; Wiernsperger, N.; Lagarde, M.; El Bawab, S. Glucosamine induces cell-cycle arrest and hypertrophy of mesangial cells: Implication of gangliosides. Biochem. J. 2005, 388, 537–544. [Google Scholar] [CrossRef]
- Yano, N.; Suzuki, D.; Endoh, M.; Cao, T.N.; Dahdah, J.R.; Tseng, A.; Stabila, J.P.; McGonnigal, B.G.; Padbury, J.F.; Tseng, Y.T. High ambient glucose induces angiotensin-independent AT-1 receptor activation, leading to increases in proliferation and extracellular matrix accumulation in MES-13 mesangial cells. Biochem. J. 2009, 423, 129–143. [Google Scholar] [CrossRef] [Green Version]
- Lan, T.; Wu, T.; Gou, H.; Zhang, Q.; Li, J.; Qi, C.; He, X.; Wu, P.; Wang, L. Andrographolide suppresses high glucose-induced fibronectin expression in mesangial cells via inhibiting the AP-1 pathway. J. Cell. Biochem. 2013, 114, 2562–2568. [Google Scholar] [CrossRef]
- Wolf, G.; Schroeder, R.; Zahner, G.; Stahl, R.A.; Shankland, S.J. High glucose-induced hypertrophy of mesangial cells requires p27(Kip1), an inhibitor of cyclin-dependent kinases. Am. J. Pathol. 2001, 158, 1091–1100. [Google Scholar] [CrossRef]
- Nahman, N.S.; Leonhart, K.L.; Cosio, F.G.; Hebert, C.L. Effects of high glucose on cellular proliferation and fibronectin production by cultured human mesangial cells. Kidney Int. 1992, 41, 396–402. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wang, L.; Ma, H. Betaine alleviates high glucose-induced mesangial cell proliferation by inhibiting cell proliferation and extracellular matrix deposition via the AKT/ERK1/2/p38 MAPK pathway. Mol. Med. Rep. 2019, 20, 1754–1760. [Google Scholar] [CrossRef]
- Wolf, G.; Sharma, K.; Chen, Y.; Ericksen, M.; Ziyadeh, F.N. High glucose-induced proliferation in mesangial cells is reversed by autocrine TGF-beta. Kidney Int. 1992, 42, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Zachara, N.; Akimoto, Y.; Hart, G.W. The O-GlcNAc Modification, 3rd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2015; pp. 2015–2017. [Google Scholar]
- Marshall, S.; Bacote, V.; Traxinger, R.R. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J. Biol. Chem. 1991, 266, 4706–4712. [Google Scholar] [PubMed]
- Groves, J.A.; Maduka, A.O.; O’Meally, R.N.; Cole, R.N.; Zachara, N.E. Fatty acid synthase inhibits the O-GlcNAcase during oxidative stress. J. Biol. Chem. 2017, 292, 6493–6511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, D.C.; Krause, M.W.; Hanover, J.A. O-GlcNAc cycling: Emerging roles in development and epigenetics. Semin. Cell Dev. Biol. 2010, 21, 646–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, M.; Barzilai, N.; Liu, R.; Hu, M.; Chen, W.; Rossetti, L. Role of the glucosamine pathway in fat-induced insulin resistance. J. Clin. Investig. 1997, 99, 2173–2182. [Google Scholar] [CrossRef]
- Weigert, C.; Klopfer, K.; Kausch, C.; Brodbeck, K.; Stumvoll, M.; Häring, H.U.; Schleicher, E.D. Palmitate-induced activation of the hexosamine pathway in human myotubes: Increased expression of glutamine:fructose-6-phosphate aminotransferase. Diabetes 2003, 52, 650–656. [Google Scholar] [CrossRef]
- Robinson, K.A.; Sens, D.A.; Buse, M.G. Pre-exposure to glucosamine induces insulin resistance of glucose transport and glycogen synthesis in isolated rat skeletal muscles. Study of mechanisms in muscle and in rat-1 fibroblasts overexpressing the human insulin receptor. Diabetes 1993, 42, 1333–1346. [Google Scholar] [CrossRef]
- McClain, D.A.; Lubas, W.A.; Cooksey, R.C.; Hazel, M.; Parker, G.J.; Love, D.C.; Hanover, J.A. Altered glycan-dependent signaling induces insulin resistance and hyperleptinemia. Proc. Natl. Acad. Sci. USA 2002, 99, 10695–10699. [Google Scholar] [CrossRef] [Green Version]
- Hebert, L.F.; Daniels, M.C.; Zhou, J.; Crook, E.D.; Turner, R.L.; Simmons, S.T.; Neidigh, J.L.; Zhu, J.S.; Baron, A.D.; McClain, D.A. Overexpression of glutamine:fructose-6-phosphate amidotransferase in transgenic mice leads to insulin resistance. J. Clin. Investig. 1996, 98, 930–936. [Google Scholar] [CrossRef]
- Slawson, C.; Zachara, N.E.; Vosseller, K.; Cheung, W.D.; Lane, M.D.; Hart, G.W. Perturbations in O-linked β-N-acetylglucosamine protein modification cause severe defects in mitotic progression and cytokinesis. J. Biol. Chem. 2005, 280, 32944–32956. [Google Scholar] [CrossRef] [PubMed]
- Fisi, V.; Kátai, E.; Orbán, J.; Dossena, S.; Miseta, A.; Nagy, T. O-Linked N-Acetylglucosamine Transiently Elevates in HeLa Cells during Mitosis. Molecules 2018, 23, 1275. [Google Scholar] [CrossRef] [PubMed]
- Delporte, A.; De Zaeytijd, J.; De Storme, N.; Azmi, A.; Geelen, D.; Smagghe, G.; Guisez, Y.; Van Damme, E.J.M. Cell cycle-dependent O-GlcNAc modification of tobacco histones and their interaction with the tobacco lectin. Plant Physiol. Biochem. PPB 2014, 83, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Olivier-Van Stichelen, S.; Drougat, L.; Dehennaut, V.; El Yazidi-Belkoura, I.; Guinez, C.; Mir, A.M.; Michalski, J.C.; Vercoutter-Edouart, A.-S.; Lefebvre, T. Serum-stimulated cell cycle entry promotes ncOGT synthesis required for cyclin D expression. Oncogenesis 2012, 1, e36. [Google Scholar] [CrossRef] [PubMed]
- Sakabe, K.; Wang, Z.; Hart, G.W. Beta-N-acetylglucosamine (O-GlcNAc) is part of the histone code. Proc. Natl. Acad. Sci. USA 2010, 107, 19915–19920. [Google Scholar] [CrossRef] [PubMed]
- Forma, E.; Jóźwiak, P.; Bryś, M.; Krześlak, A. The potential role of O-GlcNAc modification in cancer epigenetics. Cell. Mol. Biol. Lett. 2014, 19, 438–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qie, S.; Diehl, J.A. Cyclin D1, cancer progression, and opportunities in cancer treatment. J. Mol. Med. (Berl.) 2016, 94, 1313–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, M.; Qiu, Z.; Zhang, S.; Fan, X.; Cai, X.; Xu, B.; Li, X.; Zhou, J.; Zhang, X.; Chu, Y.; et al. Elevated O-GlcNAcylation promotes gastric cancer cells proliferation by modulating cell cycle related proteins and ERK 1/2 signaling. Oncotarget 2016, 7, 61390–61402. [Google Scholar] [CrossRef] [Green Version]
- Sayat, R.; Leber, B.; Grubac, V.; Wiltshire, L.; Persad, S. O-GlcNAc-glycosylation of beta-catenin regulates its nuclear localization and transcriptional activity. Exp. Cell Res. 2008, 314, 2774–2787. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.H.; Suh, H.N.; Kim, M.O.; Han, H.J. Glucosamine-induced reduction of integrin β4 and plectin complex stimulates migration and proliferation in mouse embryonic stem cells. Stem Cells Dev. 2013, 22, 2975–2989. [Google Scholar] [CrossRef]
- Fardini, Y.; Dehennaut, V.; Lefebvre, T.; Issad, T. O-GlcNAcylation: A New Cancer Hallmark? Front. Endocrinol. (Lausanne) 2013, 4, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Udeshi, N.D.; Slawson, C.; Compton, P.D.; Sakabe, K.; Cheung, W.D.; Shabanowitz, J.; Hunt, D.F.; Hart, G.W. Extensive crosstalk between O-GlcNAcylation and phosphorylation regulates cytokinesis. Sci. Signal. 2010, 3. [Google Scholar] [CrossRef] [PubMed]
- Sur, S.; Agrawal, D.K. Phosphatases and kinases regulating CDC25 activity in the cell cycle: Clinical implications of CDC25 overexpression and potential treatment strategies. Mol. Cell. Biochem. 2016, 416, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, Y.; Lampkin, T. Targeting Polo-like kinase in cancer therapy. Clin. Cancer Res. 2010, 16, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Colicino, E.G.; Hehnly, H. Regulating a key mitotic regulator, polo-like kinase 1 (PLK1). Cytoskeleton (Hoboken) 2018, 75, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Lanza, C.; Tan, E.P.; Zhang, Z.; Machacek, M.; Brinker, A.E.; Azuma, M.; Slawson, C. Reduced O-GlcNAcase expression promotes mitotic errors and spindle defects. Cell Cycle 2016, 15, 1363–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slawson, C.; Lakshmanan, T.; Knapp, S.; Hart, G.W. A mitotic GlcNAcylation/phosphorylation signaling complex alters the posttranslational state of the cytoskeletal protein vimentin. Mol. Biol. Cell 2008, 19, 4130–4140. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.P.; Caro, S.; Potnis, A.; Lanza, C.; Slawson, C. O-linked N-acetylglucosamine cycling regulates mitotic spindle organization. J. Biol. Chem. 2013, 288, 27085–27099. [Google Scholar] [CrossRef] [PubMed]
- Magescas, J.; Sengmanivong, L.; Viau, A.; Mayeux, A.; Dang, T.; Burtin, M.; Nilsson, U.J.; Leffler, H.; Poirier, F.; Terzi, F.; et al. Spindle pole cohesion requires glycosylation-mediated localization of NuMA. Sci. Rep. 2017, 7, 1474. [Google Scholar] [CrossRef]
- Sakabe, K.; Hart, G.W. O-GlcNAc transferase regulates mitotic chromatin dynamics. J. Biol. Chem. 2010, 285, 34460–34468. [Google Scholar] [CrossRef]
- Leturcq, M.; Mortuaire, M.; Hardivillé, S.; Schulz, C.; Lefebvre, T.; Vercoutter-Edouart, A.S. O-GlcNAc transferase associates with the MCM2-7 complex and its silencing destabilizes MCM-MCM interactions. Cell. Mol. Life Sci. 2018, 75, 4321–4339. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Geng, Q.; Ding, Y.; Liao, J.; Dong, M.Q.; Xu, X.; Li, J. O-GlcNAcylation Antagonizes Phosphorylation of CDH1 (CDC20 Homologue 1). J. Biol. Chem. 2016, 291, 12136–12144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozcan, S.; Andrali, S.S.; Cantrell, J.E.L. Modulation of transcription factor function by O-GlcNAc modification. Biochim. Biophys. Acta 2010, 1799, 353–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donovan, K.; Alekseev, O.; Qi, X.; Cho, W.; Azizkhan-Clifford, J. O-GlcNAc modification of transcription factor Sp1 mediates hyperglycemia-induced VEGF-A upregulation in retinal cells. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7862–7873. [Google Scholar] [CrossRef] [PubMed]
- Wells, L.; Slawson, C.; Hart, G.W. The E2F-1 associated retinoblastoma-susceptibility gene product is modified by O-GlcNAc. Amino Acids 2011, 40, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Liu, F.; Tao, T.; Zhang, D.; Liu, X.; Zhu, G.; Xu, Z.; Ni, R.; Shen, A. Modification of p27 with O-linked N-acetylglucosamine regulates cell proliferation in hepatocellular carcinoma. Mol. Carcinog. 2017, 56, 258–271. [Google Scholar] [CrossRef]
- Wang, L.; Chen, S.; Zhang, Z.; Zhang, J.; Mao, S.; Zheng, J.; Xuan, Y.; Liu, M.; Cai, K.; Zhang, W.; et al. Suppressed OGT expression inhibits cell proliferation while inducing cell apoptosis in bladder cancer. BMC Cancer 2018, 18, 1141. [Google Scholar] [CrossRef]
- Dias, W.B.; Hart, G.W. O-GlcNAc modification in diabetes and Alzheimer’s disease. Mol. Biosyst. 2007, 3, 766–772. [Google Scholar] [CrossRef]
- Banerjee, P.S.; Lagerlöf, O.; Hart, G.W. Roles of O-GlcNAc in chronic diseases of aging. Mol. Aspects Med. 2016, 51, 1–15. [Google Scholar] [CrossRef]
- Hanover, J.A.; Chen, W.; Bond, M.R. O-GlcNAc in cancer: An Oncometabolism-fueled vicious cycle. J. Bioenerg. Biomembr. 2018, 50, 155–173. [Google Scholar] [CrossRef]
- Chatham, J.C.; Marchase, R.B. Protein O-GlcNAcylation: A critical regulator of the cellular response to stress. Curr. Signal Transduct. Ther. 2010, 5, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Hanover, J.A.; Lai, Z.; Lee, G.; Lubas, W.A.; Sato, S.M. Elevated O-linked N-acetylglucosamine metabolism in pancreatic beta-cells. Arch. Biochem. Biophys. 1999, 362, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Miyazaki, J.; Hart, G.W. The transcription factor PDX-1 is post-translationally modified by O-linked N-acetylglucosamine and this modification is correlated with its DNA binding activity and insulin secretion in min6 beta-cells. Arch. Biochem. Biophys. 2003, 415, 155–163. [Google Scholar] [CrossRef]
- Durning, S.P.; Flanagan-Steet, H.; Prasad, N.; Wells, L. O-Linked β- N -acetylglucosamine (O-GlcNAc) Acts as a Glucose Sensor to Epigenetically Regulate the Insulin Gene in Pancreatic Beta Cells. J. Biol. Chem. 2016, 291, 2107–2118. [Google Scholar] [CrossRef] [PubMed]
- Filhoulaud, G.; Guillemain, G.; Scharfmann, R. The hexosamine biosynthesis pathway is essential for pancreatic beta cell development. J. Biol. Chem. 2009, 284, 24583–24594. [Google Scholar] [CrossRef] [PubMed]
- Guillemain, G.; Filhoulaud, G.; Da Silva-Xavier, G.; Rutter, G.A.; Scharfmann, R. Glucose is necessary for embryonic pancreatic endocrine cell differentiation. J. Biol. Chem. 2007, 282, 15228–15237. [Google Scholar] [CrossRef] [PubMed]
- Vanderford, N.L.; Andrali, S.S.; Ozcan, S. Glucose induces MafA expression in pancreatic beta cell lines via the hexosamine biosynthetic pathway. J. Biol. Chem. 2007, 282, 1577–1584. [Google Scholar] [CrossRef]
- Liu, K.; Paterson, A.J.; Chin, E.; Kudlow, J.E. Glucose stimulates protein modification by O-linked GlcNAc in pancreatic beta cells: Linkage of O-linked GlcNAc to beta cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 2820–2825. [Google Scholar] [CrossRef]
- Zachara, N.E.; Hart, G.W. O-GlcNAc a sensor of cellular state: The role of nucleocytoplasmic glycosylation in modulating cellular function in response to nutrition and stress. Biochim. Biophys. Acta 2004, 1673, 13–28. [Google Scholar] [CrossRef]
- De Queiroz, R.M.; Carvalho, E.; Dias, W.B. O-GlcNAcylation: The Sweet Side of the Cancer. Front. Oncol. 2014, 4, 132. [Google Scholar] [CrossRef] [Green Version]
- Lynch, T.P.; Reginato, M.J. O-GlcNAc transferase: A sweet new cancer target. Cell Cycle 2011, 10, 1712–1713. [Google Scholar] [CrossRef] [PubMed]
- Harosh-Davidovich, S.B.; Khalaila, I. O-GlcNAcylation affects β-catenin and E-cadherin expression, cell motility and tumorigenicity of colorectal cancer. Exp. Cell Res. 2018, 364, 42–49. [Google Scholar] [CrossRef] [PubMed]
- De Queiroz, R.M.; Madan, R.; Chien, J.; Dias, W.B.; Slawson, C. Changes in O-Linked N-Acetylglucosamine (O-GlcNAc) Homeostasis Activate the p53 Pathway in Ovarian Cancer Cells. J. Biol. Chem. 2016, 291, 18897–18914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Y.; Gao, J.; Han, C.; Zhang, X.; Liu, H.; Ma, L.; Sun, X.; Yu, W. O-GlcNAcylation is increased in prostate cancer tissues and enhances malignancy of prostate cancer cells. Mol. Med. Rep. 2014, 10, 897–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Wang, L.; Liu, J.; Zhang, P.; An, M.; Han, C.; Li, Y.; Guan, X.; Zhang, K. O-GlcNAcylation modulates Bmi-1 protein stability and potential oncogenic function in prostate cancer. Oncogene 2017, 36, 6293–6305. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Vocadlo, D.J.; Vosseller, K. Hyper-O-GlcNAcylation is anti-apoptotic and maintains constitutive NF-κB activity in pancreatic cancer cells. J. Biol. Chem. 2013, 288, 15121–15130. [Google Scholar] [CrossRef]
- Shin, H.; Cha, H.-J.; Na, K.; Lee, M.J.; Cho, J.Y.; Kim, C.Y.; Kim, E.K.; Kang, C.M.; Kim, H.; Paik, Y.K. O -GlcNAcylation of the Tumor Suppressor FOXO3 Triggers Aberrant Cancer Cell Growth. Cancer Res. 2018, 78, 1214–1224. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.S.; Gupta, V.K.; Dauer, P.; Kesh, K.; Hadad, R.; Giri, B.; Chandra, A.; Dudeja, V.; Slawson, C.; Banerjee, S.; et al. O-GlcNAc modification of oncogenic transcription factor Sox2 promotes protein stability and regulates self-renewal in pancreatic cancer. bioRxiv 2018, 345223. [Google Scholar] [CrossRef]
- Kanwal, S.; Fardini, Y.; Pagesy, P.; N’Tumba-Byn, T.; Pierre-Eugène, C.; Masson, E.; Hampe, C.; Issad, T. O-GlcNAcylation-Inducing Treatments Inhibit Estrogen Receptor α Expression and Confer Resistance to 4-OH-Tamoxifen in Human Breast Cancer-Derived MCF-7 Cells. PLoS ONE 2013, 8, 1–17. [Google Scholar] [CrossRef]
- Mi, W.; Gu, Y.; Han, C.; Liu, H.; Fan, Q.; Zhang, X.; Cong, Q.; Yu, W. O-GlcNAcylation is a novel regulator of lung and colon cancer malignancy. Biochim. Biophys. Acta 2011, 1812, 514–519. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Tomic, J.; Wen, F.; Shaha, S.; Bahlo, A.; Harrison, R.; Dennis, J.W.; Williams, R.; Gross, B.J.; Walker, S.; et al. Aberrant O-GlcNAcylation characterizes chronic lymphocytic leukemia. Leukemia 2010, 24, 1588–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asthana, A.; Ramakrishnan, P.; Vicioso, Y.; Zhang, K.; Parameswaran, R. Hexosamine Biosynthetic Pathway Inhibition Leads to AML Cell Differentiation and Cell Death. Mol. Cancer Ther. 2018, 17, 2226–2237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, A.; Kim, S.H.; Kim, M.J.; Choi, M.Y.; Kang, S.S.; Cho, G.J.; Kim, Y.S.; Choi, J.Y.; Choi, W.S. O-GlcNAcylation of NF-κB Promotes Lung Metastasis of Cervical Cancer Cells via Upregulation of CXCR4 Expression. Mol. Cells 2017, 40, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Lima, V.V.; Dela Justina, V.; Dos Passos, R.R.; Volpato, G.T.; Souto, P.C.S.; San Martin, S.; Giachini, F.R. O-GlcNAc Modification During Pregnancy: Focus on Placental Environment. Front. Physiol. 2018, 9, 1263. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wells, L.; Comer, F.I.; Parker, G.J.; Hart, G.W. Dynamic O-glycosylation of nuclear and cytosolic proteins: Cloning and characterization of a neutral, cytosolic beta-N-acetylglucosaminidase from human brain. J. Biol. Chem. 2001, 276, 9838–9845. [Google Scholar] [CrossRef]
- Shafi, R.; Iyer, S.P.; Ellies, L.G.; O’Donnell, N.; Marek, K.W.; Chui, D.; Hart, G.W.; Marth, J.D. The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc. Natl. Acad. Sci. USA 2000, 97, 5735–5739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.R.; Song, M.; Lee, H.; Jeon, Y.; Choi, E.J.; Jang, H.J.; Moon, H.Y.; Byun, H.Y.; Kim, E.K.; Kim, D.H.; et al. O-GlcNAcase is essential for embryonic development and maintenance of genomic stability. Aging Cell 2012, 11, 439–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefebvre, T.; Baert, F.; Bodart, J.F.; Flament, S.; Michalski, J.C.; Vilain, J.P. Modulation of O-GlcNAc glycosylation during xenopus oocyte maturation. J. Cell. Biochem. 2004, 93, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Dehennaut, V.; Lefebvre, T.; Sellier, C.; Leroy, Y.; Gross, B.; Walker, S.; Cacan, R.; Michalski, J.-C.; Vilain, J.P.; Bodart, J.F. O-linked N-acetylglucosaminyltransferase inhibition prevents G2/M transition in Xenopus laevis oocytes. J. Biol. Chem. 2007, 282, 12527–12536. [Google Scholar] [CrossRef] [PubMed]
- Dehennaut, V.; Hanoulle, X.; Bodart, J.F.; Vilain, J.P.; Michalski, J.C.; Landrieu, I.; Lippens, G.; Lefebvre, T. Microinjection of recombinant O-GlcNAc transferase potentiates Xenopus oocytes M-phase entry. Biochem. Biophys. Res. Commun. 2008, 369, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.T.; Romar, R.; Pavone, M.E.; Soriano-Úbeda, C.; Zhang, J.; Slawson, C.; Duncan, F.E. Disruption of O-GlcNAc homeostasis during mammalian oocyte meiotic maturation impacts fertilization. Mol. Reprod. Dev. 2019, 86, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Moley, K.H.; Vaughn, W.K.; DeCherney, A.H.; Diamond, M.P. Effect of diabetes mellitus on mouse pre-implantation embryo development. J. Reprod. Fertil. 1991, 93, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Pantaleon, M.; Tan, H.Y.; Kafer, G.R.; Kaye, P.L. Toxic effects of hyperglycemia are mediated by the hexosamine signaling pathway and o-linked glycosylation in early mouse embryos. Biol. Reprod. 2010, 82, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Andres, L.M.; Blong, I.W.; Evans, A.C.; Rumachik, N.G.; Yamaguchi, T.; Pham, N.D.; Thompson, P.; Kohler, J.J.; Bertozzi, C.R. Chemical Modulation of Protein O-GlcNAcylation via OGT Inhibition Promotes Human Neural Cell Differentiation. ACS Chem. Biol. 2017, 12, 2030–2039. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.T.; Kim, H.; Lu, W.; He, Q.; Liu, D.; Goodell, M.A.; Wan, M.; Songyang, Z. Ten-eleven translocation 1 (Tet1) is regulated by O-linked N-acetylglucosamine transferase (Ogt) for target gene repression in mouse embryonic stem cells. J. Biol. Chem. 2013, 288, 20776–20784. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Cao, L.; Reece, E.A.; Zhao, Z. Impact of protein O-GlcNAcylation on neural tube malformation in diabetic embryopathy. Sci. Rep. 2017, 7, 11107. [Google Scholar] [CrossRef] [PubMed]
- Parween, S.; Varghese, D.S.; Ardah, M.T.; Prabakaran, A.D.; Mensah-Brown, E.; Emerald, B.S.; Ansari, S.A. Higher O-GlcNAc Levels Are Associated with Defects in Progenitor Proliferation and Premature Neuronal Differentiation during in-Vitro Human Embryonic Cortical Neurogenesis. Front. Cell. Neurosci. 2017, 11, 415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, T.P.; Ferrer, C.M.; Jackson, S.R.; Shahriari, K.S.; Vosseller, K.; Reginato, M.J. Critical role of O-Linked β-N-acetylglucosamine transferase in prostate cancer invasion, angiogenesis, and metastasis. J. Biol. Chem. 2012, 287, 11070–11081. [Google Scholar] [CrossRef]
- Zibrova, D.; Vandermoere, F.; Göransson, O.; Peggie, M.; Mariño, K.V.; Knierim, A.; Spengler, K.; Weigert, C.; Viollet, B.; Morrice, N.A.; et al. GFAT1 phosphorylation by AMPK promotes VEGF-induced angiogenesis. Biochem. J. 2017, 474, 983–1001. [Google Scholar] [CrossRef]
- Luo, B.; Soesanto, Y.; McClain, D.A. Protein modification by O-linked GlcNAc reduces angiogenesis by inhibiting Akt activity in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 651–657. [Google Scholar] [CrossRef]
- Huang, S.M.; Wu, C.S.; Chiu, M.H.; Yang, H.J.; Chen, G.S.; Lan, C.C.E. High-glucose environment induced intracellular O-GlcNAc glycosylation and reduced galectin-7 expression in keratinocytes: Implications on impaired diabetic wound healing. J. Dermatol. Sci. 2017, 87, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Frykberg, R.G.; Banks, J. Challenges in the Treatment of Chronic Wounds. Adv. Wound Care 2015, 4, 560–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Runager, K.; Bektas, M.; Berkowitz, P.; Rubenstein, D.S. Targeting O-glycosyltransferase (OGT) to promote healing of diabetic skin wounds. J. Biol. Chem. 2014, 289, 5462–5466. [Google Scholar] [CrossRef] [PubMed]
- Waltenberger, J.; Lange, J.; Kranz, A. Vascular endothelial growth factor-A-induced chemotaxis of monocytes is attenuated in patients with diabetes mellitus: A potential predictor for the individual capacity to develop collaterals. Circulation 2000, 102, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Baudoin, L.; Issad, T. O-GlcNAcylation and Inflammation: A Vast Territory to Explore. Front. Endocrinol. (Lausanne) 2014, 5, 235. [Google Scholar] [CrossRef] [PubMed]
- De Jesus, T.; Shukla, S.; Ramakrishnan, P. Too sweet to resist: Control of immune cell function by O-GlcNAcylation. Cell. Immunol. 2018, 333, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Machacek, M.; Saunders, H.; Zhang, Z.; Tan, E.P.; Li, J.; Li, T.; Villar, M.T.; Artigues, A.; Lydic, T.; Cork, G.; et al. Elevated O-GlcNAcylation enhances pro-inflammatory Th17 function by altering the intracellular lipid microenvironment. J. Biol. Chem. 2019, 294, 8973–8990. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.L.; Chiang, M.F.; Hsu, P.H.; Tsai, D.Y.; Hung, K.H.; Wang, Y.H.; Angata, T.; Lin, K.I. O-GlcNAcylation is required for B cell homeostasis and antibody responses. Nat. Commun. 2017, 8, 1854. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, L.A.; Gao, Y.; Hou, X.; Lauvau, G.; Yates, J.R.; Wu, P. Profiling of Protein O-GlcNAcylation in Murine CD8+ Effector- and Memory-like T Cells. ACS Chem. Biol. 2017, 12, 3031–3038. [Google Scholar] [CrossRef]
- Golks, A.; Guerini, D. The O-linked N-acetylglucosamine modification in cellular signalling and the immune system. Protein modifications: Beyond the usual suspects review series. EMBO Rep. 2008, 9, 748–753. [Google Scholar] [CrossRef]
- Springhorn, C.; Matsha, T.E.; Erasmus, R.T.; Essop, M.F. Exploring leukocyte O-GlcNAcylation as a novel diagnostic tool for the earlier detection of type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2012, 97, 4640–4649. [Google Scholar] [CrossRef] [PubMed]
- Myslicki, J.P.; Shearer, J.; Hittel, D.S.; Hughey, C.C.; Belke, D.D. O-GlcNAc modification is associated with insulin sensitivity in the whole blood of healthy young adult males. Diabetol. Metab. Syndr. 2014, 6, 96. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhou, P.; Li, X.; Shi, Q.; Li, D.; Ju, X. Bitterness in sugar: O-GlcNAcylation aggravates pre-B acute lymphocytic leukemia through glycolysis via the PI3K/Akt/c-Myc pathway. Am. J. Cancer Res. 2017, 7, 1337–1349. [Google Scholar] [PubMed]
- Ballotari, P.; Vicentini, M.; Manicardi, V.; Gallo, M.; Chiatamone Ranieri, S.; Greci, M.; Rossi, G.P. Diabetes and risk of cancer incidence: Results from a population-based cohort study in northern Italy. BMC Cancer 2017, 17, 703. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, W.A.; Jackson, S.; Law, R.E. Control of Vascular Cell Proliferation and Migration by PPAR: A new approach to the macrovascular complications of diabetes. Diabetes Care 2001, 24, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, C.M.; Lynch, T.P.; Sodi, V.L.; Falcone, J.N.; Schwab, L.P.; Peacock, D.L.; Vocadlo, D.J.; Seagroves, T.N.; Reginato, M.J. O-GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of the HIF-1 pathway. Mol. Cell 2014, 54, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Slawson, C.; Hart, G.W. O-GlcNAc signalling: Implications for cancer cell biology. Nat. Rev. Cancer 2011, 11, 678–684. [Google Scholar] [CrossRef]
- Liu, Y.; Ren, Y.; Cao, Y.; Huang, H.; Wu, Q.; Li, W.; Wu, S.; Zhang, J. Discovery of a Low Toxicity O-GlcNAc Transferase (OGT) Inhibitor by Structure-based Virtual Screening of Natural Products. Sci. Rep. 2017, 7, 12334. [Google Scholar] [CrossRef]
- Liu, T.W.; Zandberg, W.F.; Gloster, T.M.; Deng, L.; Murray, K.D.; Shan, X.; Vocadlo, D.J. Metabolic Inhibitors of O-GlcNAc Transferase That Act In Vivo Implicate Decreased O-GlcNAc Levels in Leptin-Mediated Nutrient Sensing. Angew. Chem. Int. Ed. 2018, 57, 7644–7648. [Google Scholar] [CrossRef]
- Dorfmueller, H.C.; Borodkin, V.S.; Schimpl, M.; Zheng, X.; Kime, R.; Read, K.D.; Van Aalten, D.M.F. Cell-penetrant, nanomolar O-GlcNAcase inhibitors selective against lysosomal hexosaminidases. Chem. Biol. 2010, 17, 1250–1255. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Cancer Type | Change in O-GlcNAc | Proposed Effects | References |
|---|---|---|---|
| Colorectal cc. | increase | Increased cell migration by up-regulating of β-catenin and E-cadherin levels. | [236] |
| Ovarian cc. | decrease | Loss of stability and nuclear translocation of tumor suppressor p53. | [237] |
| Prostate cc. | increase | Increased cell migration by down-regulating E-cadherin levels (contradicting data found in [236]). | [238] |
| Prostate cc. | increase | Promotes Bmi-1 stability and its oncogenic activity. | [239] |
| Pancreatic cc. | increase | Increased oncogenic NF-κB transcriptional activity. O-GlcNAc modified FOXO3 suppresses p21 thus cell cycle is accelerated. Stabilization of oncogenic transcription factor Sox2 by O-GlcNAc modification. | [240] [241] [242] |
| Breast cc. | increase | Tamoxifen resistance by reducing expression level of estrogen receptor alpha. | [243] |
| Lung and colon cc. | increase | Increased invasion and enhanced anchorage-independent growth | [244] |
| CLL | increase | p53, c-myc and Akt were O-GlcNAc modification. O-GlcNAc levels did not correlate with the clinical aggressiveness of CLL. | [245] |
| AML | increase | Increased cell proliferation and sustained undifferentiated state. | [246] |
| Lung metastasis of cervical cc. | increase | O-GlcNAc modification of NF-κB upregulates CXCR4 chemokine receptor. | [247] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagy, T.; Fisi, V.; Frank, D.; Kátai, E.; Nagy, Z.; Miseta, A. Hyperglycemia-Induced Aberrant Cell Proliferation; A Metabolic Challenge Mediated by Protein O-GlcNAc Modification. Cells 2019, 8, 999. https://doi.org/10.3390/cells8090999
Nagy T, Fisi V, Frank D, Kátai E, Nagy Z, Miseta A. Hyperglycemia-Induced Aberrant Cell Proliferation; A Metabolic Challenge Mediated by Protein O-GlcNAc Modification. Cells. 2019; 8(9):999. https://doi.org/10.3390/cells8090999
Chicago/Turabian StyleNagy, Tamás, Viktória Fisi, Dorottya Frank, Emese Kátai, Zsófia Nagy, and Attila Miseta. 2019. "Hyperglycemia-Induced Aberrant Cell Proliferation; A Metabolic Challenge Mediated by Protein O-GlcNAc Modification" Cells 8, no. 9: 999. https://doi.org/10.3390/cells8090999
APA StyleNagy, T., Fisi, V., Frank, D., Kátai, E., Nagy, Z., & Miseta, A. (2019). Hyperglycemia-Induced Aberrant Cell Proliferation; A Metabolic Challenge Mediated by Protein O-GlcNAc Modification. Cells, 8(9), 999. https://doi.org/10.3390/cells8090999