Context-Dependent Strategies for Enhanced Genome Editing of Genodermatoses


Abstract
:1. Introduction
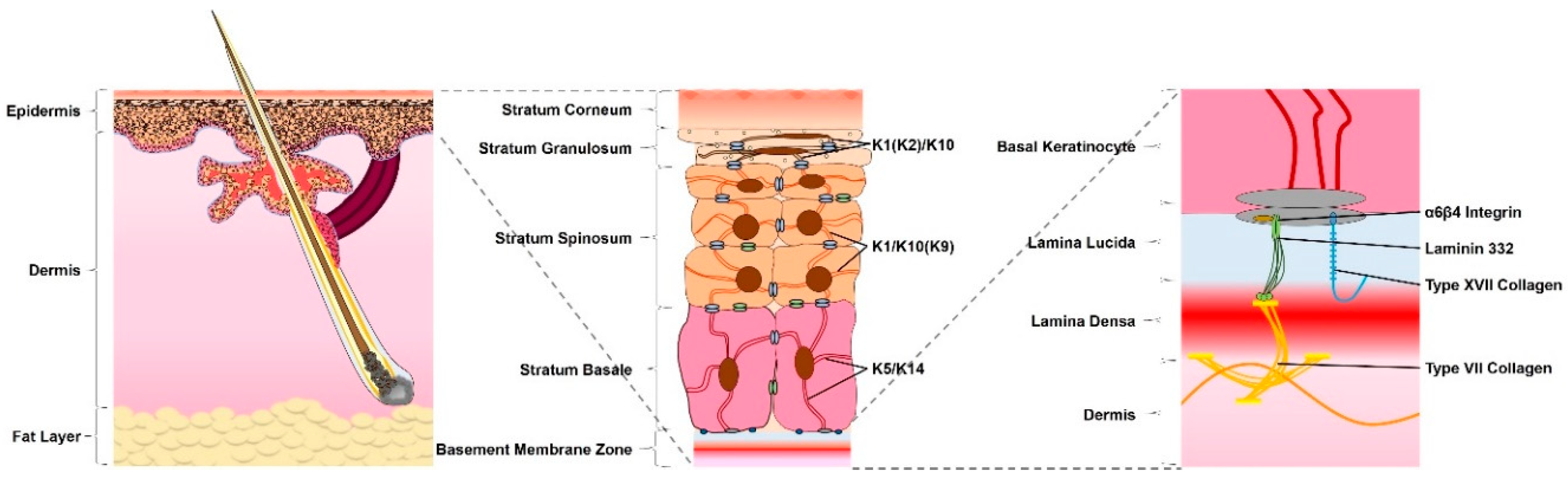
1.1. The Anatomy of the Skin
1.2. Gene Therapies for Genodermatoses
1.3. DNA Repair Pathways
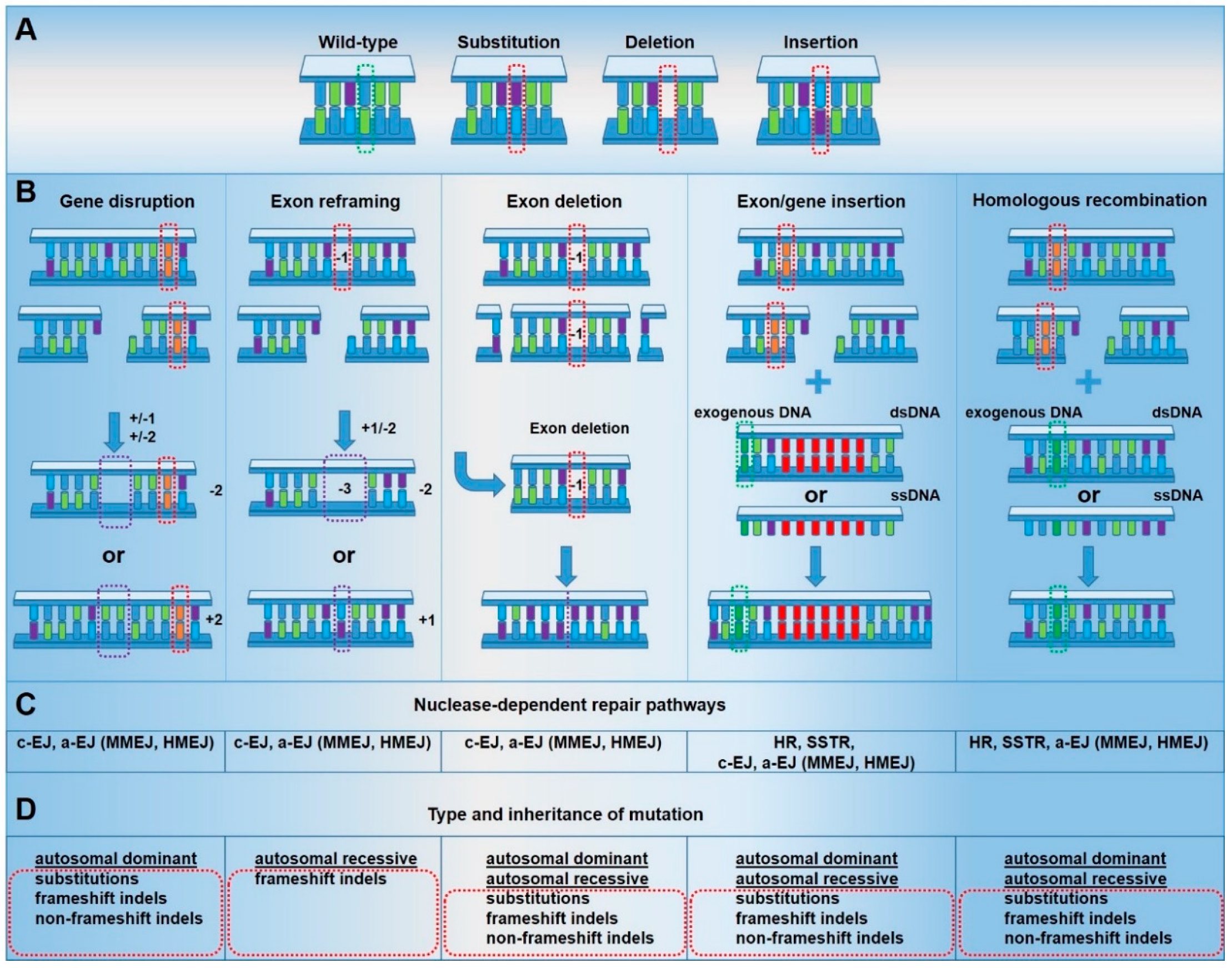
1.4. Genome Editing Strategies for Genodermatoses
2. Genome Editing Applications for Genodermatoses
2.1. Gene Disruption
2.2. Exon Reframing
2.3. Exon Deletion
2.4. Exon/Gene Insertion
2.5. Homologous Recombination
3. Future Prospects of Gene Editing in Genodermatoses
Author Contributions
Funding
Conflicts of Interest
References
- Rognoni, E.; Watt, F.M. Skin Cell Heterogeneity in Development, Wound Healing, and Cancer. Trends Cell Biol. 2018, 28, 709–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, Y.C.; Li, L.; Fuchs, E. Emerging interactions between skin stem cells and their niches. Nat. Med. 2014, 20, 847–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, E. Cell biology: More than skin deep. J. Cell Biol. 2015, 209, 629–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Has, C.; Bruckner-Tuderman, L. Molecular and diagnostic aspects of genetic skin fragility. J. Dermatol. Sci. 2006, 44, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Simpson, C.L.; Patel, D.M.; Green, K.J. Deconstructing the skin: Cytoarchitectural determinants of epidermal morphogenesis. Nat. Rev. Mol. Cell Biol. 2011, 12, 565–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.K.; Lin, H.H.; Harn, H.I.; Hughes, M.W.; Tang, M.J.; Yang, C.C. Mechanical forces in skin disorders. J. Dermatol. Sci. 2018, 90, 232–240. [Google Scholar] [CrossRef] [Green Version]
- Feramisco, J.D.; Sadreyev, R.I.; Murray, M.L.; Grishin, N.V.; Tsao, H. Phenotypic and genotypic analyses of genetic skin disease through the Online Mendelian Inheritance in Man (OMIM) database. J. Investig. Dermatol. 2009, 129, 2628–2636. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, J.; Schubert, S.; Emmert, S. Xeroderma pigmentosum: Diagnostic procedures, interdisciplinary patient care, and novel therapeutic approaches. J. Dtsch. Dermatol. Ges. 2014, 12, 867–872. [Google Scholar] [CrossRef] [Green Version]
- Mavilio, F.; Pellegrini, G.; Ferrari, S.; Di, N.F.; Di, I.E.; Recchia, A.; Maruggi, G.; Ferrari, G.; Provasi, E.; Bonini, C.; et al. Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat. Med. 2006, 12, 1397–1402. [Google Scholar] [CrossRef]
- Bauer, J.W.; Koller, J.; Murauer, E.M.; De, R.L.; Enzo, E.; Carulli, S.; Bondanza, S.; Recchia, A.; Muss, W.; Diem, A.; et al. Closure of a large chronic wound through transplantation of gene-corrected epidermal stem cells. J. Investig. Dermatol. 2016, 137, 778–781. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, T.; Rothoeft, T.; Teig, N.; Bauer, J.W.; Pellegrini, G.; DeRosa, L.; Scaglione, D.; Reichelt, J.; Klausegger, A.; Kneisz, D.; et al. Regeneration of the entire human epidermis using transgenic stem cells. Nature 2017, 551, 327–332. [Google Scholar] [CrossRef] [PubMed]
- March, O.P.; Reichelt, J.; Koller, U. Gene editing for skin diseases: Designer nucleases as tools for gene therapy of skin fragility disorders. Exp. Physiol. 2018, 103, 449–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Overbeek, M.; Capurso, D.; Carter, M.M.; Thompson, M.S.; Frias, E.; Russ, C.; Reece-Hoyes, J.S.; Nye, C.; Gradia, S.; Vidal, B.; et al. DNA Repair Profiling Reveals Nonrandom Outcomes at Cas9-Mediated Breaks. Mol. Cell 2016, 63, 633–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkman, E.K.; Chen, T.; de Haas, M.; Holland, H.A.; Akhtar, W.; van Steensel, B. Kinetics and Fidelity of the Repair of Cas9-Induced Double-Strand DNA Breaks. Mol. Cell 2018, 70, 801–813. [Google Scholar] [CrossRef] [Green Version]
- Her, J.; Bunting, S.F. How cells ensure correct repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10502–10511. [Google Scholar] [CrossRef] [Green Version]
- Danner, E.; Bashir, S.; Yumlu, S.; Wurst, W.; Wefers, B.; Kuhn, R. Control of gene editing by manipulation of DNA repair mechanisms. Mamm. Genome 2017, 28, 262–274. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell. Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Javadekar, S.M.; Pandey, M.; Srivastava, M.; Kumari, R.; Raghavan, S.C. Homology and enzymatic requirements of microhomology-dependent alternative end joining. Cell Death Dis. 2015, 6, e1697. [Google Scholar] [CrossRef] [Green Version]
- McVey, M.; Lee, S.E. MMEJ repair of double-strand breaks (director’s cut): Deleted sequences and alternative endings. Trends Genet. 2008, 24, 529–538. [Google Scholar] [CrossRef] [Green Version]
- Kowalczykowski, S.C. An Overview of the Molecular Mechanisms of Recombinational DNA Repair. Cold Spring Harb. Perspect. Biol. 2015, 7, a016410. [Google Scholar] [CrossRef] [Green Version]
- Sfeir, A.; Symington, L.S. Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem. Sci. 2015, 40, 701–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodgers, K.; McVey, M. Error-Prone Repair of DNA Double-Strand Breaks. J. Cell Physiol. 2016, 231, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousefzadeh, M.J.; Wyatt, D.W.; Takata, K.; Mu, Y.; Hensley, S.C.; Tomida, J.; Bylund, G.O.; Doublie, S.; Johansson, E.; Ramsden, D.A.; et al. Mechanism of suppression of chromosomal instability by DNA polymerase POLQ. PLoS Genet. 2014, 10, e1004654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schimmel, J.; Kool, H.; van Schendel, R.; Tijsterman, M. Mutational signatures of non-homologous and polymerase theta-mediated end-joining in embryonic stem cells. EMBO J. 2017, 36, 3634–3649. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, D.N.; Haber, J.E. Repair of a Site-Specific DNA Cleavage: Old-School Lessons for Cas9-Mediated Gene Editing. ACS Chem. Biol. 2018, 13, 397–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karanam, K.; Kafri, R.; Loewer, A.; Lahav, G. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol. Cell 2012, 47, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Bizard, A.H.; Hickson, I.D. The dissolution of double Holliday junctions. Cold Spring Harb. Perspect. Biol. 2014, 6, a016477. [Google Scholar] [CrossRef]
- Wyatt, H.D.; West, S.C. Holliday junction resolvases. Cold Spring Harb. Perspect. Biol. 2014, 6, a023192. [Google Scholar] [CrossRef] [Green Version]
- Morrical, S.W. DNA-pairing and annealing processes in homologous recombination and homology-directed repair. Cold Spring Harb. Perspect. Biol. 2015, 7, a016444. [Google Scholar] [CrossRef]
- Hustedt, N.; Durocher, D. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2016, 19, 1–9. [Google Scholar] [CrossRef]
- Shinkuma, S.; Guo, Z.; Christiano, A.M. Site-specific genome editing for correction of induced pluripotent stem cells derived from dominant dystrophic epidermolysis bullosa. Proc. Natl. Acad. Sci. USA 2016, 113, 5676–5681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aushev, M.; Koller, U.; Mussolino, C.; Cathomen, T.; Reichelt, J. Traceless Targeting and Isolation of Gene- Edited Immortalized Keratinocytes from Epidermolysis Bullosa Simplex Patients. Mol. Ther. Methods Clin. Dev. 2017, 6, 112–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luan, X.R.; Chen, X.L.; Tang, Y.X.; Zhang, J.Y.; Gao, X.; Ke, H.P.; Lin, Z.Y.; Zhang, X.N. CRISPR/Cas9-Mediated Treatment Ameliorates the Phenotype of the Epidermolytic Palmoplantar Keratoderma-like Mouse. Mol. Ther. Nucleic Acids 2018, 12, 220–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- March, O.P.; Lettner, T.; Klausegger, A.; Ablinger, M.; Kocher, T.; Hainzl, S.; Peking, P.; Lackner, N.; Rajan, N.; Hofbauer, J.P.; et al. Gene Editing-Mediated Disruption of Epidermolytic Ichthyosis-Associated KRT10 Alleles Restores Filament Stability in Keratinocytes. J. Investig. Dermatol. 2019, 139, 1699–1710. [Google Scholar] [CrossRef] [Green Version]
- Chamorro, C.; Mencia, A.; Almarza, D.; Duarte, B.; Buning, H.; Sallach, J.; Hausser, I.; Del, R.M.; Larcher, F.; Murillas, R. Gene Editing for the Efficient Correction of a Recurrent COL7A1 Mutation in Recessive Dystrophic Epidermolysis Bullosa Keratinocytes. Mol. Ther. Nucleic Acids 2016, 5, e307. [Google Scholar] [CrossRef] [Green Version]
- Mencia, A.; Chamorro, C.; Bonafont, J.; Duarte, B.; Holguin, A.; Illera, N.; Llames, S.G.; Escamez, M.J.; Hausser, I.; Del Rio, M.; et al. Deletion of a Pathogenic Mutation-Containing Exon of COL7A1 Allows Clonal Gene Editing Correction of RDEB Patient Epidermal Stem Cells. Mol. Ther. Nucleic Acids 2018, 11, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Takashima, S.; Shinkuma, S.; Fujita, Y.; Nomura, T.; Ujiie, H.; Natsuga, K.; Iwata, H.; Nakamura, H.; Vorobyev, A.; Abe, R.; et al. Efficient Gene Reframing Therapy for Recessive Dystrophic Epidermolysis Bullosa with CRISPR/Cas9. J. Investig. Dermatol. 2019, 139, 1711–1721. [Google Scholar] [CrossRef]
- Wu, W.; Lu, Z.; Li, F.; Wang, W.; Qian, N.; Duan, J.; Zhang, Y.; Wang, F.; Chen, T. Efficient in vivo gene editing using ribonucleoproteins in skin stem cells of recessive dystrophic epidermolysis bullosa mouse model. Proc. Natl. Acad. Sci. USA 2017, 114, 1660–1665. [Google Scholar] [CrossRef] [Green Version]
- Bonafont, J.; Mencia, A.; Garcia, M.; Torres, R.; Rodriguez, S.; Carretero, M.; Chacon-Solano, E.; Modamio-Hoybjor, S.; Marinas, L.; Leon, C.; et al. Clinically Relevant Correction of Recessive Dystrophic Epidermolysis Bullosa by Dual sgRNA CRISPR/Cas9-Mediated Gene Editing. Mol. Ther. 2019, 27, 986–998. [Google Scholar] [CrossRef]
- Coluccio, A.; Miselli, F.; Lombardo, A.; Marconi, A.; Malagoli Tagliazucchi, G.; Goncalves, M.A.; Pincelli, C.; Maruggi, G.; Del Rio, M.; Naldini, L.; et al. Targeted gene addition in human epithelial stem cells by zinc-finger nuclease-mediated homologous recombination. Mol. Ther. 2013, 21, 1695–1704. [Google Scholar] [CrossRef] [Green Version]
- Osborn, M.J.; Starker, C.G.; McElroy, A.N.; Webber, B.R.; Riddle, M.J.; Xia, L.; DeFeo, A.P.; Gabriel, R.; Schmidt, M.; von Kalle, C.; et al. TALEN-based gene correction for epidermolysis bullosa. Mol. Ther. 2013, 21, 1151–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastiano, V.; Zhen, H.H.; Haddad, B.; Bashkirova, E.; Melo, S.P.; Wang, P.; Leung, T.L.; Siprashvili, Z.; Tichy, A.; Li, J.; et al. Human COL7A1-corrected induced pluripotent stem cells for the treatment of recessive dystrophic epidermolysis bullosa. Sci. Transl. Med. 2014, 6, 264ra163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webber, B.R.; Osborn, M.; McElroy, A.; Twaroski, K.; Lonetree, C.L.; DeFeo, A.P.; Xia, L.; Eide, C.; Lees, C.J.; Riddle, M.J.; et al. CRISPR/Cas9-based genetic correction for recessive dystrophic epidermolysis bullosa. NPJ Regener. Med. 2016, 1, 16014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hainzl, S.; Peking, P.; Kocher, T.; Murauer, E.M.; Larcher, F.; Del Rio, M.; Duarte, B.; Steiner, M.; Klausegger, A.; Bauer, J.W.; et al. COL7A1 Editing via CRISPR/Cas9 in Recessive Dystrophic Epidermolysis Bullosa. Mol. Ther. 2017, 25, 2573–2584. [Google Scholar] [CrossRef] [Green Version]
- Benati, D.; Miselli, F.; Cocchiarella, F.; Patrizi, C.; Carretero, M.; Baldassarri, S.; Ammendola, V.; Has, C.; Colloca, S.; Del Rio, M.; et al. CRISPR/Cas9-Mediated In Situ Correction of LAMB3 Gene in Keratinocytes Derived from a Junctional Epidermolysis Bullosa Patient. Mol. Ther. 2018, 26, 2592–2603. [Google Scholar] [CrossRef] [Green Version]
- Osborn, M.J.; Lees, C.J.; McElroy, A.N.; Merkel, S.C.; Eide, C.R.; Mathews, W.; Feser, C.J.; Tschann, M.; McElmury, R.T.; Webber, B.R.; et al. CRISPR/Cas9-Based Cellular Engineering for Targeted Gene Overexpression. Int. J. Mol. Sci. 2018, 19, 946. [Google Scholar] [CrossRef] [Green Version]
- Dupuy, A.; Valton, J.; Leduc, S.; Armier, J.; Galetto, R.; Gouble, A.; Lebuhotel, C.; Stary, A.; Paques, F.; Duchateau, P.; et al. Targeted gene therapy of xeroderma pigmentosum cells using meganuclease and TALEN. PLoS ONE 2013, 8, e78678. [Google Scholar] [CrossRef]
- Izmiryan, A.; Danos, O.; Hovnanian, A. Meganuclease-Mediated COL7A1 Gene Correction for Recessive Dystrophic Epidermolysis Bullosa. J. Investig. Dermatol. 2016, 136, 872–875. [Google Scholar] [CrossRef]
- Kocher, T.; Peking, P.; Klausegger, A.; Murauer, E.M.; Hofbauer, J.P.; Wally, V.; Lettner, T.; Hainzl, S.; Ablinger, M.; Bauer, J.W.; et al. Cut and Paste: Efficient Homology-Directed Repair of a Dominant Negative KRT14 Mutation via CRISPR/Cas9 Nickases. Mol. Ther. 2017, 25, 2585–2598. [Google Scholar] [CrossRef] [Green Version]
- Izmiryan, A.; Ganier, C.; Bovolenta, M.; Schmitt, A.; Mavilio, F.; Hovnanian, A. Ex Vivo COL7A1 Correction for Recessive Dystrophic Epidermolysis Bullosa Using CRISPR/Cas9 and Homology-Directed Repair. Mol. Ther. Nucleic Acids 2018, 12, 554–567. [Google Scholar] [CrossRef]
- Kocher, T.; Wagner, R.N.; Klausegger, A.; Guttmann-Gruber, C.; Hainzl, S.; Bauer, J.W.; Reichelt, J.; Koller, U. Improved Double-Nicking Strategies for COL7A1-Editing by Homologous Recombination. Mol. Ther. Nucleic Acids 2019, 18, 496–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborn, M.J.; Newby, G.A.; McElroy, A.N.; Knipping, F.; Nielsen, S.C.; Riddle, M.J.; Xia, L.; Chen, W.; Eide, C.R.; Webber, B.R.; et al. Base editor correction of COL7A1 in recessive dystrophic epidermolysis bullosa patient-derived fibroblasts and iPSCs. J. Investig. Dermatol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Santiago, Y.; Chan, E.; Liu, P.Q.; Orlando, S.; Zhang, L.; Urnov, F.D.; Holmes, M.C.; Guschin, D.; Waite, A.; Miller, J.C.; et al. Targeted gene knockout in mammalian cells by using engineered zinc-finger nucleases. Proc. Natl. Acad. Sci. USA 2008, 105, 5809–5814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, E. Keratins and the skin. Annu. Rev. Cell Dev. Biol. 1995, 11, 123–153. [Google Scholar] [CrossRef]
- Lee, C.H.; Coulombe, P.A. Self-organization of keratin intermediate filaments into cross-linked networks. J. Cell Biol. 2009, 186, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Syder, A.J.; Yu, Q.C.; Letai, A.; Paller, A.S.; Fuchs, E. The genetic basis of epidermolytic hyperkeratosis: A disorder of differentiation-specific epidermal keratin genes. Cell 1992, 70, 811–819. [Google Scholar] [CrossRef]
- Chipev, C.C.; Korge, B.P.; Markova, N.; Bale, S.J.; DiGiovanna, J.J.; Compton, J.G.; Steinert, P.M. A leucine—Proline mutation in the H1 subdomain of keratin 1 causes epidermolytic hyperkeratosis. Cell 1992, 70, 821–828. [Google Scholar] [CrossRef]
- Rothnagel, J.A.; Dominey, A.M.; Dempsey, L.D.; Longley, M.A.; Greenhalgh, D.A.; Gagne, T.A.; Huber, M.; Frenk, E.; Hohl, D.; Roop, D.R. Mutations in the rod domains of keratins 1 and 10 in epidermolytic hyperkeratosis. Science 1992, 257, 1128–1130. [Google Scholar] [CrossRef]
- Fine, J.D.; Bruckner-Tuderman, L.; Eady, R.A.J.; Bauer, E.A.; Bauer, J.W.; Has, C.; Heagerty, A.; Hintner, H.; Hovnanian, A.; Jonkman, M.F.; et al. Inherited epidermolysis bullosa: Updated recommendations on diagnosis and classification. J. Am. Acad. Dermatol. 2014, 70, 1103–1126. [Google Scholar] [CrossRef]
- Terheyden, P.; Grimberg, G.; Hausser, I.; Rose, C.; Korge, B.P.; Krieg, T.; Arin, M.J. Recessive epidermolytic hyperkeratosis caused by a previously unreported termination codon mutation in the keratin 10 gene. J. Investig. Dermatol. 2009, 129, 2721–2723. [Google Scholar] [CrossRef] [Green Version]
- Arin, M.J.; Oji, V.; Emmert, S.; Hausser, I.; Traupe, H.; Krieg, T.; Grimberg, G. Expanding the keratin mutation database: Novel and recurrent mutations and genotype–phenotype correlations in 28 patients with epidermolytic ichthyosis. Br. J. Dermatol. 2011, 164, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Molla, K.A.; Yang, Y. Predicting CRISPR/Cas9-Induced Mutations for Precise Genome Editing. Trends Biotechnol. 2019. [Google Scholar] [CrossRef]
- Ousterout, D.G.; Perez-Pinera, P.; Thakore, P.I.; Kabadi, A.M.; Brown, M.T.; Qin, X.; Fedrigo, O.; Mouly, V.; Tremblay, J.P.; Gersbach, C.A. Reading frame correction by targeted genome editing restores dystrophin expression in cells from Duchenne muscular dystrophy patients. Mol. Ther. 2013, 21, 1718–1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amoasii, L.; Long, C.; Li, H.; Mireault, A.A.; Shelton, J.M.; Sanchez-Ortiz, E.; McAnally, J.R.; Bhattacharyya, S.; Schmidt, F.; Grimm, D.; et al. Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci. Transl. Med. 2017, 9, eaan8081. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Long, C.; Li, H.; McAnally, J.R.; Baskin, K.K.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Sci. Adv. 2017, 3, e1602814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bornert, O.; Peking, P.; Bremer, J.; Koller, U.; van den Akker, P.C.; Aartsma-Rus, A.; Pasmooij, A.M.; Murauer, E.M.; Nystrom, A. RNA-based therapies for genodermatoses. Exp. Dermatol. 2017, 26, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brusasco, A.; Tadini, G.; Cambiaghi, S.; Ermacora, E.; Grimalt, R.; Caputo, R. A case of congenital reticular ichthyosiform erythroderma--ichthyosis ‘en confettis’. Dermatology 1994, 188, 40–45. [Google Scholar] [CrossRef]
- Long, C.; McAnally, J.R.; Shelton, J.M.; Mireault, A.A.; Bassel-Duby, R.; Olson, E.N. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science 2014, 345, 1184–1188. [Google Scholar] [CrossRef] [Green Version]
- Li, H.L.; Fujimoto, N.; Sasakawa, N.; Shirai, S.; Ohkame, T.; Sakuma, T.; Tanaka, M.; Amano, N.; Watanabe, A.; Sakurai, H.; et al. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Rep. 2015, 4, 143–154. [Google Scholar] [CrossRef] [Green Version]
- Ousterout, D.G.; Kabadi, A.M.; Thakore, P.I.; Majoros, W.H.; Reddy, T.E.; Gersbach, C.A. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat. Commun. 2015, 6, 6244. [Google Scholar] [CrossRef] [Green Version]
- Iyombe-Engembe, J.P.; Ouellet, D.L.; Barbeau, X.; Rousseau, J.; Chapdelaine, P.; Lague, P.; Tremblay, J.P. Efficient Restoration of the Dystrophin Gene Reading Frame and Protein Structure in DMD Myoblasts Using the CinDel Method. Mol. Ther. Nucleic Acids 2016, 5, e283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, Y.L.; Bassel-Duby, R.; Olson, E.N. CRISPR Correction of Duchenne Muscular Dystrophy. Annu. Rev. Med. 2019, 70, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Owens, D.D.G.; Caulder, A.; Frontera, V.; Harman, J.R.; Allan, A.J.; Bucakci, A.; Greder, L.; Codner, G.F.; Hublitz, P.; McHugh, P.J.; et al. Microhomologies are prevalent at Cas9-induced larger deletions. Nucleic Acids Res. 2019, 47, 7402–7417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, M.; Sawamura, D.; Nishie, W.; Sakai, K.; McMillan, J.R.; Akiyama, M.; Shimizu, H. Targeted skipping of a single exon harboring a premature termination codon mutation: Implications and potential for gene correction therapy for selective dystrophic epidermolysis bullosa patients. J. Investig. Dermatol. 2006, 126, 2614–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christiano, A.M.; Hoffman, G.G.; Chung-Honet, L.C.; Lee, S.; Cheng, W.; Uitto, J.; Greenspan, D.S. Structural organization of the human type VII collagen gene (COL7A1), composed of more exons than any previously characterized gene. Genomics 1994, 21, 169–179. [Google Scholar] [CrossRef]
- Turczynski, S.; Titeux, M.; Tonasso, L.; Decha, A.; Ishida-Yamamoto, A.; Hovnanian, A. Targeted Exon Skipping Restores Type VII Collagen Expression and Anchoring Fibril Formation in an In Vivo RDEB Model. J. Investig. Dermatol. 2016, 136, 2387–2395. [Google Scholar] [CrossRef] [Green Version]
- Bremer, J.; Bornert, O.; Nystrom, A.; Gostynski, A.; Jonkman, M.F.; artsma-Rus, A.; van den Akker, P.C.; Pasmooij, A.M. Antisense Oligonucleotide-mediated Exon Skipping as a Systemic Therapeutic Approach for Recessive Dystrophic Epidermolysis Bullosa. Mol. Ther. Nucleic Acids 2016, 5, e379. [Google Scholar] [CrossRef]
- Van den Akker, P.C.; Jonkman, M.F.; Rengaw, T.; Bruckner-Tuderman, L.; Has, C.; Bauer, J.W.; Klausegger, A.; Zambruno, G.; Castiglia, D.; Mellerio, J.E.; et al. The international dystrophic epidermolysis bullosa patient registry: An online database of dystrophic epidermolysis bullosa patients and their COL7A1 mutations. Hum. Mutat. 2011, 32, 1100–1107. [Google Scholar] [CrossRef]
- Wertheim-Tysarowska, K.; Sobczynska-Tomaszewska, A.; Kowalewski, C.; Skronski, M.; Swieckowski, G.; Kutkowska-Kazmierczak, A.; Wozniak, K.; Bal, J. The COL7A1 mutation database. Hum. Mutat. 2012, 33, 327–331. [Google Scholar] [CrossRef]
- Escamez, M.J.; Garcia, M.; Cuadrado-Corrales, N.; Llames, S.G.; Charlesworth, A.; De Luca, N.; Illera, N.; Sanchez-Jimeno, C.; Holguin, A.; Duarte, B.; et al. The first COL7A1 mutation survey in a large Spanish dystrophic epidermolysis bullosa cohort: C.6527insC disclosed as an unusually recurrent mutation. Br. J. Dermatol. 2010, 163, 155–161. [Google Scholar]
- Yao, X.; Wang, X.; Hu, X.; Liu, Z.; Liu, J.; Zhou, H.; Shen, X.; Wei, Y.; Huang, Z.; Ying, W.; et al. Homology-mediated end joining-based targeted integration using CRISPR/Cas9. Cell Res. 2017, 27, 801–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Janssen, J.M.; Liu, J.; Maggio, I.; t Jong, A.E.J.; Mikkers, H.M.M.; Goncalves, M. In trans paired nicking triggers seamless genome editing without double-stranded DNA cutting. Nat. Commun. 2017, 8, 657. [Google Scholar] [CrossRef]
- Zhang, J.P.; Li, X.L.; Li, G.H.; Chen, W.; Arakaki, C.; Botimer, G.D.; Baylink, D.; Zhang, L.; Wen, W.; Fu, Y.W.; et al. Efficient precise knockin with a double cut HDR donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Biol. 2017, 18, 35. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.W.; Arbab, M.; Hsu, J.Y.; Worstell, D.; Culbertson, S.J.; Krabbe, O.; Cassa, C.A.; Liu, D.R.; Gifford, D.K.; Sherwood, R.I. Predictable and precise template-free CRISPR editing of pathogenic variants. Nature 2018, 563, 646–651. [Google Scholar] [CrossRef]
- Chakrabarti, A.M.; Henser-Brownhill, T.; Monserrat, J.; Poetsch, A.R.; Luscombe, N.M.; Scaffidi, P. Target-Specific Precision of CRISPR-Mediated Genome Editing. Mol. Cell 2019, 73, 699–713. [Google Scholar] [CrossRef] [Green Version]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Nishida, K.; Arazoe, T.; Yachie, N.; Banno, S.; Kakimoto, M.; Tabata, M.; Mochizuki, M.; Miyabe, A.; Araki, M.; Hara, K.Y.; et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 2016, 353, aaf8729. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Koblan, L.W.; Doman, J.L.; Wilson, C.; Levy, J.M.; Tay, T.; Newby, G.A.; Maianti, J.P.; Raguram, A.; Liu, D.R. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 2018, 36, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef]
- Grünewald, J.; Zhou, R.; Garcia, S.P.; Iyer, S.; Lareau, C.A.; Aryee, M.J.; Joung, J.K. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature 2019, 569, 433–437. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Zhao, C.; Zheng, X.; Qu, W.; Li, G.; Li, X.; Miao, Y.L.; Han, X.; Liu, X.; Li, Z.; Ma, Y.; et al. CRISPR-offinder: A CRISPR guide RNA design and off-target searching tool for user-defined protospacer adjacent motif. Int. J. Biol. Sci. 2017, 13, 1470–1478. [Google Scholar] [CrossRef] [Green Version]
- Porteus, M.H. A New Class of Medicines through DNA Editing. N. Engl. J. Med. 2019, 380, 947–959. [Google Scholar] [CrossRef]
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018, 24, 939–946. [Google Scholar] [CrossRef]
- Geisinger, J.M.; Stearns, T. CRISPR/Cas9 Treatment Causes Extended TP53-Dependent Cell Cycle Arrest In Human Cells. bioRxiv 2019, 604538. [Google Scholar] [CrossRef] [Green Version]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Cullot, G.; Boutin, J.; Toutain, J.; Prat, F.; Pennamen, P.; Rooryck, C.; Teichmann, M.; Rousseau, E.; Lamrissi-Garcia, I.; Guyonnet-Duperat, V.; et al. CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations. Nat. Commun. 2019, 10, 1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Targeting Strategy (Nuclease) | Gene Target (Mutation) | Mutation Type | Dominant vs. Recessive | Disease | Ex Vivo vs. In Vivo | Pub. |
|---|---|---|---|---|---|---|
| Gene Knockout (TALEN, CRISPR/Cas9) | COL7A1 (c.8068_8084delinsGA) | Non-Frameshift Indel | Dominant | DDEB | Ex Vivo | [31] |
| Gene Knockout (TALEN) | KRT5 (c.556G > T, c.1424A > G) | Missense, Missense | Dominant, Dominant | EBS | Ex Vivo | [32] |
| Gene Knockout (CRISPR/Cas9) | KRT9 (c.434delAinsGGCT) | Non-Frameshift Indel | Dominant | EPPK | In Vivo | [33] |
| Gene Knockout (TALEN) | KRT10 (c.1333G > A, c.481_486delTTGGAC) | Non-Frameshift Indel, Missense | Dominant, Dominant | EI | Ex Vivo | [34] |
| Gene Reframing (TALEN) | COL7A1 (c.6527insC) | Frameshift Indel | Recessive | RDEB | Ex Vivo | [35] |
| Gene Reframing (TALEN) | COL7A1 (c.6527insC) | Frameshift Indel | Recessive | RDEB | Ex Vivo | [36] |
| Gene Reframing (CRISPR/Cas9) | COL7A1 (c.5819delC) | Frameshift Indel | Recessive | RDEB | Ex Vivo | [37] |
| Exon Deletion (Dual CRISPR/Cas9) | COL7A1 (c.6485G > A) | Nonsense | Recessive | RDEB | In Vivo | [38] |
| Exon Deletion (Dual CRISPR/Cas9) | COL7A1 (c.6527insC) | Frameshift Indel | Recessive | RDEB | Ex Vivo | [39] |
| Exon/Gene Insertion (ZFN) | AAVS1 (N/A) | N/A | N/A | N/A | Ex Vivo | [40] |
| Exon/Gene Insertion (TALEN) | COL7A1 (c.1837C > T) | Nonsense | Recessive | RDEB | Ex Vivo | [41] |
| Exon/Gene Insertion (ZFN, TALEN, CRISPR/Cas9) | COL7A1 (c.356_357delCA, c.90delC) | Frameshift Indel Frameshift Indel | Recessive | RDEB | Ex Vivo | [42] |
| Exon/Gene Insertion (TALEN) | COL7A1 (c.6527insC) | Frameshift Indel | Recessive | RDEB | Ex Vivo | [35] |
| Exon/Gene Insertion (CRISPR/Cas9) | COL7A1 (c.4317delC) | Frameshift Indel | Recessive | RDEB | Ex Vivo | [43] |
| Exon/Gene Insertion (CRISPR/Cas9) | COL7A1 (c.6527insC) | Frameshift Indel | Recessive | RDEB | Ex Vivo | [44] |
| Exon/Gene Insertion (CRISPR/Cas9) | LAMB3 (c.1945dupG, c.1903C > T) | Frameshift Indel Nonsense | Recessive | JEB | Ex Vivo | [45] |
| Exon/Gene Insertion (CRISPR/Cas9) | AAVS1 (N/A) | N/A | Recessive | RDEB | Ex Vivo | [46] |
| Homologous Recombination (Meganuclease, TALEN) | XPC (c.1643_1644delTG) | Frameshift Indel | Recessive | XP | Ex Vivo | [47] |
| Homologous Recombination (Meganuclease) | COL7A1 (c.189delG, c.425A > G) | Frameshift Indel Splice Site Mut. | Recessive | RDEB | Ex Vivo | [48] |
| Homologous Recombination (Dual CRISPR/Cas9 D10A nicking) | KRT14 (c.1231G > A) | Missense | Dominant | EBS | Ex Vivo | [49] |
| Homologous Recombination (CRISPR/Cas9) | COL7A1 (c.6527insC) | Frameshift Indel | Recessive | RDEB | Ex Vivo | [44] |
| Homologous Recombination (CRISPR/Cas9) | COL7A1 (c.189delG) | Frameshift Indel | Recessive | RDEB | Ex Vivo | [50] |
| Homologous Recombination (Dual CRISPR/Cas9 D10A nicking) | COL7A1 (c.425A > G) | Splice Site Mut. | Recessive | RDEB | Ex Vivo | [51] |
| Base Editing (ABE) | COL7A1 (c.553C > T, c.1573C > T) | Nonsense | Recessive, Recessive | RDEB | Ex Vivo | [52] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
March, O.P.; Kocher, T.; Koller, U. Context-Dependent Strategies for Enhanced Genome Editing of Genodermatoses. Cells 2020, 9, 112. https://doi.org/10.3390/cells9010112
March OP, Kocher T, Koller U. Context-Dependent Strategies for Enhanced Genome Editing of Genodermatoses. Cells. 2020; 9(1):112. https://doi.org/10.3390/cells9010112
Chicago/Turabian StyleMarch, Oliver Patrick, Thomas Kocher, and Ulrich Koller. 2020. "Context-Dependent Strategies for Enhanced Genome Editing of Genodermatoses" Cells 9, no. 1: 112. https://doi.org/10.3390/cells9010112
APA StyleMarch, O. P., Kocher, T., & Koller, U. (2020). Context-Dependent Strategies for Enhanced Genome Editing of Genodermatoses. Cells, 9(1), 112. https://doi.org/10.3390/cells9010112