Mitochondrial Gene Expression and Beyond—Novel Aspects of Cellular Physiology


{kind=link}
{kind=link}
Abstract
:1. Introduction: The Role of Mitochondria within the Cell
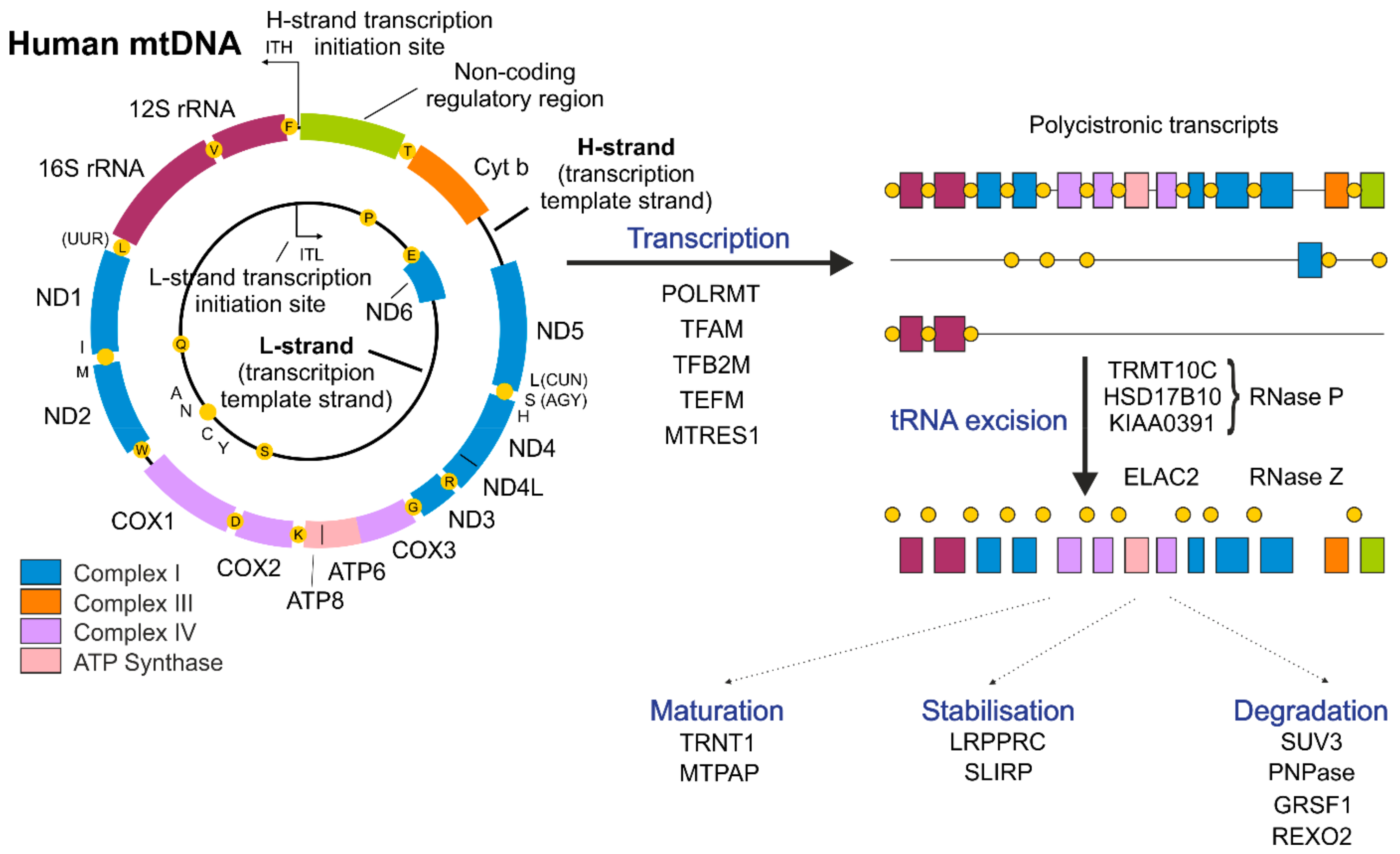
2. Mitochondrial Genome—A Simple Molecule with Unorthodox Organization
3. Mitochondrial Transcription
4. Post-Transcriptional Regulation of mtRNAs
4.1. Degradation of mtRNAs
4.2. Mitochondrial RNA-Binding Proteins (mtRBPs)
4.3. mtRNA Modifications
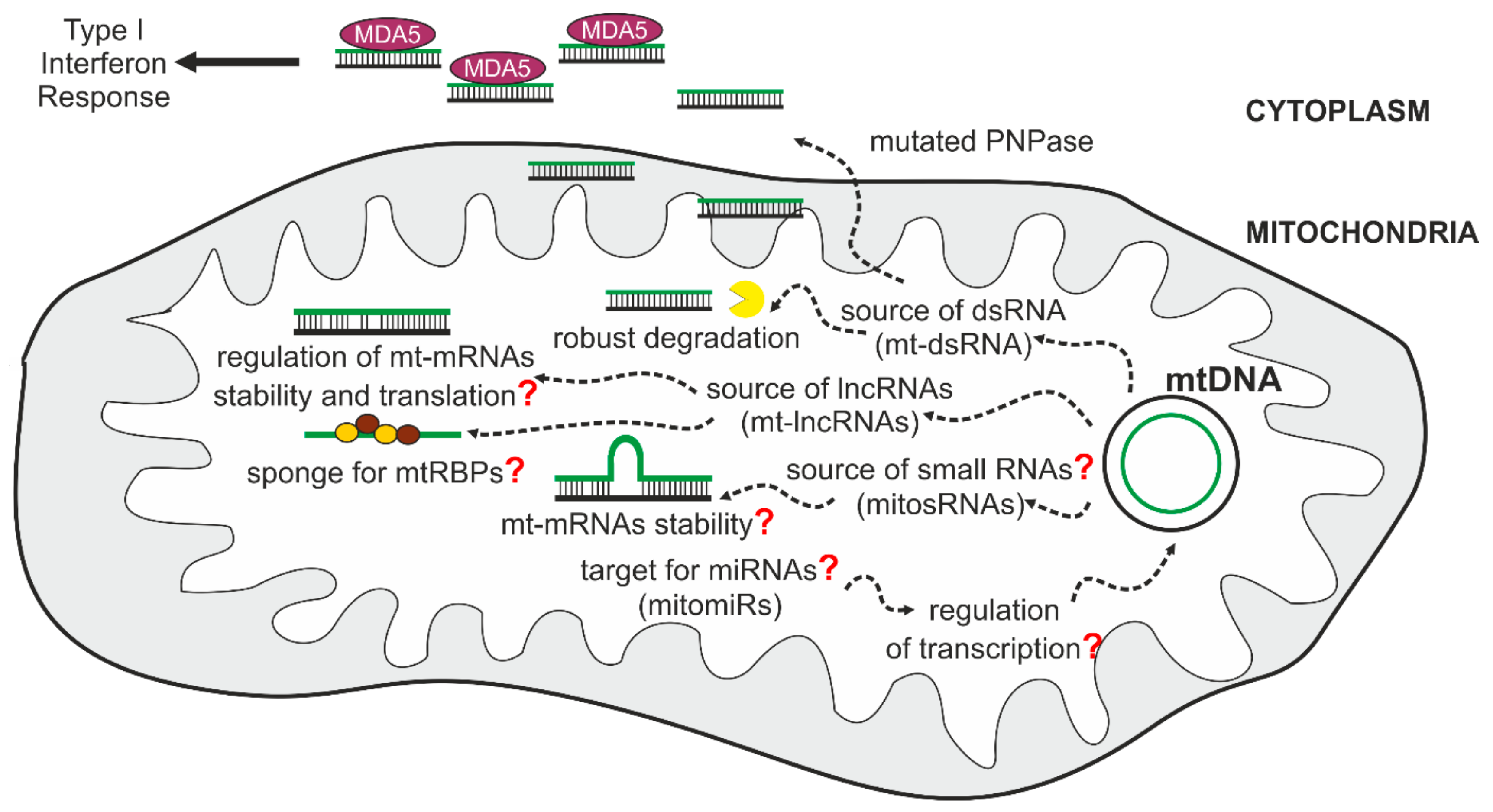
5. Mitochondrial Non-Coding Transcripts and Their Implications on Human Health
5.1. Mitochondrial Double-Stranded RNA (mt-dsRNA) and the Innate Immune Response
5.2. Mitochondrial Long Non-Coding RNAs (mt-lncRNAs)
6. Emerging Aspects of mtDNA Maintenance and Expression
6.1. mtDNA Polymerases and mtDNA Repair
6.2. mtDNA Editing
6.3. RNA Import into Mitochondria
6.4. Small RNAs in Mitochondrial Biology
6.5. 37 or More?
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ATP | adenosine triphosphate |
| dsRNA | double-stranded RNA |
| G4 | G-quadruplex |
| H-strand | heavy-strand |
| IFN | interferon |
| ITH | heavy-strand transcription initiation site |
| ITL | light-strand transcription initiation site |
| L-strand | light-strand |
| lncRNA | long non-coding RNA |
| MDP | mitochondria-derived peptide |
| miRNA | microRNA |
| mitomiRs | mitochondrial microRNAs |
| mitosRNAs | mitochondrial genome-encoded small RNAs |
| mtDNA | mitochondrial DNA |
| mtRNA | mitochondrial RNA |
| NCR | non-coding regulatory region |
| ncRNA | non-coding RNA |
| PAMP | pathogen-associated molecular pattern |
| RBP | RNA-binding protein |
| sORF | small Open Reading Frame |
| sRNA | small RNA |
References
- Brown, G.C. Control of respiration and ATP synthesis in mammalian mitochondria and cells. Biochem. J. 1992, 284, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.-M.; Jones, D.P. Redox compartmentalization in eukaryotic cells. Biochim. Biophys. Acta 2008, 1780, 1273–1290. [Google Scholar] [CrossRef] [Green Version]
- Titov, D.V.; Cracan, V.; Goodman, R.P.; Peng, J.; Grabarek, Z.; Mootha, V.K. Complementation of mitochondrial electron transport chain by manipulation of the NAD+/NADH ratio. Science 2016, 352, 231–235. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Kepp, O.; Trojel-Hansen, C.; Kroemer, G. Mitochondrial control of cellular life, stress, and death. Circ. Res. 2012, 111, 1198–1207. [Google Scholar] [CrossRef] [Green Version]
- Pizzo, P.; Drago, I.; Filadi, R.; Pozzan, T. Mitochondrial Ca2+ homeostasis: Mechanism, role, and tissue specificities. Pflugers Arch. 2012, 464, 3–17. [Google Scholar] [CrossRef]
- Brand, M.D.; Orr, A.L.; Perevoshchikova, I.V.; Quinlan, C.L. The role of mitochondrial function and cellular bioenergetics in ageing and disease. Br. J. Dermatol. 2013, 169 (Suppl. 2), 1–8. [Google Scholar] [CrossRef] [Green Version]
- Maiese, K. The bright side of reactive oxygen species: Lifespan extension without cellular demise. J. Transl. Sci. 2016, 2, 185–187. [Google Scholar] [CrossRef] [Green Version]
- Arnoult, D.; Soares, F.; Tattoli, I.; Girardin, S.E. Mitochondria in innate immunity. EMBO Rep. 2011, 12, 901–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandhir, R.; Halder, A.; Sunkaria, A. Mitochondria as a centrally positioned hub in the innate immune response. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1090–1097. [Google Scholar] [CrossRef] [PubMed]
- Czarnecka, A.M.; Golik, P.; Bartnik, E. Mitochondrial DNA mutations in human neoplasia. J. Appl. Genet. 2006, 47, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Copeland, W.C.; Wachsman, J.T.; Johnson, F.M.; Penta, J.S. Mitochondrial DNA alterations in cancer. Cancer Invest. 2002, 20, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V. Mitochondrial diseases. Lancet 2012, 379, 1825–1834. [Google Scholar] [CrossRef]
- Nicholls, T.J.; Rorbach, J.; Minczuk, M. Mitochondria: Mitochondrial RNA metabolism and human disease. Int. J. Biochem. Cell Biol. 2013, 45, 845–849. [Google Scholar] [CrossRef] [PubMed]
- Vafai, S.B.; Mootha, V.K. Mitochondrial disorders as windows into an ancient organelle. Nature 2012, 491, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef]
- Rusecka, J.; Kaliszewska, M.; Bartnik, E.; Tońska, K. Nuclear genes involved in mitochondrial diseases caused by instability of mitochondrial DNA. J. Appl. Genet. 2018, 59, 43–57. [Google Scholar] [CrossRef] [Green Version]
- Taanman, J.W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta 1999, 1410, 103–123. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Silva, P.; Enriquez, J.A.; Montoya, J. Replication and transcription of mammalian mitochondrial DNA. Exp. Physiol. 2003, 88, 41–56. [Google Scholar] [CrossRef]
- Rorbach, J.; Minczuk, M. The post-transcriptional life of mammalian mitochondrial RNA. Biochem. J. 2012, 444, 357–373. [Google Scholar] [CrossRef] [Green Version]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016, 44, D1251–D1257. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, O.; Pfanner, N.; Meisinger, C. Mitochondrial protein import: From proteomics to functional mechanisms. Nat. Rev. Mol. Cell Biol. 2010, 11, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Van Haute, L.; Pearce, S.F.; Powell, C.A.; D’Souza, A.R.; Nicholls, T.J.; Minczuk, M. Mitochondrial transcript maturation and its disorders. J. Inherit. Metab. Dis. 2015, 38, 655–680. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Oyaizu, Y.; Oyaizu, H.; Olsen, G.J.; Woese, C.R. Mitochondrial origins. Proc. Natl. Acad. Sci. USA 1985, 82, 4443–4447. [Google Scholar] [CrossRef] [Green Version]
- Gray, M.W.; Burger, G.; Lang, B.F. Mitochondrial evolution. Science 1999, 283, 1476–1481. [Google Scholar] [CrossRef] [Green Version]
- Martijn, J.; Vosseberg, J.; Guy, L.; Offre, P.; Ettema, T.J.G. Deep mitochondrial origin outside the sampled alphaproteobacteria. Nature 2018, 557, 101–105. [Google Scholar] [CrossRef]
- Nass, M.M.; Nass, S. Intramitochondrial fibers with DNA characteristics. I. fixation and electron staining reactions. J. Cell Biol. 1963, 19, 593–611. [Google Scholar] [CrossRef] [Green Version]
- Anderson, S. Shotgun DNA sequencing using cloned DNase I-generated fragments. Nucleic Acids Res. 1981, 9, 3015–3027. [Google Scholar] [CrossRef] [Green Version]
- Bonekamp, N.A.; Larsson, N.-G. SnapShot: Mitochondrial Nucleoid. Cell 2018, 172, 388–388.e1. [Google Scholar] [CrossRef]
- Garrido, N.; Griparic, L.; Jokitalo, E.; Wartiovaara, J.; van der Bliek, A.M.; Spelbrink, J.N. Composition and dynamics of human mitochondrial nucleoids. Mol. Biol. Cell 2003, 14, 1583–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Bogenhagen, D.F. Human mitochondrial DNA nucleoids are linked to protein folding machinery and metabolic enzymes at the mitochondrial inner membrane. J. Biol. Chem. 2006, 281, 25791–25802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kukat, C.; Wurm, C.A.; Spåhr, H.; Falkenberg, M.; Larsson, N.-G.; Jakobs, S. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc. Natl. Acad. Sci. USA 2011, 108, 13534–13539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kukat, C.; Davies, K.M.; Wurm, C.A.; Spåhr, H.; Bonekamp, N.A.; Kühl, I.; Joos, F.; Polosa, P.L.; Park, C.B.; Posse, V.; et al. Cross-strand binding of TFAM to a single mtDNA molecule forms the mitochondrial nucleoid. Proc. Natl. Acad. Sci. USA 2015, 112, 11288–11293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekstrand, M.I.; Falkenberg, M.; Rantanen, A.; Park, C.B.; Gaspari, M.; Hultenby, K.; Rustin, P.; Gustafsson, C.M.; Larsson, N.-G. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum. Mol. Genet. 2004, 13, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Kuroiwa, T. Organization of multiple nucleoids and DNA molecules in mitochondria of a human cell. Exp. Cell Res. 1991, 196, 137–140. [Google Scholar] [CrossRef]
- Legros, F.; Malka, F.; Frachon, P.; Lombès, A.; Rojo, M. Organization and dynamics of human mitochondrial DNA. J. Cell Sci. 2004, 117, 2653–2662. [Google Scholar] [CrossRef] [Green Version]
- Iborra, F.J.; Kimura, H.; Cook, P.R. The functional organization of mitochondrial genomes in human cells. BMC Biol. 2004, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Jenuth, J.P.; Peterson, A.C.; Fu, K.; Shoubridge, E.A. Random genetic drift in the female germline explains the rapid segregation of mammalian mitochondrial DNA. Nat. Genet. 1996, 14, 146–151. [Google Scholar] [CrossRef]
- Ishihara, T.; Ban-Ishihara, R.; Maeda, M.; Matsunaga, Y.; Ichimura, A.; Kyogoku, S.; Aoki, H.; Katada, S.; Nakada, K.; Nomura, M.; et al. Dynamics of mitochondrial DNA nucleoids regulated by mitochondrial fission is essential for maintenance of homogeneously active mitochondria during neonatal heart development. Mol. Cell. Biol. 2015, 35, 211–223. [Google Scholar] [CrossRef] [Green Version]
- Kvist, L.; Martens, J.; Nazarenko, A.A.; Orell, M. Paternal leakage of mitochondrial DNA in the great tit (Parus major). Mol. Biol. Evol. 2003, 20, 243–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cree, L.M.; Samuels, D.C.; de Sousa Lopes, S.C.; Rajasimha, H.K.; Wonnapinij, P.; Mann, J.R.; Dahl, H.-H.M.; Chinnery, P.F. A reduction of mitochondrial DNA molecules during embryogenesis explains the rapid segregation of genotypes. Nat. Genet. 2008, 40, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Shoubridge, E.A. Mitochondrial DNA segregation in the developing embryo. Hum. Reprod. 2000, 15 (Suppl. 2), 229–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, M.; Vissing, J. Paternal inheritance of mitochondrial DNA. N. Engl. J. Med. 2002, 347, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Valencia, C.A.; Zhang, J.; Lee, N.-C.; Slone, J.; Gui, B.; Wang, X.; Li, Z.; Dell, S.; Brown, J.; et al. Biparental Inheritance of Mitochondrial DNA in Humans. Proc. Natl. Acad. Sci. USA 2018, 115, 13039–13044. [Google Scholar] [CrossRef] [Green Version]
- Pohjoismäki, J.L.O.; Wanrooij, S.; Hyvärinen, A.K.; Goffart, S.; Holt, I.J.; Spelbrink, J.N.; Jacobs, H.T. Alterations to the expression level of mitochondrial transcription factor A, TFAM, modify the mode of mitochondrial DNA replication in cultured human cells. Nucleic Acids Res. 2006, 34, 5815–5828. [Google Scholar] [CrossRef]
- Pohjoismäki, J.L.O.; Goffart, S.; Tyynismaa, H.; Willcox, S.; Ide, T.; Kang, D.; Suomalainen, A.; Karhunen, P.J.; Griffith, J.D.; Holt, I.J.; et al. Human heart mitochondrial DNA is organized in complex catenated networks containing abundant four-way junctions and replication forks. J. Biol. Chem. 2009, 284, 21446–21457. [Google Scholar] [CrossRef] [Green Version]
- Tuppen, H.A.L.; Blakely, E.L.; Turnbull, D.M.; Taylor, R.W. Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta 2010, 1797, 113–128. [Google Scholar] [CrossRef] [Green Version]
- Holt, I.J.; Reyes, A. Human mitochondrial DNA replication. Cold Spring Harb. Perspect. Biol. 2012, 4, a012971. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, C.M.; Falkenberg, M.; Larsson, N.-G. Maintenance and Expression of Mammalian Mitochondrial DNA. Annu. Rev. Biochem. 2016, 85, 133–160. [Google Scholar] [CrossRef]
- Young, M.J.; Copeland, W.C. Human mitochondrial DNA replication machinery and disease. Curr. Opin. Genet. Dev. 2016, 38, 52–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Copeland, W.C.; Longley, M.J. Mitochondrial genome maintenance in health and disease. DNA Repair 2014, 19, 190–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attardi, G.; Chomyn, A.; King, M.P.; Kruse, B.; Polosa, P.L.; Murdter, N.N. Regulation of mitochondrial gene expression in mammalian cells. Biochem. Soc. Trans. 1990, 18, 509–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fish, J.; Raule, N.; Attardi, G. Discovery of a major D-loop replication origin reveals two modes of human mtDNA synthesis. Science 2004, 306, 2098–2101. [Google Scholar] [CrossRef] [Green Version]
- Aloni, Y.; Attardi, G. Expression of the mitochondrial genome in HeLa cells. II. Evidence for complete transcription of mitochondrial DNA. J. Mol. Biol. 1971, 55, 251–267. [Google Scholar] [CrossRef]
- Hillen, H.S.; Temiakov, D.; Cramer, P. Structural basis of mitochondrial transcription. Nat. Struct. Mol. Biol. 2018, 25, 754–765. [Google Scholar] [CrossRef]
- Fisher, R.P.; Clayton, D.A. A transcription factor required for promoter recognition by human mitochondrial RNA polymerase. Accurate initiation at the heavy- and light-strand promoters dissected and reconstituted in vitro. J. Biol. Chem. 1985, 260, 11330–11338. [Google Scholar]
- Morozov, Y.I.; Parshin, A.V.; Agaronyan, K.; Cheung, A.C.M.; Anikin, M.; Cramer, P.; Temiakov, D. A model for transcription initiation in human mitochondria. Nucleic Acids Res. 2015, 43, 3726–3735. [Google Scholar] [CrossRef] [Green Version]
- Hillen, H.S.; Morozov, Y.I.; Sarfallah, A.; Temiakov, D.; Cramer, P. Structural Basis of Mitochondrial Transcription Initiation. Cell 2017, 171, 1072.e10–1081.e10. [Google Scholar] [CrossRef] [Green Version]
- Minczuk, M.; He, J.; Duch, A.M.; Ettema, T.J.; Chlebowski, A.; Dzionek, K.; Nijtmans, L.G.J.; Huynen, M.A.; Holt, I.J. TEFM (c17orf42) is necessary for transcription of human mtDNA. Nucleic Acids Res. 2011, 39, 4284–4299. [Google Scholar] [CrossRef] [Green Version]
- Posse, V.; Shahzad, S.; Falkenberg, M.; Hällberg, B.M.; Gustafsson, C.M. TEFM is a potent stimulator of mitochondrial transcription elongation in vitro. Nucleic Acids Res. 2015, 43, 2615–2624. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Xue, C.; Long, M.; Jia, H.; Xue, G.; Du, S.; Coello, Y.; Ishibashi, T. TEFM Enhances Transcription Elongation by Modifying mtRNAP Pausing Dynamics. Biophys. J. 2018, 115, 2295–2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agaronyan, K.; Morozov, Y.I.; Anikin, M.; Temiakov, D. Mitochondrial biology. Replication-transcription switch in human mitochondria. Science 2015, 347, 548–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillen, H.S.; Parshin, A.V.; Agaronyan, K.; Morozov, Y.I.; Graber, J.J.; Chernev, A.; Schwinghammer, K.; Urlaub, H.; Anikin, M.; Cramer, P.; et al. Mechanism of Transcription Anti-termination in Human Mitochondria. Cell 2017, 171, 1082–1093.e13. [Google Scholar] [CrossRef]
- Jiang, S.; Koolmeister, C.; Misic, J.; Siira, S.; Kühl, I.; Silva Ramos, E.; Miranda, M.; Jiang, M.; Posse, V.; Lytovchenko, O.; et al. TEFM regulates both transcription elongation and RNA processing in mitochondria. EMBO Rep. 2019, 20, e48101. [Google Scholar] [CrossRef]
- Kruse, B.; Narasimhan, N.; Attardi, G. Termination of transcription in human mitochondria: Identification and purification of a DNA binding protein factor that promotes termination. Cell 1989, 58, 391–397. [Google Scholar] [CrossRef]
- Yakubovskaya, E.; Mejia, E.; Byrnes, J.; Hambardjieva, E.; Garcia-Diaz, M. Helix unwinding and base flipping enable human MTERF1 to terminate mitochondrial transcription. Cell 2010, 141, 982–993. [Google Scholar] [CrossRef] [Green Version]
- Terzioglu, M.; Ruzzenente, B.; Harmel, J.; Mourier, A.; Jemt, E.; López, M.D.; Kukat, C.; Stewart, J.B.; Wibom, R.; Meharg, C.; et al. MTERF1 binds mtDNA to prevent transcriptional interference at the light-strand promoter but is dispensable for rRNA gene transcription regulation. Cell Metab. 2013, 17, 618–626. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Posse, V.; Zhu, X.; Hyvärinen, A.K.; Jacobs, H.T.; Falkenberg, M.; Gustafsson, C.M. Mitochondrial transcription termination factor 1 directs polar replication fork pausing. Nucleic Acids Res. 2016, 44, 5732–5742. [Google Scholar] [CrossRef] [Green Version]
- Linder, T.; Park, C.B.; Asin-Cayuela, J.; Pellegrini, M.; Larsson, N.-G.; Falkenberg, M.; Samuelsson, T.; Gustafsson, C.M. A family of putative transcription termination factors shared amongst metazoans and plants. Curr. Genet. 2005, 48, 265–269. [Google Scholar] [CrossRef]
- Pellegrini, M.; Asin-Cayuela, J.; Erdjument-Bromage, H.; Tempst, P.; Larsson, N.-G.; Gustafsson, C.M. MTERF2 is a nucleoid component in mammalian mitochondria. Biochim. Biophys. Acta 2009, 1787, 296–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cámara, Y.; Asin-Cayuela, J.; Park, C.B.; Metodiev, M.D.; Shi, Y.; Ruzzenente, B.; Kukat, C.; Habermann, B.; Wibom, R.; Hultenby, K.; et al. MTERF4 regulates translation by targeting the methyltransferase NSUN4 to the mammalian mitochondrial ribosome. Cell Metab. 2011, 13, 527–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wredenberg, A.; Lagouge, M.; Bratic, A.; Metodiev, M.D.; Spåhr, H.; Mourier, A.; Freyer, C.; Ruzzenente, B.; Tain, L.; Grönke, S.; et al. MTERF3 regulates mitochondrial ribosome biogenesis in invertebrates and mammals. PLoS Genet. 2013, 9, e1003178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, T.I.; Kanki, T.; Muta, T.; Ukaji, K.; Abe, Y.; Nakayama, H.; Takio, K.; Hamasaki, N.; Kang, D. Human mitochondrial DNA is packaged with TFAM. Nucleic Acids Res. 2003, 31, 1640–1645. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, B.A.; Durisic, N.; Mativetsky, J.M.; Costantino, S.; Hancock, M.A.; Grutter, P.; Shoubridge, E.A. The mitochondrial transcription factor TFAM coordinates the assembly of multiple DNA molecules into nucleoid-like structures. Mol. Biol. Cell 2007, 18, 3225–3236. [Google Scholar] [CrossRef] [PubMed]
- Shadel, G.S.; Clayton, D.A. Mitochondrial DNA maintenance in vertebrates. Annu. Rev. Biochem. 1997, 66, 409–435. [Google Scholar] [CrossRef]
- Campbell, C.T.; Kolesar, J.E.; Kaufman, B.A. Mitochondrial transcription factor A regulates mitochondrial transcription initiation, DNA packaging, and genome copy number. Biochim. Biophys. Acta 2012, 1819, 921–929. [Google Scholar] [CrossRef]
- Larsson, N.G.; Wang, J.; Wilhelmsson, H.; Oldfors, A.; Rustin, P.; Lewandoski, M.; Barsh, G.S.; Clayton, D.A. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 1998, 18, 231–236. [Google Scholar] [CrossRef]
- Stiles, A.R.; Simon, M.T.; Stover, A.; Eftekharian, S.; Khanlou, N.; Wang, H.L.; Magaki, S.; Lee, H.; Partynski, K.; Dorrani, N.; et al. Mutations in TFAM, encoding mitochondrial transcription factor A, cause neonatal liver failure associated with mtDNA depletion. Mol. Genet. Metab. 2016, 119, 91–99. [Google Scholar] [CrossRef]
- Kang, I.; Chu, C.T.; Kaufman, B.A. The mitochondrial transcription factor TFAM in neurodegeneration: Emerging evidence and mechanisms. FEBS Lett. 2018, 592, 793–811. [Google Scholar] [CrossRef] [Green Version]
- Kotrys, A.V.; Cysewski, D.; Czarnomska, S.D.; Pietras, Z.; Borowski, L.S.; Dziembowski, A.; Szczesny, R.J. Quantitative proteomics revealed C6orf203/MTRES1 as a factor preventing stress-induced transcription deficiency in human mitochondria. Nucleic Acids Res. 2019, 47, 7502–7517. [Google Scholar] [CrossRef] [PubMed]
- Serre, V.; Rozanska, A.; Beinat, M.; Chretien, D.; Boddaert, N.; Munnich, A.; Rötig, A.; Chrzanowska-Lightowlers, Z.M. Mutations in mitochondrial ribosomal protein MRPL12 leads to growth retardation, neurological deterioration and mitochondrial translation deficiency. Biochim. Biophys. Acta 2013, 1832, 1304–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surovtseva, Y.V.; Shutt, T.E.; Cotney, J.; Cimen, H.; Chen, S.Y.; Koc, E.C.; Shadel, G.S. Mitochondrial ribosomal protein L12 selectively associates with human mitochondrial RNA polymerase to activate transcription. Proc. Natl. Acad. Sci. USA 2011, 108, 17921–17926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Cotney, J.; Shadel, G.S. Human mitochondrial ribosomal protein MRPL12 interacts directly with mitochondrial RNA polymerase to modulate mitochondrial gene expression. J. Biol. Chem. 2007, 282, 12610–12618. [Google Scholar] [CrossRef] [Green Version]
- Nouws, J.; Goswami, A.V.; Bestwick, M.; McCann, B.J.; Surovtseva, Y.V.; Shadel, G.S. Mitochondrial Ribosomal Protein L12 Is Required for POLRMT Stability and Exists as Two Forms Generated by Alternative Proteolysis during Import. J. Biol. Chem. 2016, 291, 989–997. [Google Scholar] [CrossRef] [Green Version]
- Barchiesi, A.; Vascotto, C. Transcription, Processing, and Decay of Mitochondrial RNA in Health and Disease. Int. J. Mol. Sci. 2019, 20, 2221. [Google Scholar] [CrossRef] [Green Version]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Holzmann, J.; Frank, P.; Löffler, E.; Bennett, K.L.; Gerner, C.; Rossmanith, W. RNase P without RNA: Identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell 2008, 135, 462–474. [Google Scholar] [CrossRef] [Green Version]
- Brzezniak, L.K.; Bijata, M.; Szczesny, R.J.; Stepien, P.P. Involvement of human ELAC2 gene product in 3’ end processing of mitochondrial tRNAs. RNA Biol. 2011, 8, 616–626. [Google Scholar] [CrossRef] [Green Version]
- Mercer, T.R.; Neph, S.; Dinger, M.E.; Crawford, J.; Smith, M.A.; Shearwood, A.-M.J.; Haugen, E.; Bracken, C.P.; Rackham, O.; Stamatoyannopoulos, J.A.; et al. The human mitochondrial transcriptome. Cell 2011, 146, 645–658. [Google Scholar] [CrossRef] [Green Version]
- Szczesny, R.J.; Borowski, L.S.; Brzezniak, L.K.; Dmochowska, A.; Gewartowski, K.; Bartnik, E.; Stepien, P.P. Human mitochondrial RNA turnover caught in flagranti: Involvement of hSuv3p helicase in RNA surveillance. Nucleic Acids Res. 2010, 38, 279–298. [Google Scholar] [CrossRef] [PubMed]
- Piechota, J.; Tomecki, R.; Gewartowski, K.; Szczesny, R.; Dmochowska, A.; Kudła, M.; Dybczyńska, L.; Stepien, P.P.; Bartnik, E. Differential stability of mitochondrial mRNA in HeLa cells. Acta Biochim. Pol. 2006, 53, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Szczesny, R.J.; Borowski, L.S.; Malecki, M.; Wojcik, M.A.; Stepien, P.P.; Golik, P. RNA degradation in yeast and human mitochondria. Biochim. Biophys. Acta 2012, 1819, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Borowski, L.S.; Dziembowski, A.; Hejnowicz, M.S.; Stepien, P.P.; Szczesny, R.J. Human mitochondrial RNA decay mediated by PNPase-hSuv3 complex takes place in distinct foci. Nucleic Acids Res. 2013, 41, 1223–1240. [Google Scholar] [CrossRef]
- Pereira, M.; Mason, P.; Szczesny, R.J.; Maddukuri, L.; Dziwura, S.; Jedrzejczak, R.; Paul, E.; Wojcik, A.; Dybczynska, L.; Tudek, B.; et al. Interaction of human SUV3 RNA/DNA helicase with BLM helicase; loss of the SUV3 gene results in mouse embryonic lethality. Mech. Ageing Dev. 2007, 128, 609–617. [Google Scholar] [CrossRef]
- Wang, G.; Chen, H.-W.; Oktay, Y.; Zhang, J.; Allen, E.L.; Smith, G.M.; Fan, K.C.; Hong, J.S.; French, S.W.; McCaffery, J.M.; et al. PNPASE regulates RNA import into mitochondria. Cell 2010, 142, 456–467. [Google Scholar] [CrossRef] [Green Version]
- Minczuk, M.; Piwowarski, J.; Papworth, M.A.; Awiszus, K.; Schalinski, S.; Dziembowski, A.; Dmochowska, A.; Bartnik, E.; Tokatlidis, K.; Stepien, P.P.; et al. Localisation of the human hSuv3p helicase in the mitochondrial matrix and its preferential unwinding of dsDNA. Nucleic Acids Res. 2002, 30, 5074–5086. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.L.; Wang, Y.-T.; Yang, W.-Z.; Hsiao, Y.-Y.; Yuan, H.S. Crystal structure of human polynucleotide phosphorylase: Insights into its domain function in RNA binding and degradation. Nucleic Acids Res. 2012, 40, 4146–4157. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.D.-H.; Shu, Z.; Lieser, S.A.; Chen, P.-L.; Lee, W.-H. Human mitochondrial SUV3 and polynucleotide phosphorylase form a 330-kDa heteropentamer to cooperatively degrade double-stranded RNA with a 3’-to-5’ directionality. J. Biol. Chem. 2009, 284, 20812–20821. [Google Scholar] [CrossRef] [Green Version]
- Jedrzejczak, R.; Wang, J.; Dauter, M.; Szczesny, R.J.; Stepien, P.P.; Dauter, Z. Human Suv3 protein reveals unique features among SF2 helicases. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 988–996. [Google Scholar] [CrossRef]
- Aloni, Y.; Attardi, G. Symmetrical in vivo transcription of mitochondrial DNA in HeLa cells. Proc. Natl. Acad. Sci. USA 1971, 68, 1757–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chujo, T.; Ohira, T.; Sakaguchi, Y.; Goshima, N.; Nomura, N.; Nagao, A.; Suzuki, T. LRPPRC/SLIRP suppresses PNPase-mediated mRNA decay and promotes polyadenylation in human mitochondria. Nucleic Acids Res. 2012, 40, 8033–8047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, Y.-T.; Barrientos, A. The Human Mitochondrial DEAD-Box Protein DDX28 Resides in RNA Granules and Functions in Mitoribosome Assembly. Cell Rep. 2015, 10, 854–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jourdain, A.A.; Koppen, M.; Rodley, C.D.; Maundrell, K.; Gueguen, N.; Reynier, P.; Guaras, A.M.; Enriquez, J.A.; Anderson, P.; Simarro, M.; et al. A mitochondria-specific isoform of FASTK is present in mitochondrial RNA granules and regulates gene expression and function. Cell Rep. 2015, 10, 1110–1121. [Google Scholar] [CrossRef] [Green Version]
- Bruni, F.; Gramegna, P.; Oliveira, J.M.A.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M.A. REXO2 is an oligoribonuclease active in human mitochondria. PLoS ONE 2013, 8, e64670. [Google Scholar] [CrossRef] [Green Version]
- Pietras, Z.; Wojcik, M.A.; Borowski, L.S.; Szewczyk, M.; Kulinski, T.M.; Cysewski, D.; Stepien, P.P.; Dziembowski, A.; Szczesny, R.J. Dedicated surveillance mechanism controls G-quadruplex forming non-coding RNAs in human mitochondria. Nat. Commun. 2018, 9, 2558. [Google Scholar] [CrossRef] [Green Version]
- Baughman, J.M.; Nilsson, R.; Gohil, V.M.; Arlow, D.H.; Gauhar, Z.; Mootha, V.K. A computational screen for regulators of oxidative phosphorylation implicates SLIRP in mitochondrial RNA homeostasis. PLoS Genet. 2009, 5, e1000590. [Google Scholar] [CrossRef] [Green Version]
- Sasarman, F.; Brunel-Guitton, C.; Antonicka, H.; Wai, T.; Shoubridge, E.A. LSFC Consortium LRPPRC and SLIRP interact in a ribonucleoprotein complex that regulates posttranscriptional gene expression in mitochondria. Mol. Biol. Cell 2010, 21, 1315–1323. [Google Scholar] [CrossRef]
- Siira, S.J.; Spåhr, H.; Shearwood, A.-M.J.; Ruzzenente, B.; Larsson, N.-G.; Rackham, O.; Filipovska, A. LRPPRC-mediated folding of the mitochondrial transcriptome. Nat. Commun. 2017, 8, 1532. [Google Scholar] [CrossRef] [Green Version]
- Ruzzenente, B.; Metodiev, M.D.; Wredenberg, A.; Bratic, A.; Park, C.B.; Cámara, Y.; Milenkovic, D.; Zickermann, V.; Wibom, R.; Hultenby, K.; et al. LRPPRC is necessary for polyadenylation and coordination of translation of mitochondrial mRNAs. EMBO J. 2012, 31, 443–456. [Google Scholar] [CrossRef] [Green Version]
- Mootha, V.K.; Lepage, P.; Miller, K.; Bunkenborg, J.; Reich, M.; Hjerrild, M.; Delmonte, T.; Villeneuve, A.; Sladek, R.; Xu, F.; et al. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc. Natl. Acad. Sci. USA 2003, 100, 605–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, J.; Wang, L.; Ren, X.; Zhang, Y.; Zhang, H. LRPPRC: A Multifunctional Protein Involved in Energy Metabolism and Human Disease. Front. Physiol. 2019, 10, 595. [Google Scholar] [CrossRef] [PubMed]
- Lagouge, M.; Mourier, A.; Lee, H.J.; Spåhr, H.; Wai, T.; Kukat, C.; Silva Ramos, E.; Motori, E.; Busch, J.D.; Siira, S.; et al. SLIRP Regulates the Rate of Mitochondrial Protein Synthesis and Protects LRPPRC from Degradation. PLoS Genet. 2015, 11, e1005423. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Wilusz, J. GRSF-1: A poly(A)+ mRNA binding protein which interacts with a conserved G-rich element. Nucleic Acids Res. 1994, 22, 2334–2343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonicka, H.; Sasarman, F.; Nishimura, T.; Paupe, V.; Shoubridge, E.A. The mitochondrial RNA-binding protein GRSF1 localizes to RNA granules and is required for posttranscriptional mitochondrial gene expression. Cell Metab. 2013, 17, 386–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jourdain, A.A.; Koppen, M.; Wydro, M.; Rodley, C.D.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M.; Martinou, J.-C. GRSF1 regulates RNA processing in mitochondrial RNA granules. Cell Metab. 2013, 17, 399–410. [Google Scholar] [CrossRef] [Green Version]
- Pietras, Z.; Wojcik, M.A.; Borowski, L.S.; Szewczyk, M.; Kulinski, T.M.; Cysewski, D.; Stepien, P.P.; Dziembowski, A.; Szczesny, R.J. Controlling the mitochondrial antisense—role of the SUV3-PNPase complex and its co-factor GRSF1 in mitochondrial RNA surveillance. Mol. Cell. Oncol. 2018, 5, e1516452. [Google Scholar] [CrossRef] [Green Version]
- Gopalakrishna, S.; Pearce, S.F.; Dinan, A.M.; Schober, F.A.; Cipullo, M.; Spåhr, H.; Khawaja, A.; Maffezzini, C.; Freyer, C.; Wredenberg, A.; et al. C6orf203 is an RNA-binding protein involved in mitochondrial protein synthesis. Nucleic Acids Res. 2019, 47, 9386–9399. [Google Scholar] [CrossRef]
- Simarro, M.; Gimenez-Cassina, A.; Kedersha, N.; Lazaro, J.-B.; Adelmant, G.O.; Marto, J.A.; Rhee, K.; Tisdale, S.; Danial, N.; Benarafa, C.; et al. Fast kinase domain-containing protein 3 is a mitochondrial protein essential for cellular respiration. Biochem. Biophys. Res. Commun. 2010, 401, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Antonicka, H.; Shoubridge, E.A. Mitochondrial RNA Granules Are Centers for Posttranscriptional RNA Processing and Ribosome Biogenesis. Cell Rep. 2015, 10, 920–932. [Google Scholar] [CrossRef] [Green Version]
- Popow, J.; Alleaume, A.-M.; Curk, T.; Schwarzl, T.; Sauer, S.; Hentze, M.W. FASTKD2 is an RNA-binding protein required for mitochondrial RNA processing and translation. RNA 2015, 21, 1873–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jourdain, A.A.; Popow, J.; de la Fuente, M.A.; Martinou, J.-C.; Anderson, P.; Simarro, M. The FASTK family of proteins: Emerging regulators of mitochondrial RNA biology. Nucleic Acids Res. 2017, 45, 10941–10947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomecki, R.; Dmochowska, A.; Gewartowski, K.; Dziembowski, A.; Stepien, P.P. Identification of a novel human nuclear-encoded mitochondrial poly(A) polymerase. Nucleic Acids Res. 2004, 32, 6001–6014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagaike, T.; Suzuki, T.; Katoh, T.; Ueda, T. Human mitochondrial mRNAs are stabilized with polyadenylation regulated by mitochondria-specific poly(A) polymerase and polynucleotide phosphorylase. J. Biol. Chem. 2005, 280, 19721–19727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wydro, M.; Bobrowicz, A.; Temperley, R.J.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M. Targeting of the cytosolic poly(A) binding protein PABPC1 to mitochondria causes mitochondrial translation inhibition. Nucleic Acids Res. 2010, 38, 3732–3742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rorbach, J.; Nicholls, T.J.J.; Minczuk, M. PDE12 removes mitochondrial RNA poly(A) tails and controls translation in human mitochondria. Nucleic Acids Res. 2011, 39, 7750–7763. [Google Scholar] [CrossRef] [Green Version]
- Temperley, R.J.; Wydro, M.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M. Human mitochondrial mRNAs--like members of all families, similar but different. Biochim. Biophys. Acta 2010, 1797, 1081–1085. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, J.M.; Woellhaf, M.W.; Bonnefoy, N. Control of protein synthesis in yeast mitochondria: The concept of translational activators. Biochim. Biophys. Acta 2013, 1833, 286–294. [Google Scholar] [CrossRef] [Green Version]
- Weraarpachai, W.; Antonicka, H.; Sasarman, F.; Seeger, J.; Schrank, B.; Kolesar, J.E.; Lochmüller, H.; Chevrette, M.; Kaufman, B.A.; Horvath, R.; et al. Mutation in TACO1, encoding a translational activator of COX I, results in cytochrome c oxidase deficiency and late-onset Leigh syndrome. Nat. Genet. 2009, 41, 833–837. [Google Scholar] [CrossRef]
- Safra, M.; Sas-Chen, A.; Nir, R.; Winkler, R.; Nachshon, A.; Bar-Yaacov, D.; Erlacher, M.; Rossmanith, W.; Stern-Ginossar, N.; Schwartz, S. The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature 2017, 551, 251–255. [Google Scholar] [CrossRef]
- Li, X.; Xiong, X.; Zhang, M.; Wang, K.; Chen, Y.; Zhou, J.; Mao, Y.; Lv, J.; Yi, D.; Chen, X.-W.; et al. Base-Resolution Mapping Reveals Distinct m1A Methylome in Nuclear- and Mitochondrial-Encoded Transcripts. Mol. Cell 2017, 68, 993–1005.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlile, T.M.; Rojas-Duran, M.F.; Zinshteyn, B.; Shin, H.; Bartoli, K.M.; Gilbert, W.V. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 2014, 515, 143–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhu, P.; Ma, S.; Song, J.; Bai, J.; Sun, F.; Yi, C. Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nat. Chem. Biol. 2015, 11, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Nagaike, T.; Suzuki, T.; Tomari, Y.; Takemoto-Hori, C.; Negayama, F.; Watanabe, K.; Ueda, T. Identification and characterization of mammalian mitochondrial tRNA nucleotidyltransferases. J. Biol. Chem. 2001, 276, 40041–40049. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Nagao, A.; Suzuki, T. Human mitochondrial tRNAs: Biogenesis, function, structural aspects, and diseases. Annu. Rev. Genet. 2011, 45, 299–329. [Google Scholar] [CrossRef]
- Rackham, O.; Mercer, T.R.; Filipovska, A. The human mitochondrial transcriptome and the RNA-binding proteins that regulate its expression. Wiley Interdiscip. Rev. RNA 2012, 3, 675–695. [Google Scholar] [CrossRef]
- Pearce, S.F.; Rebelo-Guiomar, P.; D’Souza, A.R.; Powell, C.A.; Van Haute, L.; Minczuk, M. Regulation of Mammalian Mitochondrial Gene Expression: Recent Advances. Trends Biochem. Sci. 2017, 42, 625–639. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.G.; Rudler, D.L.; Rackham, O.; Filipovska, A. Is mitochondrial gene expression coordinated or stochastic? Biochem. Soc. Trans. 2018, 46, 1239–1246. [Google Scholar] [CrossRef]
- White, E.; Schlackow, M.; Kamieniarz-Gdula, K.; Proudfoot, N.J.; Gullerova, M. Human nuclear Dicer restricts the deleterious accumulation of endogenous double-stranded RNA. Nat. Struct. Mol. Biol. 2014, 21, 552–559. [Google Scholar] [CrossRef] [Green Version]
- Gantier, M.P. Processing of double-stranded RNA in mammalian cells: A direct antiviral role? J. Interferon Cytokine Res. 2014, 34, 469–477. [Google Scholar] [CrossRef]
- Sen, G.C. Biological functions of double-stranded RNA and its binding proteins. J. Interferon Cytokine Res. 2014, 34, 413–414. [Google Scholar] [CrossRef] [PubMed]
- Svoboda, P. Renaissance of mammalian endogenous RNAi. FEBS Lett. 2014, 588, 2550–2556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Park, J.; Kim, S.; Kim, M.; Kang, M.-G.; Kwak, C.; Kang, M.; Kim, B.; Rhee, H.-W.; Kim, V.N. PKR Senses Nuclear and Mitochondrial Signals by Interacting with Endogenous Double-Stranded RNAs. Mol. Cell 2018, 71, 1051–1063.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhir, A.; Dhir, S.; Borowski, L.S.; Jimenez, L.; Teitell, M.; Rötig, A.; Crow, Y.J.; Rice, G.I.; Duffy, D.; Tamby, C.; et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 2018, 560, 238–242. [Google Scholar] [CrossRef]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [Green Version]
- Medzhitov, R. Recognition of microorganisms and activation of the immune response. Nature 2007, 449, 819–826. [Google Scholar] [CrossRef]
- Belgnaoui, S.M.; Paz, S.; Hiscott, J. Orchestrating the interferon antiviral response through the mitochondrial antiviral signaling (MAVS) adapter. Curr. Opin. Immunol. 2011, 23, 564–572. [Google Scholar] [CrossRef]
- Rius, R.; Van Bergen, N.J.; Compton, A.G.; Riley, L.G.; Kava, M.P.; Balasubramaniam, S.; Amor, D.J.; Fanjul-Fernandez, M.; Cowley, M.J.; Fahey, M.C.; et al. Clinical Spectrum and Functional Consequences Associated with Bi-Allelic Pathogenic PNPT1 Variants. J. Clin. Med. 2019, 8, 2020. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.E.; Hughes, J.; Gu, Q.; Behdenna, A.; Singer, J.B.; Dennis, T.; Orton, R.J.; Varela, M.; Gifford, R.J.; Wilson, S.J.; et al. Fundamental properties of the mammalian innate immune system revealed by multispecies comparison of type I interferon responses. PLoS Biol. 2017, 15, e2004086. [Google Scholar] [CrossRef]
- Munday, D.C.; Hiscox, J.A.; Barr, J.N. Quantitative proteomic analysis of A549 cells infected with human respiratory syncytial virus subgroup B using SILAC coupled to LC-MS/MS. Proteomics 2010, 10, 4320–4334. [Google Scholar] [CrossRef]
- Crow, Y.J.; Manel, N. Aicardi-Goutières syndrome and the type I interferonopathies. Nat. Rev. Immunol. 2015, 15, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Pajak, A.; Laine, I.; Clemente, P.; El-Fissi, N.; Schober, F.A.; Maffezzini, C.; Calvo-Garrido, J.; Wibom, R.; Filograna, R.; Dhir, A.; et al. Defects of mitochondrial RNA turnover lead to the accumulation of double-stranded RNA in vivo. PLoS Genet. 2019, 15, e1008240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiatrek, D.M.; Candela, M.E.; Sedmík, J.; Oppelt, J.; Keegan, L.P.; O’Connell, M.A. Activation of innate immunity by mitochondrial dsRNA in mouse cells lacking p53 protein. RNA 2019, 25, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Rackham, O.; Shearwood, A.-M.J.; Mercer, T.R.; Davies, S.M.K.; Mattick, J.S.; Filipovska, A. Long noncoding RNAs are generated from the mitochondrial genome and regulated by nuclear-encoded proteins. RNA 2011, 17, 2085–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villegas, J.; Burzio, V.; Villota, C.; Landerer, E.; Martinez, R.; Santander, M.; Martinez, R.; Pinto, R.; Vera, M.I.; Boccardo, E.; et al. Expression of a novel non-coding mitochondrial RNA in human proliferating cells. Nucleic Acids Res. 2007, 35, 7336–7347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burzio, V.A.; Villota, C.; Villegas, J.; Landerer, E.; Boccardo, E.; Villa, L.L.; Martínez, R.; Lopez, C.; Gaete, F.; Toro, V.; et al. Expression of a family of noncoding mitochondrial RNAs distinguishes normal from cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 9430–9434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidaurre, S.; Fitzpatrick, C.; Burzio, V.A.; Briones, M.; Villota, C.; Villegas, J.; Echenique, J.; Oliveira-Cruz, L.; Araya, M.; Borgna, V.; et al. Down-regulation of the antisense mitochondrial non-coding RNAs (ncRNAs) is a unique vulnerability of cancer cells and a potential target for cancer therapy. J. Biol. Chem. 2014, 289, 27182–27198. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.-C.; Yamada, K.A.; Patel, A.Y.; Topkara, V.K.; George, I.; Cheema, F.H.; Ewald, G.A.; Mann, D.L.; Nerbonne, J.M. Deep RNA sequencing reveals dynamic regulation of myocardial noncoding RNAs in failing human heart and remodeling with mechanical circulatory support. Circulation 2014, 129, 1009–1021. [Google Scholar] [CrossRef]
- Kumarswamy, R.; Bauters, C.; Volkmann, I.; Maury, F.; Fetisch, J.; Holzmann, A.; Lemesle, G.; de Groote, P.; Pinet, F.; Thum, T. Circulating long noncoding RNA, LIPCAR, predicts survival in patients with heart failure. Circ. Res. 2014, 114, 1569–1575. [Google Scholar] [CrossRef] [Green Version]
- Dorn, G.W. LIPCAR: A mitochondrial lnc in the noncoding RNA chain? Circ. Res. 2014, 114, 1548–1550. [Google Scholar] [CrossRef]
- Clayton, D.A. Transcription and replication of mitochondrial DNA. Hum. Reprod. 2000, 15 (Suppl. 2), 11–17. [Google Scholar] [CrossRef] [PubMed]
- Krasich, R.; Copeland, W.C. DNA polymerases in the mitochondria: A critical review of the evidence. Front. Biosci. 2017, 22, 692–709. [Google Scholar]
- García-Gómez, S.; Reyes, A.; Martínez-Jiménez, M.I.; Chocrón, E.S.; Mourón, S.; Terrados, G.; Powell, C.; Salido, E.; Méndez, J.; Holt, I.J.; et al. PrimPol, an archaic primase/polymerase operating in human cells. Mol. Cell 2013, 52, 541–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudd, S.G.; Bianchi, J.; Doherty, A.J. PrimPol-A new polymerase on the block. Mol. Cell. Oncol. 2014, 1, e960754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sykora, P.; Kanno, S.; Akbari, M.; Kulikowicz, T.; Baptiste, B.A.; Leandro, G.S.; Lu, H.; Tian, J.; May, A.; Becker, K.A.; et al. DNA Polymerase Beta Participates in Mitochondrial DNA Repair. Mol. Cell. Biol. 2017, 37, e00237-17. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, B.A.; Van Houten, B. POLB: A new role of DNA polymerase beta in mitochondrial base excision repair. DNA Repair 2017, 60, A1–A5. [Google Scholar] [CrossRef]
- Singh, B.; Li, X.; Owens, K.M.; Vanniarajan, A.; Liang, P.; Singh, K.K. Human REV3 DNA Polymerase Zeta Localizes to Mitochondria and Protects the Mitochondrial Genome. PLoS ONE 2015, 10, e0140409. [Google Scholar] [CrossRef] [Green Version]
- Wisnovsky, S.; Jean, S.R.; Kelley, S.O. Mitochondrial DNA repair and replication proteins revealed by targeted chemical probes. Nat. Chem. Biol. 2016, 12, 567–573. [Google Scholar] [CrossRef]
- Longley, M.J.; Ropp, P.A.; Lim, S.E.; Copeland, W.C. Characterization of the native and recombinant catalytic subunit of human DNA polymerase gamma: Identification of residues critical for exonuclease activity and dideoxynucleotide sensitivity. Biochemistry 1998, 37, 10529–10539. [Google Scholar] [CrossRef]
- Spelbrink, J.N.; Toivonen, J.M.; Hakkaart, G.A.; Kurkela, J.M.; Cooper, H.M.; Lehtinen, S.K.; Lecrenier, N.; Back, J.W.; Speijer, D.; Foury, F.; et al. In vivo functional analysis of the human mitochondrial DNA polymerase POLG expressed in cultured human cells. J. Biol. Chem. 2000, 275, 24818–24828. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, R.; Wiederhold, L.; Szczesny, B.; Boldogh, I.; Hazra, T.K.; Izumi, T.; Mitra, S. Identification and characterization of mitochondrial abasic (AP)-endonuclease in mammalian cells. Nucleic Acids Res. 2006, 34, 2067–2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbari, M.; Keijzers, G.; Maynard, S.; Scheibye-Knudsen, M.; Desler, C.; Hickson, I.D.; Bohr, V.A. Overexpression of DNA ligase III in mitochondria protects cells against oxidative stress and improves mitochondrial DNA base excision repair. DNA Repair 2014, 16, 44–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barchiesi, A.; Wasilewski, M.; Chacinska, A.; Tell, G.; Vascotto, C. Mitochondrial translocation of APE1 relies on the MIA pathway. Nucleic Acids Res. 2015, 43, 5451–5464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szymanski, M.R.; Yu, W.; Gmyrek, A.M.; White, M.A.; Molineux, I.J.; Lee, J.C.; Yin, Y.W. A domain in human EXOG converts apoptotic endonuclease to DNA-repair exonuclease. Nat. Commun. 2017, 8, 14959. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Sampath, H. Mitochondrial DNA Integrity: Role in Health and Disease. Cells 2019, 8, 100. [Google Scholar] [CrossRef] [Green Version]
- Gammage, P.A.; Moraes, C.T.; Minczuk, M. Mitochondrial Genome Engineering: The Revolution May Not Be CRISPR-Ized. Trends Genet. 2018, 34, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Nissanka, N.; Minczuk, M.; Moraes, C.T. Mechanisms of Mitochondrial DNA Deletion Formation. Trends Genet. 2019, 35, 235–244. [Google Scholar] [CrossRef]
- Srivastava, S.; Moraes, C.T. Manipulating mitochondrial DNA heteroplasmy by a mitochondrially targeted restriction endonuclease. Hum. Mol. Genet. 2001, 10, 3093–3099. [Google Scholar] [CrossRef]
- Srivastava, S.; Moraes, C.T. Double-strand breaks of mouse muscle mtDNA promote large deletions similar to multiple mtDNA deletions in humans. Hum. Mol. Genet. 2005, 14, 893–902. [Google Scholar] [CrossRef] [Green Version]
- Bacman, S.R.; Williams, S.L.; Garcia, S.; Moraes, C.T. Organ-specific shifts in mtDNA heteroplasmy following systemic delivery of a mitochondria-targeted restriction endonuclease. Gene Ther. 2010, 17, 713–720. [Google Scholar] [CrossRef]
- Minczuk, M.; Papworth, M.A.; Miller, J.C.; Murphy, M.P.; Klug, A. Development of a single-chain, quasi-dimeric zinc-finger nuclease for the selective degradation of mutated human mitochondrial DNA. Nucleic Acids Res. 2008, 36, 3926–3938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.; Ocampo, A.; Suzuki, K.; Luo, J.; Bacman, S.R.; Williams, S.L.; Sugawara, A.; Okamura, D.; Tsunekawa, Y.; Wu, J.; et al. Selective elimination of mitochondrial mutations in the germline by genome editing. Cell 2015, 161, 459–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gammage, P.A.; Rorbach, J.; Vincent, A.I.; Rebar, E.J.; Minczuk, M. Mitochondrially targeted ZFNs for selective degradation of pathogenic mitochondrial genomes bearing large-scale deletions or point mutations. EMBO Mol. Med. 2014, 6, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Gammage, P.A.; Gaude, E.; Van Haute, L.; Rebelo-Guiomar, P.; Jackson, C.B.; Rorbach, J.; Pekalski, M.L.; Robinson, A.J.; Charpentier, M.; Concordet, J.-P.; et al. Near-complete elimination of mutant mtDNA by iterative or dynamic dose-controlled treatment with mtZFNs. Nucleic Acids Res. 2016, 44, 7804–7816. [Google Scholar] [CrossRef]
- Bacman, S.R.; Williams, S.L.; Pinto, M.; Peralta, S.; Moraes, C.T. Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nat. Med. 2013, 19, 1111–1113. [Google Scholar] [CrossRef]
- Gammage, P.A.; Viscomi, C.; Simard, M.-L.; Costa, A.S.H.; Gaude, E.; Powell, C.A.; Van Haute, L.; McCann, B.J.; Rebelo-Guiomar, P.; Cerutti, R.; et al. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat. Med. 2018, 24, 1691–1695. [Google Scholar] [CrossRef]
- Bacman, S.R.; Kauppila, J.H.K.; Pereira, C.V.; Nissanka, N.; Miranda, M.; Pinto, M.; Williams, S.L.; Larsson, N.-G.; Stewart, J.B.; Moraes, C.T. MitoTALEN reduces mutant mtDNA load and restores tRNAAla levels in a mouse model of heteroplasmic mtDNA mutation. Nat. Med. 2018, 24, 1696–1700. [Google Scholar] [CrossRef]
- Wang, G.; Shimada, E.; Zhang, J.; Hong, J.S.; Smith, G.M.; Teitell, M.A.; Koehler, C.M. Correcting human mitochondrial mutations with targeted RNA import. Proc. Natl. Acad. Sci. USA 2012, 109, 4840–4845. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.-W.; Rainey, R.N.; Balatoni, C.E.; Dawson, D.W.; Troke, J.J.; Wasiak, S.; Hong, J.S.; McBride, H.M.; Koehler, C.M.; Teitell, M.A.; et al. Mammalian polynucleotide phosphorylase is an intermembrane space RNase that maintains mitochondrial homeostasis. Mol. Cell. Biol. 2006, 26, 8475–8487. [Google Scholar] [CrossRef] [Green Version]
- Tarassov, I.; Entelis, N.; Martin, R.P. An intact protein translocating machinery is required for mitochondrial import of a yeast cytoplasmic tRNA. J. Mol. Biol. 1995, 245, 315–323. [Google Scholar] [CrossRef]
- Kolesnikova, O.A.; Entelis, N.S.; Jacquin-Becker, C.; Goltzene, F.; Chrzanowska-Lightowlers, Z.M.; Lightowlers, R.N.; Martin, R.P.; Tarassov, I. Nuclear DNA-encoded tRNAs targeted into mitochondria can rescue a mitochondrial DNA mutation associated with the MERRF syndrome in cultured human cells. Hum. Mol. Genet. 2004, 13, 2519–2534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comte, C.; Tonin, Y.; Heckel-Mager, A.-M.; Boucheham, A.; Smirnov, A.; Auré, K.; Lombès, A.; Martin, R.P.; Entelis, N.; Tarassov, I. Mitochondrial targeting of recombinant RNAs modulates the level of a heteroplasmic mutation in human mitochondrial DNA associated with Kearns Sayre Syndrome. Nucleic Acids Res. 2013, 41, 418–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonin, Y.; Heckel, A.-M.; Vysokikh, M.; Dovydenko, I.; Meschaninova, M.; Rötig, A.; Munnich, A.; Venyaminova, A.; Tarassov, I.; Entelis, N. Modeling of antigenomic therapy of mitochondrial diseases by mitochondrially addressed RNA targeting a pathogenic point mutation in mitochondrial DNA. J. Biol. Chem. 2014, 289, 13323–13334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, A.; Martin, J.; Agabian, N. A nuclear encoded tRNA of Trypanosoma brucei is imported into mitochondria. Mol. Cell. Biol. 1994, 14, 2317–2322. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Maréchal-Drouard, L.; Akama, K.; Small, I. Striking differences in mitochondrial tRNA import between different plant species. Mol. Gen. Genet. 1996, 252, 404–411. [Google Scholar] [CrossRef]
- Kiss, T.; Filipowicz, W. Evidence against a mitochondrial location of the 7-2/MRP RNA in mammalian cells. Cell 1992, 70, 11–16. [Google Scholar] [CrossRef]
- Jacobson, M.R.; Cao, L.G.; Wang, Y.L.; Pederson, T. Dynamic localization of RNase MRP RNA in the nucleolus observed by fluorescent RNA cytochemistry in living cells. J. Cell Biol. 1995, 131, 1649–1658. [Google Scholar] [CrossRef]
- Goldfarb, K.C.; Cech, T.R. Targeted CRISPR disruption reveals a role for RNase MRP RNA in human preribosomal RNA processing. Genes Dev. 2017, 31, 59–71. [Google Scholar] [CrossRef] [Green Version]
- Greber, B.J.; Boehringer, D.; Leitner, A.; Bieri, P.; Voigts-Hoffmann, F.; Erzberger, J.P.; Leibundgut, M.; Aebersold, R.; Ban, N. Architecture of the large subunit of the mammalian mitochondrial ribosome. Nature 2014, 505, 515–519. [Google Scholar] [CrossRef]
- Brown, A.; Amunts, A.; Bai, X.-C.; Sugimoto, Y.; Edwards, P.C.; Murshudov, G.; Scheres, S.H.W.; Ramakrishnan, V. Structure of the large ribosomal subunit from human mitochondria. Science 2014, 346, 718–722. [Google Scholar] [CrossRef] [Green Version]
- Greber, B.J.; Bieri, P.; Leibundgut, M.; Leitner, A.; Aebersold, R.; Boehringer, D.; Ban, N. Ribosome. The complete structure of the 55S mammalian mitochondrial ribosome. Science 2015, 348, 303–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.S.; Shibata, Y.; Malhotra, A.; Dutta, A. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes Dev. 2009, 23, 2639–2649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haussecker, D.; Huang, Y.; Lau, A.; Parameswaran, P.; Fire, A.Z.; Kay, M.A. Human tRNA-derived small RNAs in the global regulation of RNA silencing. RNA 2010, 16, 673–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ro, S.; Ma, H.-Y.; Park, C.; Ortogero, N.; Song, R.; Hennig, G.W.; Zheng, H.; Lin, Y.-M.; Moro, L.; Hsieh, J.-T.; et al. The mitochondrial genome encodes abundant small noncoding RNAs. Cell Res. 2013, 23, 759–774. [Google Scholar] [CrossRef] [Green Version]
- Geiger, J.; Dalgaard, L.T. Interplay of mitochondrial metabolism and microRNAs. Cell. Mol. Life Sci. 2017, 74, 631–646. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Bandiera, S.; Rüberg, S.; Girard, M.; Cagnard, N.; Hanein, S.; Chrétien, D.; Munnich, A.; Lyonnet, S.; Henrion-Caude, A. Nuclear outsourcing of RNA interference components to human mitochondria. PLoS ONE 2011, 6, e20746. [Google Scholar] [CrossRef] [Green Version]
- Barrey, E.; Saint-Auret, G.; Bonnamy, B.; Damas, D.; Boyer, O.; Gidrol, X. Pre-microRNA and mature microRNA in human mitochondria. PLoS ONE 2011, 6, e20220. [Google Scholar] [CrossRef]
- Sripada, L.; Tomar, D.; Prajapati, P.; Singh, R.; Singh, A.K.; Singh, R. Systematic analysis of small RNAs associated with human mitochondria by deep sequencing: Detailed analysis of mitochondrial associated miRNA. PLoS ONE 2012, 7, e44873. [Google Scholar] [CrossRef]
- Jagannathan, R.; Thapa, D.; Nichols, C.E.; Shepherd, D.L.; Stricker, J.C.; Croston, T.L.; Baseler, W.A.; Lewis, S.E.; Martinez, I.; Hollander, J.M. Translational Regulation of the Mitochondrial Genome Following Redistribution of Mitochondrial MicroRNA in the Diabetic Heart. Circ. Cardiovasc. Genet. 2015, 8, 785–802. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, Y.; Ito, Y.; Niikura, T.; Shao, Z.; Hata, M.; Oyama, F.; Nishimoto, I. Mechanisms of neuroprotection by a novel rescue factor humanin from Swedish mutant amyloid precursor protein. Biochem. Biophys. Res. Commun. 2001, 283, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Niikura, T.; Tajima, H.; Yasukawa, T.; Sudo, H.; Ito, Y.; Kita, Y.; Kawasumi, M.; Kouyama, K.; Doyu, M.; et al. A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer’s disease genes and Abeta. Proc. Natl. Acad. Sci. USA 2001, 98, 6336–6341. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yen, K.; Cohen, P. Humanin: A harbinger of mitochondrial-derived peptides? Trends Endocrinol. Metab. 2013, 24, 222–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Zeng, J.; Drew, B.G.; Sallam, T.; Martin-Montalvo, A.; Wan, J.; Kim, S.-J.; Mehta, H.; Hevener, A.L.; de Cabo, R.; et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 2015, 21, 443–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobb, L.J.; Lee, C.; Xiao, J.; Yen, K.; Wong, R.G.; Nakamura, H.K.; Mehta, H.H.; Gao, Q.; Ashur, C.; Huffman, D.M.; et al. Naturally occurring mitochondrial-derived peptides are age-dependent regulators of apoptosis, insulin sensitivity, and inflammatory markers. Aging 2016, 8, 796–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-J.; Xiao, J.; Wan, J.; Cohen, P.; Yen, K. Mitochondrially derived peptides as novel regulators of metabolism. J. Physiol. 2017, 595, 6613–6621. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotrys, A.V.; Szczesny, R.J. Mitochondrial Gene Expression and Beyond—Novel Aspects of Cellular Physiology. Cells 2020, 9, 17. https://doi.org/10.3390/cells9010017
Kotrys AV, Szczesny RJ. Mitochondrial Gene Expression and Beyond—Novel Aspects of Cellular Physiology. Cells. 2020; 9(1):17. https://doi.org/10.3390/cells9010017
Chicago/Turabian StyleKotrys, Anna V., and Roman J. Szczesny. 2020. "Mitochondrial Gene Expression and Beyond—Novel Aspects of Cellular Physiology" Cells 9, no. 1: 17. https://doi.org/10.3390/cells9010017
APA StyleKotrys, A. V., & Szczesny, R. J. (2020). Mitochondrial Gene Expression and Beyond—Novel Aspects of Cellular Physiology. Cells, 9(1), 17. https://doi.org/10.3390/cells9010017