Insights into Activation Mechanisms of Store-Operated TRPC1 Channels in Vascular Smooth Muscle



{kind=link}
Abstract
:1. Introduction
2. Biophysical Properties and Molecular Composition of SOCs in VSMCs
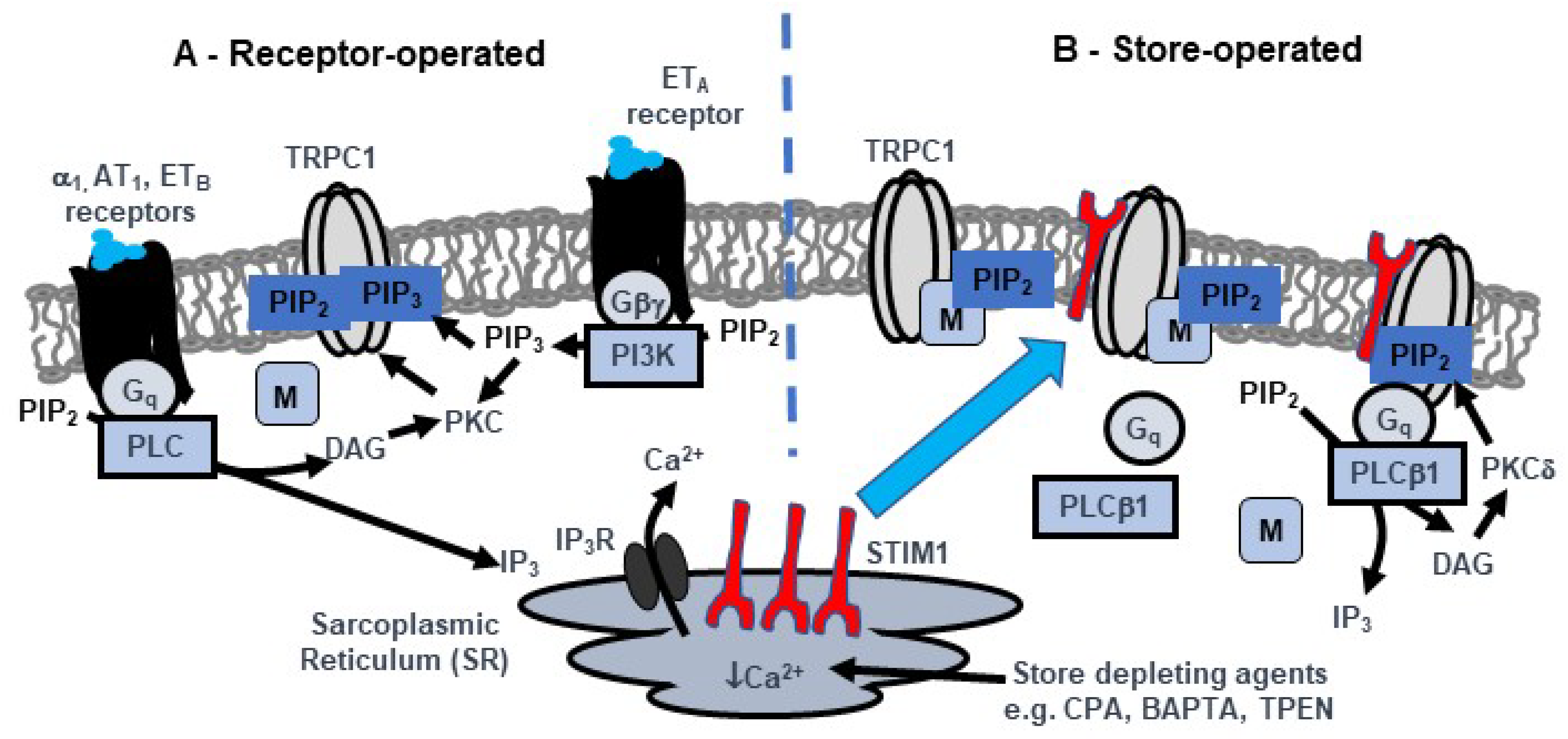
3. Activation Mechanisms of TRPC1-Based SOCs in Native Contractile VSMCs
3.1. PKC Activity and PIP2 have Obligatory Roles in Activation of TRPC1-based SOCs
3.2. Interactions Between PKC Activity and PIP2 are Regulated by MARCKS
3.3. Store Depletion Activates a Gq-PLCβ1 Pathway Involved in Activation of TRPC1-Based SOCs
3.4. STIM1 Couples Store Depletion to Gq-PLCβ1 Activity to Stimulate TRPC1 SOCs
3.5. Activation Mechanisms of TRPC1-Based SOCs are Independent of Orai1
4. Physiological Significance of TRPC1-Based SOCs Activation Pathway
5. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Albert, A.P.; Large, W.A. Store-operated Ca2+-permeable non-selective cation channels in smooth muscle cells. Cell Calcium 2003, 33, 345–356. [Google Scholar] [CrossRef]
- Albert, A.P.; Saleh, S.N.; Peppiatt-Wildman, C.M.; Large, W.A. Multiple activation mechanisms of store-operated TRPC channels in smooth muscle cells. J. Physiol. 2007, 583, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Abramowitz, J.; Birnbaumer, L. Physiology and pathophysiology of canonical transient receptor potential channels. FASEB J. 2009, 23, 297–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beech, D.J. Characteristics of transient receptor potential canonical calcium-permeable channels and their relevance to vascular physiology and disease. Circ. J. 2013, 77, 570–579. [Google Scholar] [CrossRef] [Green Version]
- Earley, S.; Brayden, J.E. Transient receptor potential channels in the vasculature. Physiol. Rev. 2015, 95, 645–690. [Google Scholar] [CrossRef] [Green Version]
- Large, W.A. Receptor-operated Ca2+-permeable nonselective cation channels in vascular smooth muscle: A physiologic perspective. J. Cardiovasc. Electrophysiol. 2002, 13, 493–501. [Google Scholar] [CrossRef]
- Albert, A.P.; Saleh, S.N.; Large, W.A. Identification of canonical transient receptor potential (TRPC) channel proteins in native vascular smooth muscle cells. Curr. Med. Chem. 2009, 16, 1158–1165. [Google Scholar] [CrossRef]
- Albert, A.P.; Large, W.A. Activation of store-operated channels by noradrenaline via protein kinase C in rabbit portal vein myocytes. J. Physiol. 2002, 544, 113–125. [Google Scholar] [CrossRef]
- Saleh, S.N.; Albert, A.P.; Peppiatt, C.M.; Large, W.A. Angiotensin II activates two cation conductances with distinct TRPC1 and TRPC6 channel properties in rabbit mesenteric artery myocytes. J. Physiol. 2006, 577, 479–495. [Google Scholar] [CrossRef]
- Shi, J.; Ju, M.; Saleh, S.N.; Albert, A.P.; Large, W.A. TRPC6 channels stimulated by angiotensin II are inhibited by TRPC1/C5 channel activity through a Ca2+- and PKC-dependent mechanism in native vascular myocytes. J. Physiol. 2010, 588, 3671–3682. [Google Scholar] [CrossRef]
- Shi, J.; Ju, M.; Large, W.A.; Albert, A.P. Pharmacological profile of phosphatidylinositol 3-kinases and related phosphatidylinositols mediating endothelin(A) receptor-operated native TRPC channels in rabbit coronary artery myocytes. Br. J. Pharmacol. 2012, 166, 2161–2175. [Google Scholar] [CrossRef] [Green Version]
- Prakriya, M.; Lewis, R.S. Store-operated calcium channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.T.; Ong, H.L.; Liu, X.; Ambudkar, I.S. Contribution and regulation of TRPC channels in store-operated Ca2+ entry. Curr. Top. Membr. 2013, 71, 149–179. [Google Scholar]
- Liao, Y.; Abramowitz, J.; Birnbaumer, L. The TRPC family of TRP channels: Roles inferred (mostly) from knockout mice and relationship to ORAI proteins. Handb. Exp. Pharmacol. 2014, 223, 1055–1075. [Google Scholar]
- Worley, P.F.; Zeng, W.; Huang, G.N.; Yuan, J.P.; Kim, J.Y.; Lee, M.G.; Muallem, S. TRPC channels as STIM1-regulated store-operated channels. Cell Calcium 2007, 42, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.P.; Kim, M.S.; Zeng, W.; Shin, D.M.; Huang, G.; Worley, P.F.; Muallem, S. TRPC channels as STIM1-regulated SOCs. Channels 2009, 3, 221–225. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.P.; Choi, S.; Hong, J.H.; Ahuja, M.; Graham, S.; Ma, R.; So, I.; Shin, D.M.; Muallem, S.; Yuan, J.P. Molecular determinants mediating gating of transient receptor potential canonical (TRPC) channels by stromal interaction molecule 1 (STIM1). J. Biol. Chem. 2014, 289, 6372–6382. [Google Scholar] [CrossRef] [Green Version]
- Asanov, A.; Sampieri, A.; Moreno, C.; Pacheco, J.; Salgado, A.; Sherry, R.; Vaca, L. Combined single channel and single molecule detection identifies subunit composition of STIM1-activated transient receptor potential canonical (TRPC) channels. Cell Calcium 2015, 57, 1–13. [Google Scholar] [CrossRef]
- Ambudkar, I.S.; de Souza, L.B.; Ong, H.L. TRPC1, Orai1, and STIM1 in SOCE: Friends in tight spaces. Cell Calcium 2017, 63, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Trepakova, E.S.; Gericke, M.; Hirakawa, Y.; Weisbrod, R.M.; Cohen, R.A.; Bolotina, V.M. Properties of a native cation channel activated by Ca2+ store depletion in vascular smooth muscle cells. J. Biol. Chem. 2001, 276, 7782–7790. [Google Scholar] [CrossRef] [Green Version]
- Albert, A.P.; Large, W.A. A Ca2+-permeable non-selective cation channel activated by depletion of internal Ca2+ stores in single rabbit portal vein myocytes. J. Physiol. 2002, 538, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Saleh, S.N.; Albert, A.P.; Peppiatt-Wildman, C.M.; Large, W.A. Diverse properties of store-operated TRPC channels activated by protein kinase C in vascular myocytes. J. Physiol. 2008, 586, 2463–2476. [Google Scholar] [CrossRef]
- Large, W.A.; Saleh, S.N.; Albert, A.P. Role of phosphoinositol 4,5-bisphosphate and diacylycerol in regulating native TRPC channel proteins in vascular smooth muscle. Cell Calcium 2009, 45, 574–582. [Google Scholar] [CrossRef]
- Albert, A.P. Gating mechanisms of canonical transient receptor potential channel proteins: Role of phosphoinositols and diacylglycerol. Adv. Exp. Med. Biol. 2011, 704, 391–411. [Google Scholar]
- Shi, J.; Ju, M.; Abramowitz, J.; Large, W.A.; Birnbaumer, L.; Albert, A.P. TRPC1 proteins confer PKC and phosphoinositol activation on native heteromeric TRPC1/C5 channels in vascular smooth muscle: Comparative study of wild-type and TRPC1−/− mice. FASEB J. 2012, 26, 409–419. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Birnbaumer, L.; Large, W.A.; Albert, A.P. Myristoylated alanine-rich C kinase substrate coordinates native TRPC1 channel activation by phosphatidylinositol 4,5-bisphosphate and protein kinase C in vascular smooth muscle. FASEB J. 2014, 28, 244–255. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Miralles, F.; Birnbaumer, L.; Large, W.A.; Albert, A.P. Store depletion induces Gαq-mediated PLCβ1 activity to stimulate TRPC1 channels in vascular smooth muscle cells. FASEB J. 2016, 30, 702–715. [Google Scholar] [CrossRef]
- Shi, J.; Miralles, F.; Birnbaumer, L.; Large, W.A.; Albert, A.P. Store-operated interactions between plasmalemmal STIM1 and TRPC1 proteins stimulate PLCβ1 to induce TRPC1 channel activation in vascular smooth muscle cells. J. Physiol. 2017, 595, 1039–1058. [Google Scholar] [CrossRef]
- Shi, J.; Miralles, F.; Kinet, J.P.; Birnbaumer, L.; Large, W.A.; Albert, A.P. Evidence that Orai1 does not contribute to store-operated TRPC1 channels in vascular smooth muscle cells. Channels 2017, 11, 329–339. [Google Scholar] [CrossRef] [Green Version]
- Rubaiy, H.N.; Ludlow, M.J.; Henrot, M.; Gaunt, H.J.; Miteva, K.; Cheung, S.Y.; Tanahashi, Y.; Hamzah, N.; Musialowski, K.E.; Blythe, N.M.; et al. Picomolar, selective, and subtype-specific small-molecule inhibition of TRPC1/4/5 channels. J. Biol. Chem. 2017, 292, 8158–8173. [Google Scholar] [CrossRef] [Green Version]
- Martin-Aragon Baudel, M.; Shi, J.; Jahan, K.S.; Large, W.A.; Albert, A.P. Obligatory role for PKCδ in PIP2-mediated activation of store-operated TRPC1 channels in vascular smooth muscle cells (manuscript in preparation). (manuscript in preparation).
- Xu, S.Z.; Beech, D.J. TrpC1 is a membrane-spanning subunit of store-operated Ca2+ channels in native vascular smooth muscle cells. Circ. Res. 2001, 88, 84–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.Z.; Boulay, G.; Flemming, R.; Beech, D.J. E3-targeted anti-TRPC5 antibody inhibits store-operated calcium entry in freshly isolated pial arterioles. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2653–H2659. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Albert, A.P.; Large, W.A. Facilitatory effect of Ins(1,4,5)P3 on store-operated Ca2+-permeable cation channels in rabbit portal vein myocytes. J. Physiol. 2005, 566, 161–171. [Google Scholar] [CrossRef]
- Berra-Romani, R.; Mazzocco-Spezzia, A.; Pulina, M.V.; Golovina, V.A. Ca2+ handling is altered when arterial myocytes progress from a contractile to a proliferative phenotype in culture. Am. J. Physiol. Cell Physiol. 2008, 295, C779–C790. [Google Scholar] [CrossRef] [Green Version]
- Potier, M.; Gonzalez, J.C.; Motiani, R.K.; Abdullaev, I.F.; Bisaillon, J.M.; Singer, H.A.; Trebak, M. Evidence for STIM1- and Orai1-dependent store-operated calcium influx through ICRAC in vascular smooth muscle cells: Role in proliferation and migration. FASEB J. 2009, 23, 2425–2437. [Google Scholar] [CrossRef] [Green Version]
- Beech, D.J. Orai1 calcium channels in the vasculature. Pflug. Arch. 2012, 463, 635–647. [Google Scholar] [CrossRef] [Green Version]
- Trebak, M. STIM/Orai signalling complexes in vascular smooth muscle. J. Physiol. 2012, 590, 4201–4208. [Google Scholar] [CrossRef]
- Li, J.; Sukumar, P.; Milligan, C.J.; Kumar, B.; Ma, Z.Y.; Munsch, C.M.; Jiang, L.H.; Porter, K.E.; Beech, D.J. Interactions, functions, and independence of plasma membrane STIM1 and TRPC1 in vascular smooth muscle cells. Circ. Res. 2008, 103, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Ng, L.C.; O′Neill, K.G.; French, D.; Airey, J.A.; Singer, C.A.; Tian, H.; Shen, X.M.; Hume, J.R. TRPC1 and STIM1 mediate capacitative Ca2+ entry in mouse pulmonary arterial smooth muscle cells. J. Physiol. 2009, 587, 2429–2442. [Google Scholar] [CrossRef]
- Ng, L.C.; Ramduny, D.; Airey, J.A.; Singer, C.A.; Keller, P.S.; Shen, X.M.; Tian, H.; Valencik, M.; Hume, J.R. Orai1 interacts with STIM1 and mediates capacitative Ca2+ entry in mouse pulmonary arterial smooth muscle cells. Am. J. Physiol. Cell Physiol. 2010, 299, C1079–C1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; McKeown, L.; Ojelabi, O.; Stacey, M.; Foster, R.; O′Regan, D.; Porter, K.E.; Beech, D.J. Nanomolar potency and selectivity of a Ca²⁺ release-activated Ca²⁺ channel inhibitor against store-operated Ca²⁺ entry and migration of vascular smooth muscle cells. Br. J. Pharmacol. 2011, 164, 382–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, A.; Kalwa, H.; Storch, U.; Mederos y Schnitzler, M.; Salannova, B.; Pinkenburg, O.; Dubrovska, G.; Essin, K.; Gollasch, M.; Birnbaumer, L.; et al. Pressure-induced and store-operated cation influx in vascular smooth muscle cells is independent of TRPC1. Pflug. Arch. 2007, 455, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Saleh, S.N.; Albert, A.P.; Large, W.A. Obligatory role for phosphatidylinositol 4,5-bisphosphate in activation of native TRPC1 store-operated channels in vascular myocytes. J. Physiol. 2009, 587, 531–540. [Google Scholar] [CrossRef] [Green Version]
- Albert, A.P.; Saleh, S.N.; Large, W.A. Inhibition of native TRPC6 channel activity by phosphatidylinositol 4,5-bisphosphate in mesenteric artery myocytes. J. Physiol. 2008, 586, 3087–3095. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Itsuki, K.; Okamura, Y.; Inoue, R.; Mori, M.X. A self-limiting regulation of vasoconstrictor-activated TRPC3/C6/C7 channels coupled to PI(4,5)P2-diacylglycerol signalling. J. Physiol. 2012, 590, 1101–1119. [Google Scholar] [CrossRef] [Green Version]
- Salamanca, D.A.; Khalil, R.A. Protein kinase C isoforms as specific targets for modulation of vascular smooth muscle function in hypertension. Biochem. Pharmacol. 2005, 70, 1537–1547. [Google Scholar] [CrossRef] [Green Version]
- Kwon, Y.; Hofmann, T.; Montell, C. Integration of phosphoinositide- and calmodulin-mediated regulation of TRPC6. Mol. Cell 2007, 25, 491–503. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Large, W.A.; Albert, A.P. Stimulation of beta-adrenoceptors inhibits store-operated channel currents via a cAMP-dependent protein kinase mechanism in rabbit portal vein myocytes. J. Physiol. 2005, 562, 395–406. [Google Scholar] [CrossRef]
- Albert, A.P.; Liu, M.; Large, W.A. Dual effect of calmodulin on store-operated Ca2+ -permeable cation channels in rabbit portal vein myocytes. Br. J. Pharmacol. 2006, 148, 1001–1011. [Google Scholar] [CrossRef]
- Chen, I.S.; Dai, Z.K.; Welsh, D.G.; Chen, I.J.; Wu, B.N. Protein kinases modulate store-operated channels in pulmonary artery smooth muscle cells. J. Biomed. Sci. 2011, 18, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logothetis, D.E.; Petrou, V.I.; Zhang, M.; Mahajan, R.; Meng, X.Y.; Adney, S.K.; Cui, M.; Baki, L. Phosphoinositide control of membrane protein function: A frontier led by studies on ion channels. Annu. Rev. Physiol. 2015, 77, 81–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaughlin, S.; Wang, J.; Gambhir, A.; Murray, D. PIP2 and proteins: Interactions, organization, and information flow. Annu. Rev. Biophys. Biomol. Struct. 2002, 31, 151–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamper, N.; Shapiro, M.S. Target-specific PIP2 signalling: How might it work? J. Physiol. 2007, 582, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Gallant, C.; You, J.Y.; Sasaki, Y.; Grabarek, Z.; Morgan, K.G. MARCKS is a major PKC-dependent regulator of calmodulin targeting in smooth muscle. J. Cell Sci. 2005, 118, 3595–3605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, K.V.; Behe, P.; Tinker, A. Monitoring changes in membrane phosphatidyylinositol 4,5-bisphoshate in living cells using a domain from the transcription factor tubby. J. Physiol. 2008, 586, 2855–2871. [Google Scholar] [CrossRef] [PubMed]
- Balla, T.; Vamai, P. Visualization of cellular phosphoinositide pools with GFP-fused protein-domains. Curr. Protoc. Cell. Biol. 2009, 42. [Google Scholar] [CrossRef] [Green Version]
- Szentpetery, Z.; Balla, A.; Kim, Y.J.; Lemmon, M.A.; Balla, T. Live cell imaging with protein domains vapable of recognizing phosphatidylinositol 4,5-bisphosphate; a comparative study. BMC Cell Biol. 2009, 10, 67. [Google Scholar] [CrossRef] [Green Version]
- Balla, T. Phosphoinositides: Tiny lipids with giant impact on cell regulation. Physiol. Rev. 2013, 93, 1019–1137. [Google Scholar] [CrossRef]
- Park, C.Y.; Shcheglovitov, A.; Dolmetsch, R. The CRAC channel activator binds and inhibits L-type voltage-gated calcium channels. Science 2010, 330, 101–105. [Google Scholar] [CrossRef]
- Wang, Y.; Deng, X.; Mancarella, S.; Hendron, E.; Eguchi, S.; Soboloff, J.; Tang, X.D.; Gill, D.L. The calcium store sensor, STIM1, reciprocally controls Orai1 and CaV1.2 channels. Science 2010, 330, 105–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jousset, H.; Rrieden, M.; Demaurex, N. STIM1 knockdown reveals that store-operated Ca2+ channels located close to sarco/endoplasmic Ca2+-ATPases (SECRA) pumps silently refull the endoplasmic tecticulum. J. Biol. Chem. 2007, 282, 11456–11464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.F.; Samakai, E.; Soboloff, J. STIM1 is required for attenuation of PMCA-mediated Ca2+ clearance during T-cell activation. EMBO J. 2012, 31, 1123–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefkimmiatis, K.; Srikanthan, M.; Maiellaro, I.; Moyer, M.P.; Curci, S.; Hofer, A.M. Store-operated cyclic AMP signalling mediated by STIM1. Nat. Cell Biol. 2009, 11, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Spassova, M.A.; Soboloff, J.; He, L.P.; Xu, W.; Dziadek, M.A.; Gill, D.L. STIM1 has a plasma membrane role in the activation of store-operated Ca2+ channels. Proc. Natl. Acad. Sci. USA 2006, 103, 4040–4045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, J.L.; Shuttleworth, T.J. Exploring the unique features of the ARC channel a store-operated Orai channel. Channels 2013, 7, 364–373. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Gwozdz, T.; Dutko-Gwozdz, J.; Bolotina, V.M. Orai1 and Ca2+-independent phospholipase A2 are required for store-operated Icat-SOC current, Ca2+ entry, and proliferation of primary vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 2012, 302, C748–C756. [Google Scholar] [CrossRef] [Green Version]
- Bolotina, V.M. Orai, STIM1 and iPLA2beta: A view from a different perspective. J. Physiol. 2008, 586, 3035–3042. [Google Scholar] [CrossRef]
- Avila-Medina, J.; Calderón-Sánchez, E.; González-Rodríguez, P.; Monje-Quiroga, F.; Rosado, J.A.; Castellano, A.; Ordonez, A.; Smani, T. Orai1 and TRPC1 Proteins Co-localize with CaV1.2 Channels to Form a Signal Complex in Vascular Smooth Muscle Cells. J. Biol. Chem. 2016, 291, 21148–21159. [Google Scholar] [CrossRef] [Green Version]
- Avila-Medina, J.; Mayoral-Gonzalez, I.; Dominguez-Rodriguez, A.; Gallardo-Castillo, I.; Ribas, J.; Ordonez, A.; Rosado, J.A.; Smani, T. The Complex Role of Store Operated Calcium Entry Pathways and Related Proteins in the Function of Cardiac, Skeletal and Vascular Smooth Muscle Cells. Front. Physiol. 2018, 9, 257. [Google Scholar] [CrossRef]
- Chakraborty, S.; Berwick, Z.C.; Bartlett, P.J.; Kumar, S.; Thomas, A.P.; Sturek, M.; Tune, J.D.; Obukhov, A.G. Bromoenol lactone inhibits voltage-gated Ca2+ and transient receptor potential canonical channels. J. Pharmacol. Exp. Ther. 2011, 339, 329–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, S.N.; Albert, A.P.; Large, W.A. Activation of native TRPC1/C5/C6 channels by endothelin-1 is mediated by both PIP3 and PIP2 in rabbit coronary artery myocytes. J. Physiol. 2009, 587, 5361–5375. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martín-Aragón Baudel, M.A.S.; Shi, J.; Large, W.A.; Albert, A.P. Insights into Activation Mechanisms of Store-Operated TRPC1 Channels in Vascular Smooth Muscle. Cells 2020, 9, 179. https://doi.org/10.3390/cells9010179
Martín-Aragón Baudel MAS, Shi J, Large WA, Albert AP. Insights into Activation Mechanisms of Store-Operated TRPC1 Channels in Vascular Smooth Muscle. Cells. 2020; 9(1):179. https://doi.org/10.3390/cells9010179
Chicago/Turabian StyleMartín-Aragón Baudel, Miguel A. S., Jian Shi, William A. Large, and Anthony P. Albert. 2020. "Insights into Activation Mechanisms of Store-Operated TRPC1 Channels in Vascular Smooth Muscle" Cells 9, no. 1: 179. https://doi.org/10.3390/cells9010179
APA StyleMartín-Aragón Baudel, M. A. S., Shi, J., Large, W. A., & Albert, A. P. (2020). Insights into Activation Mechanisms of Store-Operated TRPC1 Channels in Vascular Smooth Muscle. Cells, 9(1), 179. https://doi.org/10.3390/cells9010179