AXL Controls Directed Migration of Mesenchymal Triple-Negative Breast Cancer Cells




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Cell Lines, Cell Authentication and Cell Culture
2.2. Protein Extraction
2.3. Immunoblot
2.4. Small Interfering RNAs (siRNAs) and Transfection
2.5. Random Migration
2.6. Cell Survival Assays
2.7. Immunoprecipitation (IP)
2.8. Immunofluorescence (IF)
2.9. Wound Assays
2.10. AXL Localization Analysis
2.11. Patient Samples and Multiplexed Immunohistochemistry (IHC)
2.12. Antibodies and Staining
2.13. Statistics
3. Results
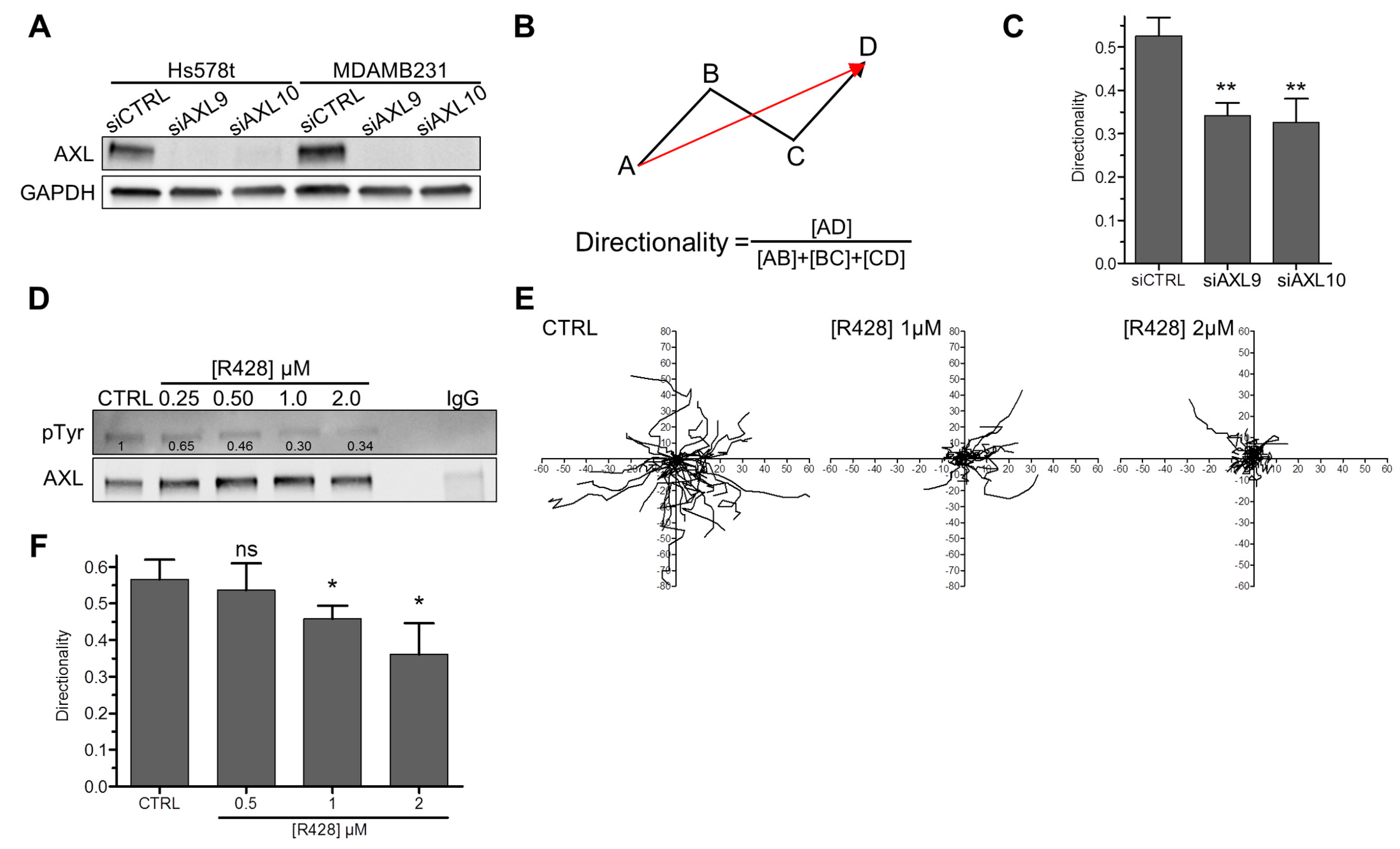
3.1. AXL Controls Directed Migration in Mesenchymal TNBC Cell Lines
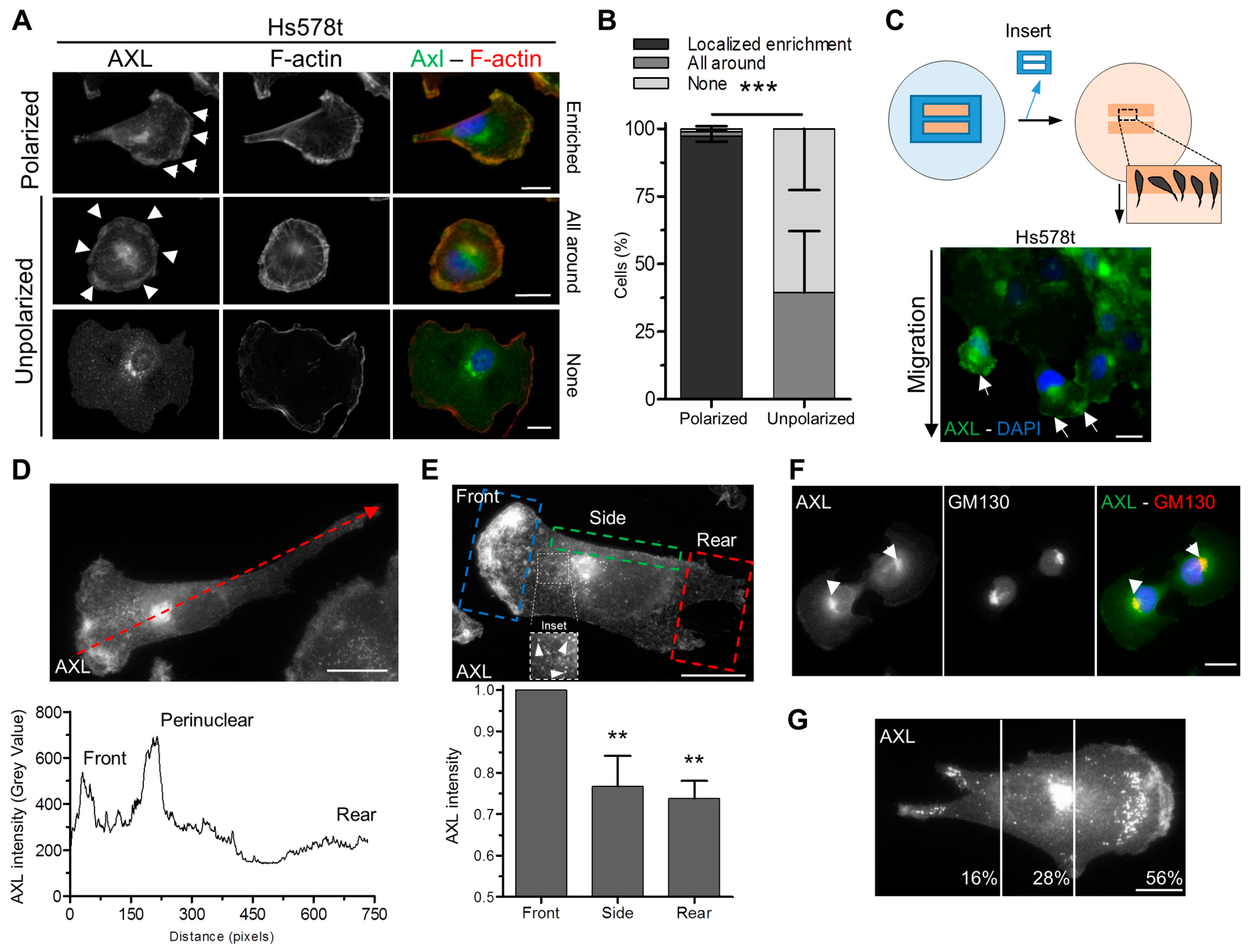
3.2. Polarized Localization of AXL at the Leading Edge and the Golgi Apparatus in Migrating Cells
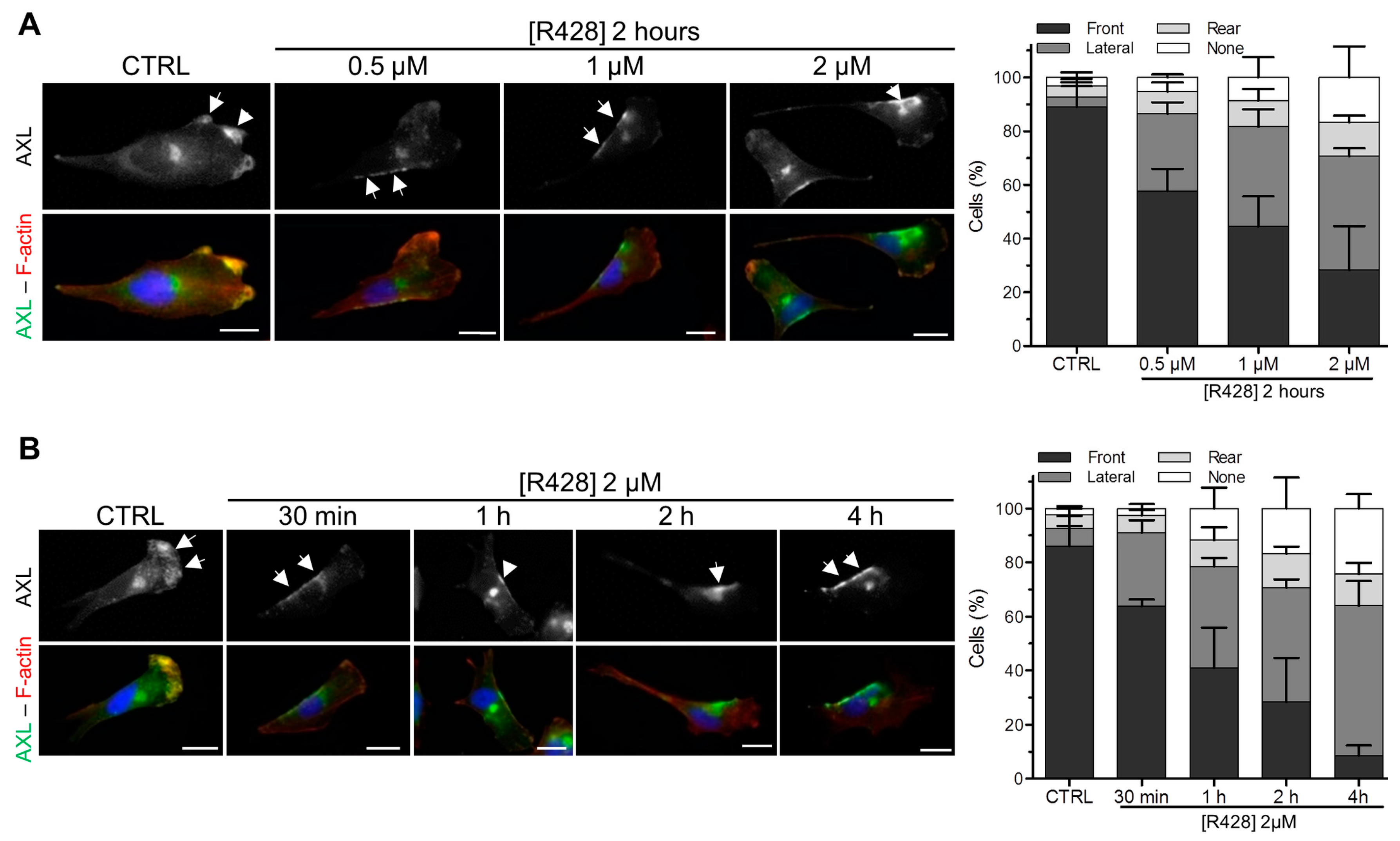
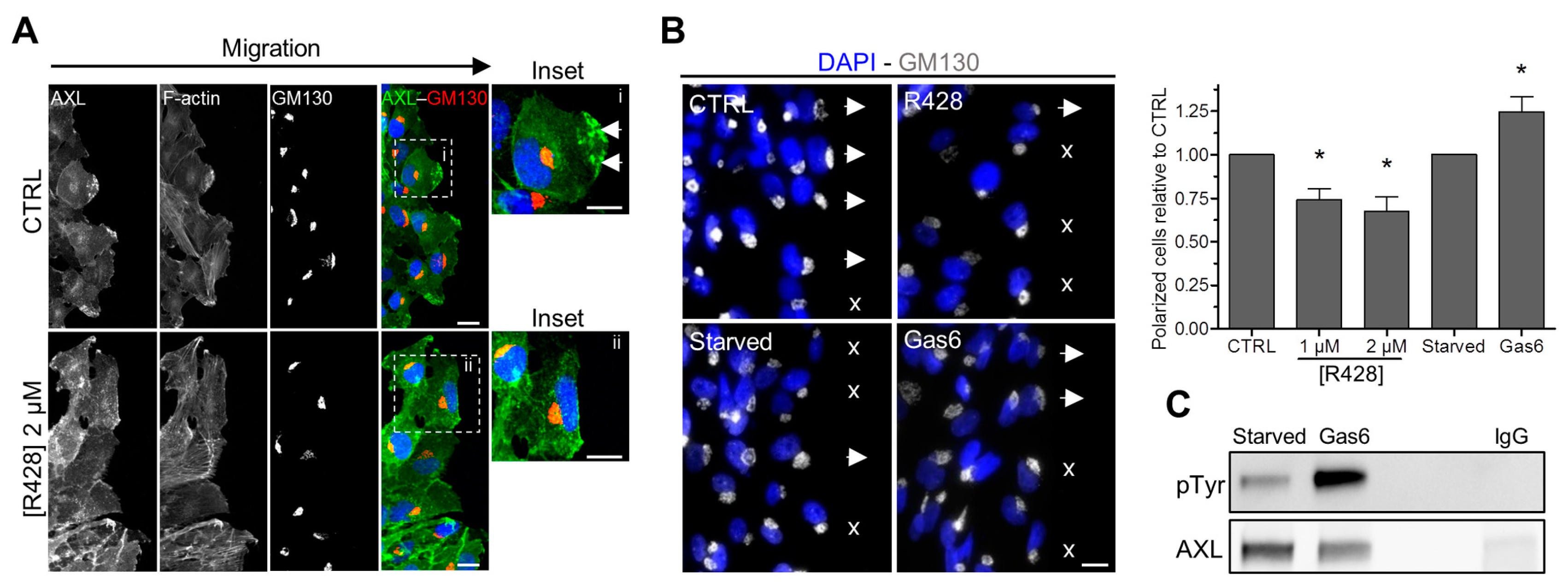
3.3. The Inhibition of AXL Disrupts Its Localization at the Leading Edge of Migrating Cells
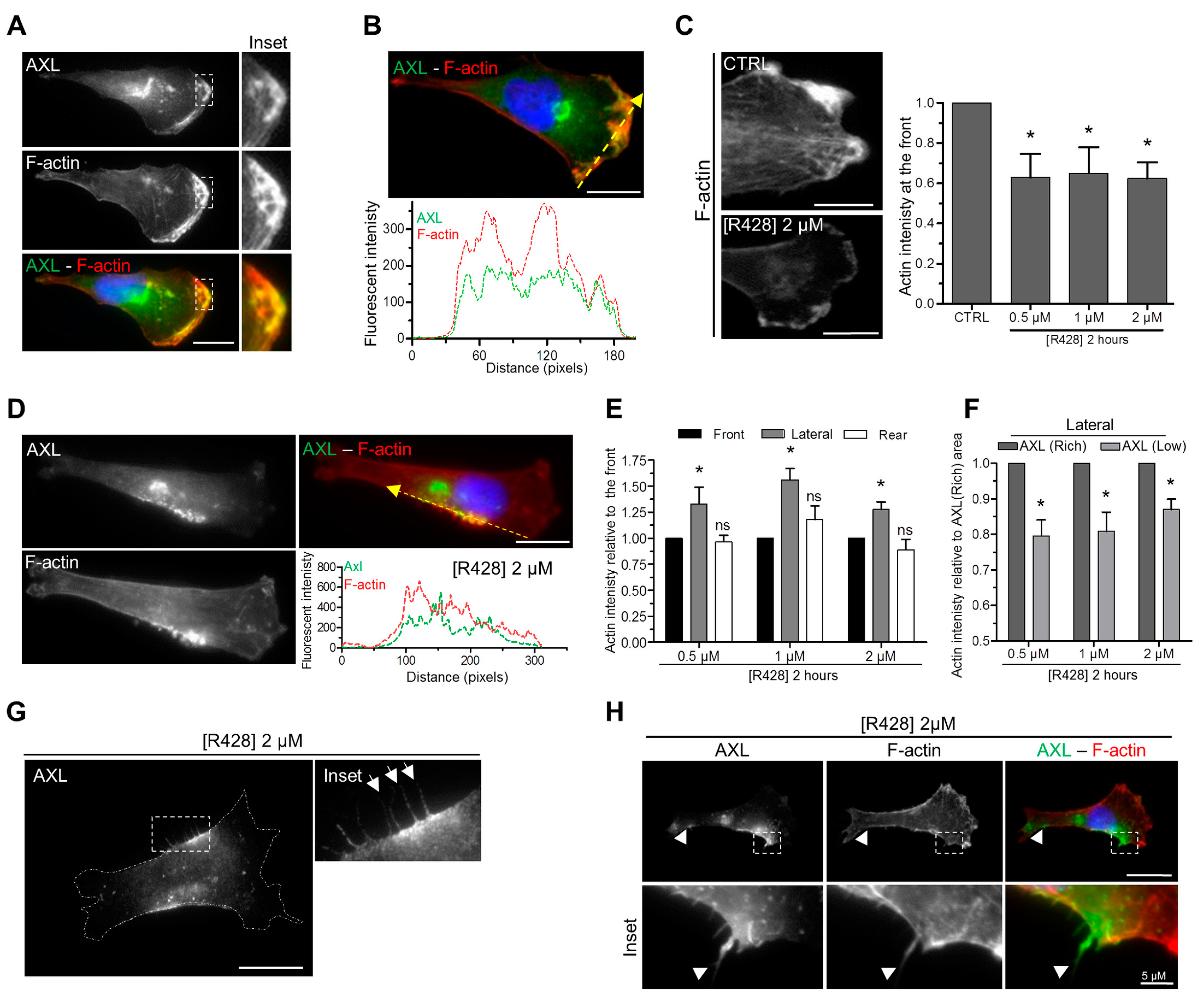
3.4. AXL Inhibition Disrupts Actin Polymerization at the Leading Edge of Migrating Cells
3.5. AXL Controls the Polarized Position of the Golgi Apparatus during Migration
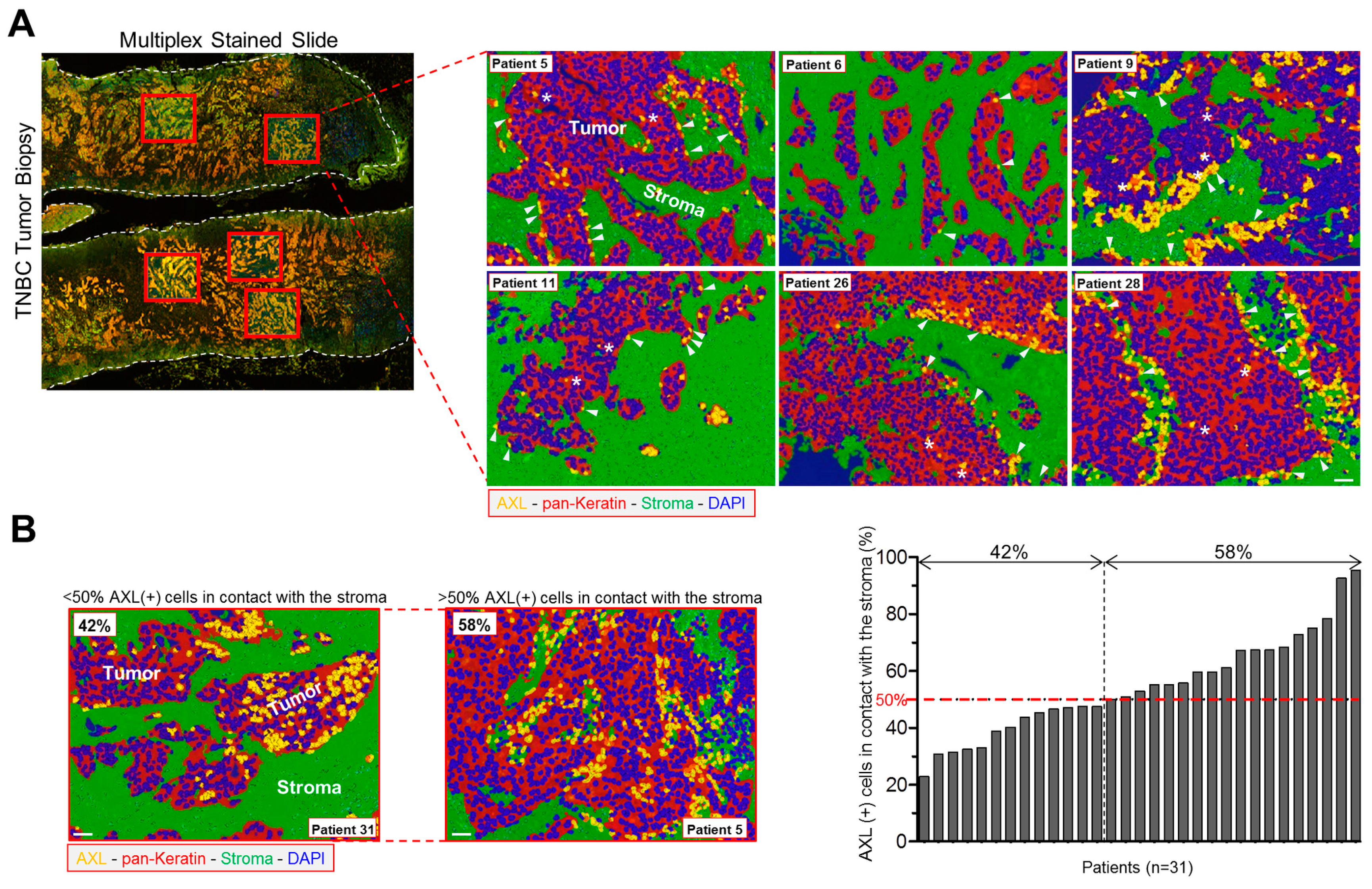
3.6. AXL-Expressing Tumor Cells Are Preferentially Located in Contact with the Stroma in Human TNBC
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AXL | AXL receptor tyrosine kinase |
| DMSO | Dimethyl Sulfoxide |
| DTT | 1:4-dithio-dl-threitol |
| EGFR | Epidermal Growth Factor Receptor 1 |
| EMT | Epithelial-to-Mesenchymal Transition |
| ER | Estrogen Receptor |
| F-actin | Filament of actin |
| FBS | Fetal Bovine Serum |
| GAS6 | Growth Arrest Specific protein 6 |
| IHC | Immunohistochemistry |
| INCa | French National Cancer Institute |
| IF | Immunofluorescence |
| IP | Immunoprecipitation |
| Lamp1 | Lysosomal Associated Membrane Protein 1 |
| MERTK | MER receptor Tyrosine Kinase |
| PR | Progesterone Receptor |
| P/S | Penicillin and streptomycin |
| Rac | Rac family of small GTPases |
| RTK | Receptor Tyrosine Kinases |
| TNBC | Triple Negative Breast Cancer |
| TYRO3 | TYRO3 receptor tyrosine kinase |
References
- Denkert, C.; Liedtke, C.; Tutt, A.; von Minckwitz, G. Molecular alterations in triple-negative breast cancer-the road to new treatment strategies. Lancet 2016, 389, 2430–2442. [Google Scholar] [CrossRef] [Green Version]
- Jitariu, A.A.; Cimpean, A.M.; Ribatti, D.; Raica, M. Triple negative breast cancer: The kiss of death. Oncotarget 2017, 8, 46652–46662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, B.D.; Jovanovic, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef] [PubMed]
- Karaayvaz, M.; Cristea, S.; Gillespie, S.M.; Patel, A.P.; Mylvaganam, R.; Luo, C.C.; Specht, M.C.; Bernstein, B.E.; Michor, F.; Ellisen, L.W. Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq. Nat. Commun. 2018, 9, 3588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.Y.; Jiang, Z.; Ben-David, Y.; Woodgett, J.R.; Zacksenhaus, E. Molecular stratification within triple-negative breast cancer subtypes. Sci. Rep. 2019, 9, 19107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.G.; Kim, S.J.; Kim, C.; Jeong, J. Molecular Classification of Triple-Negative Breast Cancer. J. Breast Cancer 2016, 19, 223–230. [Google Scholar] [CrossRef]
- Fedele, M.; Cerchia, L.; Chiappetta, G. The Epithelial-to-Mesenchymal Transition in Breast Cancer: Focus on Basal-Like Carcinomas. Cancers 2017, 9, 134. [Google Scholar] [CrossRef]
- Gu, G.; Dustin, D.; Fuqua, S.A. Targeted therapy for breast cancer and molecular mechanisms of resistance to treatment. Curr. Opin. Pharm. 2016, 31, 97–103. [Google Scholar] [CrossRef]
- Yang, F.; Wang, Y.; Li, Q.; Cao, L.; Sun, Z.; Jin, J.; Fang, H.; Zhu, A.; Li, Y.; Zhang, W.; et al. Intratumor heterogeneity predicts metastasis of triple-negative breast cancer. Carcinogenesis 2017, 38, 900–909. [Google Scholar] [CrossRef]
- Antony, J.; Thiery, J.P.; Huang, R.Y. Epithelial-to-mesenchymal transition: Lessons from development, insights into cancer and the potential of EMT-subtype based therapeutic intervention. Phys. Biol. 2019, 16, 041004. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Puisieux, A.; Pommier, R.M.; Morel, A.P.; Lavial, F. Cellular Pliancy and the Multistep Process of Tumorigenesis. Cancer Cell 2018, 33, 164–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016, 35, 645–654. [Google Scholar] [CrossRef]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Voon, D.C.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. The EMT spectrum and therapeutic opportunities. Mol. Oncol. 2017, 11, 878–891. [Google Scholar] [CrossRef] [Green Version]
- Santamaria, P.G.; Moreno-Bueno, G.; Cano, A. Contribution of Epithelial Plasticity to Therapy Resistance. J. Clin. Med. 2019, 8, 676. [Google Scholar] [CrossRef] [Green Version]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [Green Version]
- Marcucci, F.; Stassi, G.; De Maria, R. Epithelial-mesenchymal transition: A new target in anticancer drug discovery. Nat. Rev. Drug Discov. 2016, 15, 311–325. [Google Scholar] [CrossRef] [PubMed]
- O’Bryan, J.P.; Frye, R.A.; Cogswell, P.C.; Neubauer, A.; Kitch, B.; Prokop, C.; Espinosa, R., 3rd; Le Beau, M.M.; Earp, H.S.; Liu, E.T. axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol. Cell. Biol. 1991, 11, 5016–5031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef]
- Stitt, T.N.; Conn, G.; Gore, M.; Lai, C.; Bruno, J.; Radziejewski, C.; Mattsson, K.; Fisher, J.; Gies, D.R.; Jones, P.F.; et al. The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell 1995, 80, 661–670. [Google Scholar] [CrossRef] [Green Version]
- Varnum, B.C.; Young, C.; Elliott, G.; Garcia, A.; Bartley, T.D.; Fridell, Y.W.; Hunt, R.W.; Trail, G.; Clogston, C.; Toso, R.J.; et al. Axl receptor tyrosine kinase stimulated by the vitamin K-dependent protein encoded by growth-arrest-specific gene 6. Nature 1995, 373, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.S.; Miller, M.A.; Gertler, F.B.; Lauffenburger, D.A. The receptor AXL diversifies EGFR signaling and limits the response to EGFR-targeted inhibitors in triple-negative breast cancer cells. Sci. Signal. 2013, 6, ra66. [Google Scholar] [CrossRef] [Green Version]
- Rankin, E.B.; Giaccia, A.J. The Receptor Tyrosine Kinase AXL in Cancer Progression. Cancers 2016, 8, 103. [Google Scholar] [CrossRef]
- Schoumacher, M.; Burbridge, M. Key Roles of AXL and MER Receptor Tyrosine Kinases in Resistance to Multiple Anticancer Therapies. Curr. Oncol Rep. 2017, 19, 19. [Google Scholar] [CrossRef] [Green Version]
- Gjerdrum, C.; Tiron, C.; Hoiby, T.; Stefansson, I.; Haugen, H.; Sandal, T.; Collett, K.; Li, S.; McCormack, E.; Gjertsen, B.T.; et al. Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc. Natl. Acad. Sci. USA 2010, 107, 1124–1129. [Google Scholar] [CrossRef] [Green Version]
- Asiedu, M.K.; Beauchamp-Perez, F.D.; Ingle, J.N.; Behrens, M.D.; Radisky, D.C.; Knutson, K.L. AXL induces epithelial-to-mesenchymal transition and regulates the function of breast cancer stem cells. Oncogene 2014, 33, 1316–1324. [Google Scholar] [CrossRef] [Green Version]
- Paccez, J.D.; Vogelsang, M.; Parker, M.I.; Zerbini, L.F. The receptor tyrosine kinase Axl in cancer: Biological functions and therapeutic implications. Int. J. Cancer 2014, 134, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, X.; Koul, S.; Lee, C.Y.; Zhang, Z.; Halmos, B. AXL kinase as a novel target for cancer therapy. Oncotarget 2014, 5, 9546–9563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuoriluoto, K.; Haugen, H.; Kiviluoto, S.; Mpindi, J.P.; Nevo, J.; Gjerdrum, C.; Tiron, C.; Lorens, J.B.; Ivaska, J. Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene 2011, 30, 1436–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vouri, M.; Hafizi, S. TAM Receptor Tyrosine Kinases in Cancer Drug Resistance. Cancer Res. 2017, 77, 2775–2778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaltriti, M.; Elkabets, M.; Baselga, J. Molecular Pathways: AXL, a Membrane Receptor Mediator of Resistance to Therapy. Clin. Cancer Res. 2016, 22, 1313–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyette, M.A.; Duhamel, S.; Aubert, L.; Pelletier, A.; Savage, P.; Thibault, M.P.; Johnson, R.M.; Carmeliet, P.; Basik, M.; Gaboury, L.; et al. The Receptor Tyrosine Kinase AXL Is Required at Multiple Steps of the Metastatic Cascade during HER2-Positive Breast Cancer Progression. Cell Rep. 2018, 23, 1476–1490. [Google Scholar] [CrossRef]
- Antony, J.; Huang, R.Y. AXL-Driven EMT State as a Targetable Conduit in Cancer. Cancer Res. 2017, 77, 3725–3732. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.; Ye, X.; Pham, T.; Lin, E.; Chan, S.; McNamara, E.; Neve, R.M.; Belmont, L.; Koeppen, H.; Yauch, R.L.; et al. AXL inhibition sensitizes mesenchymal cancer cells to antimitotic drugs. Cancer Res. 2014, 74, 5878–5890. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Greger, J.; Shi, H.; Liu, Y.; Greshock, J.; Annan, R.; Halsey, W.; Sathe, G.M.; Martin, A.M.; Gilmer, T.M. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: Activation of AXL. Cancer Res. 2009, 69, 6871–6878. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Rankin, E.B.; Fuh, K.C.; Taylor, T.E.; Krieg, A.J.; Musser, M.; Yuan, J.; Wei, K.; Kuo, C.J.; Longacre, T.A.; Giaccia, A.J. AXL is an essential factor and therapeutic target for metastatic ovarian cancer. Cancer Res. 2010, 70, 7570–7579. [Google Scholar] [CrossRef] [Green Version]
- Creedon, H.; Gomez-Cuadrado, L.; Tarnauskaite, Z.; Balla, J.; Canel, M.; MacLeod, K.G.; Serrels, B.; Fraser, C.; Unciti-Broceta, A.; Tracey, N.; et al. Identification of novel pathways linking epithelial-to-mesenchymal transition with resistance to HER2-targeted therapy. Oncotarget 2016, 7, 11539–11552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, C.C.; Lay, J.D.; Huang, J.S.; Cheng, A.L.; Tang, J.L.; Lin, M.T.; Lai, G.M.; Chuang, S.E. Receptor tyrosine kinase AXL is induced by chemotherapy drugs and overexpression of AXL confers drug resistance in acute myeloid leukemia. Cancer Lett. 2008, 268, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, M.; Ise, N.; Omi, K.; Goishi, K.; Higashiyama, S. Cisplatin influences acquisition of resistance to molecular-targeted agents through epithelial-mesenchymal transition-like changes. Cancer Sci. 2013, 104, 904–911. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Chen, X.; He, J.; Liao, D.; Zu, X. Axl inhibitors as novel cancer therapeutic agents. Life Sci. 2018, 198, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Holland, S.J.; Pan, A.; Franci, C.; Hu, Y.; Chang, B.; Li, W.; Duan, M.; Torneros, A.; Yu, J.; Heckrodt, T.J.; et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010, 70, 1544–1554. [Google Scholar] [CrossRef] [Green Version]
- Vouri, M.; An, Q.; Birt, M.; Pilkington, G.J.; Hafizi, S. Small molecule inhibition of Axl receptor tyrosine kinase potently suppresses multiple malignant properties of glioma cells. Oncotarget 2015, 6, 16183–16197. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.X.; Knyazev, P.G.; Cheburkin, Y.V.; Sharma, K.; Knyazev, Y.P.; Orfi, L.; Szabadkai, I.; Daub, H.; Keri, G.; Ullrich, A. AXL is a potential target for therapeutic intervention in breast cancer progression. Cancer Res. 2008, 68, 1905–1915. [Google Scholar] [CrossRef] [Green Version]
- D’Alfonso, T.M.; Hannah, J.; Chen, Z.; Liu, Y.; Zhou, P.; Shin, S.J. Axl receptor tyrosine kinase expression in breast cancer. J. Clin. Pathol. 2014, 67, 690–696. [Google Scholar] [CrossRef]
- Vinet, M.; Suresh, S.; Maire, V.; Monchecourt, C.; Nemati, F.; Lesage, L.; Pierre, F.; Ye, M.; Lescure, A.; Brisson, A.; et al. Protein arginine methyltransferase 5: A novel therapeutic target for triple-negative breast cancers. Cancer Med. 2019, 8, 2414–2428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maubant, S.; Tahtouh, T.; Brisson, A.; Maire, V.; Némati, F.; Tesson, B.; Ye, M.; Rigaill, G.; Noizet, M.; Dumont, A.; et al. LRP5 regulates the expression of STK40, a new potential target in triple-negative breast cancers. Oncotarget 2018, in press. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldeyron, C.; Brisson, A.; Tesson, B.; Nemati, F.; Koundrioukoff, S.; Saliba, E.; De Koning, L.; Martel, E.; Ye, M.; Rigaill, G.; et al. TIPIN depletion leads to apoptosis in breast cancer cells. Mol. Oncol. 2015, 9, 1580–1598. [Google Scholar] [CrossRef] [PubMed]
- Maire, V.; Mahmood, F.; Rigaill, G.; Ye, M.; Brisson, A.; Nemati, F.; Gentien, D.; Tucker, G.C.; Roman-Roman, S.; Dubois, T. LRP8 is overexpressed in estrogen-negative breast cancers and a potential target for these tumors. Cancer Med. 2019, 8, 325–336. [Google Scholar] [CrossRef]
- Holland, S.J.; Powell, M.J.; Franci, C.; Chan, E.W.; Friera, A.M.; Atchison, R.E.; McLaughlin, J.; Swift, S.E.; Pali, E.S.; Yam, G.; et al. Multiple roles for the receptor tyrosine kinase axl in tumor formation. Cancer Res. 2005, 65, 9294–9303. [Google Scholar] [CrossRef] [Green Version]
- McDaniel, N.K.; Cummings, C.T.; Iida, M.; Hulse, J.; Pearson, H.E.; Vasileiadi, E.; Parker, R.E.; Orbuch, R.A.; Ondracek, O.J.; Welke, N.B.; et al. MERTK Mediates Intrinsic and Adaptive Resistance to AXL-targeting Agents. Mol. Cancer 2018, 17, 2297–2308. [Google Scholar] [CrossRef] [Green Version]
- Uribe, D.J.; Mandell, E.K.; Watson, A.; Martinez, J.D.; Leighton, J.A.; Ghosh, S.; Rothlin, C.V. The receptor tyrosine kinase AXL promotes migration and invasion in colorectal cancer. PLoS ONE 2017, 12, e0179979. [Google Scholar] [CrossRef] [Green Version]
- Maacha, S.; Hong, J.; von Lersner, A.; Zijlstra, A.; Belkhiri, A. AXL Mediates Esophageal Adenocarcinoma Cell Invasion through Regulation of Extracellular Acidification and Lysosome Trafficking. Neoplasia 2018, 20, 1008–1022. [Google Scholar] [CrossRef]
- Ridley, A.J. Rho GTPase signalling in cell migration. Curr. Opin. Cell Biol. 2015, 36, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Bear, J.E.; Haugh, J.M. Directed migration of mesenchymal cells: Where signaling and the cytoskeleton meet. Curr. Opin. Cell Biol. 2014, 30, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Pearson, G.W. Control of Invasion by Epithelial-to-Mesenchymal Transition Programs during Metastasis. J. Clin. Med. 2019, 8, 646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, H.; Pienta, K.J. Axl as a mediator of cellular growth and survival. Oncotarget 2014, 5, 8818–8852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gay, C.M.; Balaji, K.; Byers, L.A. Giving AXL the axe: Targeting AXL in human malignancy. Br. J. Cancer 2017, 116, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jia, L.; Liu, C.; Gong, Y.; Ren, D.; Wang, N.; Zhang, X.; Zhao, Y. Axl as a downstream effector of TGF-beta1 via PI3K/Akt-PAK1 signaling pathway promotes tumor invasion and chemoresistance in breast carcinoma. Tumour Biol. 2015, 36, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Bottai, G.; Raschioni, C.; Szekely, B.; Di Tommaso, L.; Szasz, A.M.; Losurdo, A.; Gyorffy, B.; Acs, B.; Torrisi, R.; Karachaliou, N.; et al. AXL-associated tumor inflammation as a poor prognostic signature in chemotherapy-treated triple-negative breast cancer patients. Npj Breast Cancer 2016, 2, 16033. [Google Scholar] [CrossRef]
- Wang, C.; Jin, H.; Wang, N.; Fan, S.; Wang, Y.; Zhang, Y.; Wei, L.; Tao, X.; Gu, D.; Zhao, F.; et al. Gas6/Axl Axis Contributes to Chemoresistance and Metastasis in Breast Cancer through Akt/GSK-3beta/beta-catenin Signaling. Theranostics 2016, 6, 1205–1219. [Google Scholar] [CrossRef]
- Leconet, W.; Chentouf, M.; du Manoir, S.; Chevalier, C.; Sirvent, A.; Ait-Arsa, I.; Busson, M.; Jarlier, M.; Radosevic-Robin, N.; Theillet, C.; et al. Therapeutic Activity of Anti-AXL Antibody against Triple-Negative Breast Cancer Patient-Derived Xenografts and Metastasis. Clin. Cancer Res. 2017, 23, 2806–2816. [Google Scholar] [CrossRef] [Green Version]
- Del Pozo Martin, Y.; Park, D.; Ramachandran, A.; Ombrato, L.; Calvo, F.; Chakravarty, P.; Spencer-Dene, B.; Derzsi, S.; Hill, C.S.; Sahai, E.; et al. Mesenchymal Cancer Cell-Stroma Crosstalk Promotes Niche Activation, Epithelial Reversion, and Metastatic Colonization. Cell Rep. 2015, 13, 2456–2469. [Google Scholar] [CrossRef] [Green Version]
- Ye, Q.H.; Zhu, W.W.; Zhang, J.B.; Qin, Y.; Lu, M.; Lin, G.L.; Guo, L.; Zhang, B.; Lin, Z.H.; Roessler, S.; et al. GOLM1 Modulates EGFR/RTK Cell-Surface Recycling to Drive Hepatocellular Carcinoma Metastasis. Cancer Cell 2016, 30, 444–458. [Google Scholar] [CrossRef] [Green Version]
- Gondi, C.S.; Rao, J.S. Cathepsin B as a cancer target. Expert Opin. Ther. Targets 2013, 17, 281–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.J.; Tang, Y.J.; Tang, Y.L.; Liang, X.H. What makes cells move: Requirements and obstacles for leader cells in collective invasion. Exp. Cell Res. 2019, 382, 111481. [Google Scholar] [CrossRef] [PubMed]
- Pallesi-Pocachard, E.; Bazellieres, E.; Viallat-Lieutaud, A.; Delgrossi, M.H.; Barthelemy-Requin, M.; Le Bivic, A.; Massey-Harroche, D. Hook2, a microtubule-binding protein, interacts with Par6alpha and controls centrosome orientation during polarized cell migration. Sci. Rep. 2016, 6, 33259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revach, O.Y.; Sandler, O.; Samuels, Y.; Geiger, B. Cross-Talk between Receptor Tyrosine Kinases AXL and ERBB3 Regulates Invadopodia Formation in Melanoma Cells. Cancer Res. 2019, 79, 2634–2648. [Google Scholar] [CrossRef] [Green Version]
- Jokela, T.A.; Engelsen, A.S.T.; Rybicka, A.; Pelissier Vatter, F.A.; Garbe, J.C.; Miyano, M.; Tiron, C.; Ferariu, D.; Akslen, L.A.; Stampfer, M.R.; et al. Microenvironment-Induced Non-sporadic Expression of the AXL and cKIT Receptors Are Related to Epithelial Plasticity and Drug Resistance. Front. Cell Dev. Biol. 2018, 6, 41. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.M.; Carron, E.C.; Mills, K.L.; Dow, A.M.; Gray, Z.; Fecca, C.R.; Lakey, M.A.; Carmeliet, P.; Kittrell, F.; Medina, D.; et al. Stromal Gas6 promotes the progression of premalignant mammary cells. Oncogene 2019, 38, 2437–2450. [Google Scholar] [CrossRef]
- Kanzaki, R.; Naito, H.; Kise, K.; Takara, K.; Eino, D.; Minami, M.; Shintani, Y.; Funaki, S.; Kawamura, T.; Kimura, T.; et al. Gas6 derived from cancer-associated fibroblasts promotes migration of Axl-expressing lung cancer cells during chemotherapy. Sci. Rep. 2017, 7, 10613. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zajac, O.; Leclere, R.; Nicolas, A.; Meseure, D.; Marchiò, C.; Vincent-Salomon, A.; Roman-Roman, S.; Schoumacher, M.; Dubois, T. AXL Controls Directed Migration of Mesenchymal Triple-Negative Breast Cancer Cells. Cells 2020, 9, 247. https://doi.org/10.3390/cells9010247
Zajac O, Leclere R, Nicolas A, Meseure D, Marchiò C, Vincent-Salomon A, Roman-Roman S, Schoumacher M, Dubois T. AXL Controls Directed Migration of Mesenchymal Triple-Negative Breast Cancer Cells. Cells. 2020; 9(1):247. https://doi.org/10.3390/cells9010247
Chicago/Turabian StyleZajac, Olivier, Renaud Leclere, André Nicolas, Didier Meseure, Caterina Marchiò, Anne Vincent-Salomon, Sergio Roman-Roman, Marie Schoumacher, and Thierry Dubois. 2020. "AXL Controls Directed Migration of Mesenchymal Triple-Negative Breast Cancer Cells" Cells 9, no. 1: 247. https://doi.org/10.3390/cells9010247
APA StyleZajac, O., Leclere, R., Nicolas, A., Meseure, D., Marchiò, C., Vincent-Salomon, A., Roman-Roman, S., Schoumacher, M., & Dubois, T. (2020). AXL Controls Directed Migration of Mesenchymal Triple-Negative Breast Cancer Cells. Cells, 9(1), 247. https://doi.org/10.3390/cells9010247