Sensor Sensibility—HIV-1 and the Innate Immune Response

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Innate Immune Sensors of HIV-1 Infection
2.1. HIV-1 RNA Sensors
2.2. Innate Immune Sensors of HIV-1 Reverse Transcription Products
3. Regulation of cGAS-STING Signaling in Response to HIV-1 Infection
3.1. Co-Factors of HIV-1-Mediated cGAS-STING Pathway Activation
3.2. Negative Regulators of HIV-1-Mediated cGAS-STING Pathway Activation
3.3. Innate Control of Cytoplasmic DNA Accumulation
3.4. HIV-1 Capsid Is a Viral Determinant of Innate Sensing of HIV-1
4. The Cell Type-Dependencies of Innate Immune Response to HIV-1 Infection
4.1. Innate Immune Response to HIV-1 Infection in DCs and Macrophages
4.2. Innate Immune Response to HIV-1 Infection in CD4+ T Cells
5. Strategies Adopted by HIV-1 to Avoid Sensing and IFN Response
5.1. Counteraction of HIV-1 Restriction Factors
5.2. Disruption of Signaling Pathway Transduction
5.3. HIV-1 PAMPs Masking
6. Summary
- Understanding the role of the innate immune response in the control of viral transmission, systemic infection, and potentially latency.
- Defining the structure, molecular features, and accessibility of the HIV-1 DNA PAMP.
- Dissecting the distinct roles of PQBP1, NONO, and other cellular co-factors in cGAS signaling activation in response to HIV-1 infection.
- Gaining a thorough mechanistic understanding of innate sensing of HIV-1 infection in CD4+ T cells.
- Mechanisms of HIV-1-mediated evasion of innate sensing.
- Harnessing knowledge of the innate response to develop novel vaccines, vaccine adjuvants, as well as prophylactic and therapeutic approaches to prevention of HIV infection.
Funding
Acknowledgments
Conflicts of Interest
References
- Samuel, C.E. Antiviral actions of interferons. Clin. Microbiol. Rev. 2001, 14, 778–809. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Sastre, A. Ten Strategies of Interferon Evasion by Viruses. Cell Host Microbe 2017, 22, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [Green Version]
- Fensterl, V.; Sen, G.C. Interferons and viral infections. Biofactors 2009, 35, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, L. Complexity of danger: The diverse nature of damage-associated molecular patterns. J. Biol. Chem. 2014, 289, 35237–35245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, H.H.; Schneider, W.M.; Rice, C.M. Interferons and viruses: An evolutionary arms race of molecular interactions. Trends Immunol. 2015, 36, 124–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colomer-Lluch, M.; Ruiz, A.; Moris, A.; Prado, J.G. Restriction Factors: From Intrinsic Viral Restriction to Shaping Cellular Immunity Against HIV-1. Front. Immunol. 2018, 9, 2876. [Google Scholar] [CrossRef] [Green Version]
- Martin-Gayo, E.; Buzon, M.J.; Ouyang, Z.; Hickman, T.; Cronin, J.; Pimenova, D.; Walker, B.D.; Lichterfeld, M.; Yu, X.G. Potent Cell-Intrinsic Immune Responses in Dendritic Cells Facilitate HIV-1-Specific T Cell Immunity in HIV-1 Elite Controllers. PLoS Pathog. 2015, 11, e1004930. [Google Scholar] [CrossRef]
- Altfeld, M.; Gale, M., Jr. Innate immunity against HIV-1 infection. Nat. Immunol. 2015, 16, 554–562. [Google Scholar] [CrossRef]
- Bergantz, L.; Subra, F.; Deprez, E.; Delelis, O.; Richetta, C. Interplay between Intrinsic and Innate Immunity during HIV Infection. Cells 2019, 8, 922. [Google Scholar] [CrossRef] [Green Version]
- Parrish, N.F.; Gao, F.; Li, H.; Giorgi, E.E.; Barbian, H.J.; Parrish, E.H.; Zajic, L.; Iyer, S.S.; Decker, J.M.; Kumar, A.; et al. Phenotypic properties of transmitted founder HIV-1. Proc. Natl. Acad. Sci. USA 2013, 110, 6626–6633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenton-May, A.E.; Dibben, O.; Emmerich, T.; Ding, H.; Pfafferott, K.; Aasa-Chapman, M.M.; Pellegrino, P.; Williams, I.; Cohen, M.S.; Gao, F.; et al. Relative resistance of HIV-1 founder viruses to control by interferon-alpha. Retrovirology 2013, 10, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, S.S.; Bibollet-Ruche, F.; Sherrill-Mix, S.; Learn, G.H.; Plenderleith, L.; Smith, A.G.; Barbian, H.J.; Russell, R.M.; Gondim, M.V.P.; Bahari, C.Y.; et al. Resistance to type 1 interferons is a major determinant of HIV-1 transmission fitness. Proc. Natl. Acad. Sci. USA 2017, 114, E590–E599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahaye, X.; Satoh, T.; Gentili, M.; Cerboni, S.; Conrad, C.; Hurbain, I.; El Marjou, A.; Lacabaratz, C.; Lelievre, J.D.; Manel, N. The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity 2013, 39, 1132–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoh, S.M.; Schneider, M.; Seifried, J.; Soonthornvacharin, S.; Akleh, R.E.; Olivieri, K.C.; De Jesus, P.D.; Ruan, C.; de Castro, E.; Ruiz, P.A.; et al. PQBP1 Is a Proximal Sensor of the cGAS-Dependent Innate Response to HIV-1. Cell 2015, 161, 1293–1305. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Wu, J.; Wu, Y.T.; Du, F.; Aroh, C.; Yan, N.; Sun, L.; Chen, Z.J. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 2013, 341, 903–906. [Google Scholar] [CrossRef] [Green Version]
- Manel, N.; Hogstad, B.; Wang, Y.; Levy, D.E.; Unutmaz, D.; Littman, D.R. A cryptic sensor for HIV-1 activates antiviral innate immunity in dendritic cells. Nature 2010, 467, 214–217. [Google Scholar] [CrossRef]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef] [Green Version]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Segeral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef]
- Rasaiyaah, J.; Tan, C.P.; Fletcher, A.J.; Price, A.J.; Blondeau, C.; Hilditch, L.; Jacques, D.A.; Selwood, D.L.; James, L.C.; Noursadeghi, M.; et al. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature 2013, 503, 402–405. [Google Scholar] [CrossRef]
- Lahaye, X.; Gentili, M.; Silvin, A.; Conrad, C.; Picard, L.; Jouve, M.; Zueva, E.; Maurin, M.; Nadalin, F.; Knott, G.J.; et al. NONO Detects the Nuclear HIV Capsid to Promote cGAS-Mediated Innate Immune Activation. Cell 2018, 175, 488–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakobsen, M.R.; Bak, R.O.; Andersen, A.; Berg, R.K.; Jensen, S.B.; Tengchuan, J.; Laustsen, A.; Hansen, K.; Ostergaard, L.; Fitzgerald, K.A.; et al. IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc. Natl. Acad. Sci. USA 2013, 110, E4571–E4580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solis, M.; Nakhaei, P.; Jalalirad, M.; Lacoste, J.; Douville, R.; Arguello, M.; Zhao, T.; Laughrea, M.; Wainberg, M.A.; Hiscott, J. RIG-I-mediated antiviral signaling is inhibited in HIV-1 infection by a protease-mediated sequestration of RIG-I. J. Virol. 2011, 85, 1224–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepelley, A.; Louis, S.; Sourisseau, M.; Law, H.K.; Pothlichet, J.; Schilte, C.; Chaperot, L.; Plumas, J.; Randall, R.E.; Si-Tahar, M.; et al. Innate sensing of HIV-infected cells. PLoS Pathog. 2011, 7, e1001284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landau, N.R. The innate immune response to HIV-1: To sense or not to sense. DNA Cell Biol. 2014, 33, 271–274. [Google Scholar] [CrossRef] [Green Version]
- Langer, S.; Hammer, C.; Hopfensperger, K.; Klein, L.; Hotter, D.; De Jesus, P.D.; Herbert, K.M.; Pache, L.; Smith, N.; van der Merwe, J.A.; et al. HIV-1 Vpu is a potent transcriptional suppressor of NF-kappaB-elicited antiviral immune responses. Elife 2019, 8. [Google Scholar] [CrossRef]
- Ringeard, M.; Marchand, V.; Decroly, E.; Motorin, Y.; Bennasser, Y. FTSJ3 is an RNA 2′-O-methyltransferase recruited by HIV to avoid innate immune sensing. Nature 2019, 565, 500–504. [Google Scholar] [CrossRef]
- Kumar, S.; Morrison, J.H.; Dingli, D.; Poeschla, E. HIV-1 Activation of Innate Immunity Depends Strongly on the Intracellular Level of TREX1 and Sensing of Incomplete Reverse Transcription Products. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Gringhuis, S.I.; Hertoghs, N.; Kaptein, T.M.; Zijlstra-Willems, E.M.; Sarrami-Forooshani, R.; Sprokholt, J.K.; van Teijlingen, N.H.; Kootstra, N.A.; Booiman, T.; van Dort, K.A.; et al. HIV-1 blocks the signaling adaptor MAVS to evade antiviral host defense after sensing of abortive HIV-1 RNA by the host helicase DDX3. Nat. Immunol. 2017, 18, 225–235. [Google Scholar] [CrossRef]
- Tomalka, J.; Ghneim, K.; Bhattacharyya, S.; Aid, M.; Barouch, D.H.; Sekaly, R.P.; Ribeiro, S.P. The sooner the better: Innate immunity as a path toward the HIV cure. Curr. Opin. Virol. 2016, 19, 85–91. [Google Scholar] [CrossRef] [Green Version]
- Muenchhoff, M.; Prendergast, A.J.; Goulder, P.J. Immunity to HIV in Early Life. Front. Immunol. 2014, 5, 391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gringhuis, S.I.; van der Vlist, M.; van den Berg, L.M.; den Dunnen, J.; Litjens, M.; Geijtenbeek, T.B. HIV-1 exploits innate signaling by TLR8 and DC-SIGN for productive infection of dendritic cells. Nat. Immunol. 2010, 11, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Meier, A.; Alter, G.; Frahm, N.; Sidhu, H.; Li, B.; Bagchi, A.; Teigen, N.; Streeck, H.; Stellbrink, H.J.; Hellman, J.; et al. MyD88-dependent immune activation mediated by human immunodeficiency virus type 1-encoded Toll-like receptor ligands. J. Virol. 2007, 81, 8180–8191. [Google Scholar] [CrossRef] [Green Version]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.Q.; Huang, Y.L.; Huang, J.; Zheng, J.L.; Qian, G.X. RIG-I detects HIV-1 infection and mediates type I interferon response in human macrophages from patients with HIV-1-associated neurocognitive disorders. Genet. Mol. Res. 2015, 14, 13799–13811. [Google Scholar] [CrossRef]
- McCauley, S.M.; Kim, K.; Nowosielska, A.; Dauphin, A.; Yurkovetskiy, L.; Diehl, W.E.; Luban, J. Intron-containing RNA from the HIV-1 provirus activates type I interferon and inflammatory cytokines. Nat. Commun. 2018, 9, 5305. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, H.; Miller, C.M.; Ettinger, C.R.; Belkina, A.C.; Snyder-Cappione, J.E.; Gummuluru, S. HIV-1 intron-containing RNA expression induces innate immune activation and T cell dysfunction. Nat. Commun. 2018, 8, 3450. [Google Scholar] [CrossRef]
- Towers, G.J.; Noursadeghi, M. Interactions between HIV-1 and the cell-autonomous innate immune system. Cell Host Microbe 2014, 16, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Doitsh, G.; Galloway, N.L.; Geng, X.; Yang, Z.; Monroe, K.M.; Zepeda, O.; Hunt, P.W.; Hatano, H.; Sowinski, S.; Munoz-Arias, I.; et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 2014, 505, 509–514. [Google Scholar] [CrossRef] [Green Version]
- Bode, C.; Fox, M.; Tewary, P.; Steinhagen, A.; Ellerkmann, R.K.; Klinman, D.; Baumgarten, G.; Hornung, V.; Steinhagen, F. Human plasmacytoid dentritic cells elicit a Type I Interferon response by sensing DNA via the cGAS-STING signaling pathway. Eur. J. Immunol. 2016, 46, 1615–1621. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregg, R.W.; Sarkar, S.N.; Shoemaker, J.E. Mathematical modeling of the cGAS pathway reveals robustness of DNA sensing to TREX1 feedback. J. Biol. 2019, 462, 148–157. [Google Scholar] [CrossRef]
- Beignon, A.S.; McKenna, K.; Skoberne, M.; Manches, O.; DaSilva, I.; Kavanagh, D.G.; Larsson, M.; Gorelick, R.J.; Lifson, J.D.; Bhardwaj, N. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J. Clin. Investig. 2005, 115, 3265–3275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decalf, J.; Desdouits, M.; Rodrigues, V.; Gobert, F.X.; Gentili, M.; Marques-Ladeira, S.; Chamontin, C.; Mougel, M.; Cunha de Alencar, B.; Benaroch, P. Sensing of HIV-1 Entry Triggers a Type I Interferon Response in Human Primary Macrophages. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kell, A.M.; Gale, M., Jr. RIG-I in RNA virus recognition. Virology 2015, 479–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto-Rifo, R.; Rubilar, P.S.; Limousin, T.; de Breyne, S.; Decimo, D.; Ohlmann, T. DEAD-box protein DDX3 associates with eIF4F to promote translation of selected mRNAs. EMBO J. 2012, 31, 3745–3756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naji, S.; Ambrus, G.; Cimermancic, P.; Reyes, J.R.; Johnson, J.R.; Filbrandt, R.; Huber, M.D.; Vesely, P.; Krogan, N.J.; Yates, J.R.; et al. Host cell interactome of HIV-1 Rev includes RNA helicases involved in multiple facets of virus production. Mol. Cell Proteomics 2012. [Google Scholar] [CrossRef] [Green Version]
- Yedavalli, V.S.; Neuveut, C.; Chi, Y.H.; Kleiman, L.; Jeang, K.T. Requirement of DDX3 DEAD box RNA helicase for HIV-1 Rev-RRE export function. Cell 2004, 119, 381–392. [Google Scholar] [CrossRef] [Green Version]
- Geijtenbeek, T.B.; van Kooyk, Y. DC-SIGN: A novel HIV receptor on DCs that mediates HIV-1 transmission. Curr. Top. Microbiol. Immunol. 2003, 276, 31–54. [Google Scholar] [CrossRef]
- Monroe, K.M.; Yang, Z.; Johnson, J.R.; Geng, X.; Doitsh, G.; Krogan, N.J.; Greene, W.C. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science 2014, 343, 428–432. [Google Scholar] [CrossRef] [Green Version]
- Stavrou, S.; Aguilera, A.N.; Blouch, K.; Ross, S.R. DDX41 Recognizes RNA/DNA Retroviral Reverse Transcripts and Is Critical for In Vivo Control of Murine Leukemia Virus Infection. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotter, D.; Bosso, M.; Jonsson, K.L.; Krapp, C.; Sturzel, C.M.; Das, A.; Littwitz-Salomon, E.; Berkhout, B.; Russ, A.; Wittmann, S.; et al. IFI16 Targets the Transcription Factor Sp1 to Suppress HIV-1 Transcription and Latency Reactivation. Cell Host Microbe 2019, 25, 858–872.e13. [Google Scholar] [CrossRef]
- Herzner, A.M.; Hagmann, C.A.; Goldeck, M.; Wolter, S.; Kubler, K.; Wittmann, S.; Gramberg, T.; Andreeva, L.; Hopfner, K.P.; Mertens, C.; et al. Sequence-specific activation of the DNA sensor cGAS by Y-form DNA structures as found in primary HIV-1 cDNA. Nat. Immunol. 2015, 16, 1025–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.S.; Lucas, S.Y.; Amon, L.M.; Skelton, S.; Nazitto, R.; Carbonetti, S.; Sather, D.N.; Littman, D.R.; Aderem, A. Reshaping of the Dendritic Cell Chromatin Landscape and Interferon Pathways during HIV Infection. Cell Host Microbe 2018, 23, 366–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Zhang, H.; Wu, X.; Ma, D.; Wu, J.; Wang, L.; Jiang, Y.; Fei, Y.; Zhu, C.; Tan, R.; et al. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 2018, 563, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Piekna-Przybylska, D.; Sharma, G.; Maggirwar, S.B.; Bambara, R.A. Deficiency in DNA damage response, a new characteristic of cells infected with latent HIV-1. Cell Cycle 2017, 16, 968–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jauregui, P.; Landau, N.R. DNA damage induces a SAMHD1-mediated block to the infection of macrophages by HIV-1. Sci. Rep. 2018, 8, 4153. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Huang, L.; Hong, Z.; Lv, Z.; Mao, Z.; Tang, Y.; Kong, X.; Li, S.; Cui, Y.; Liu, H.; et al. The E3 ubiquitin ligase RNF185 facilitates the cGAS-mediated innate immune response. PLoS Pathog. 2017, 13, e1006264. [Google Scholar] [CrossRef]
- Xia, P.; Ye, B.; Wang, S.; Zhu, X.; Du, Y.; Xiong, Z.; Tian, Y.; Fan, Z. Glutamylation of the DNA sensor cGAS regulates its binding and synthase activity in antiviral immunity. Nat. Immunol. 2016, 17, 369–378. [Google Scholar] [CrossRef]
- Hu, M.M.; Yang, Q.; Xie, X.Q.; Liao, C.Y.; Lin, H.; Liu, T.T.; Yin, L.; Shu, H.B. Sumoylation Promotes the Stability of the DNA Sensor cGAS and the Adaptor STING to Regulate the Kinetics of Response to DNA Virus. Immunity 2016, 45, 555–569. [Google Scholar] [CrossRef] [Green Version]
- Seo, G.J.; Yang, A.; Tan, B.; Kim, S.; Liang, Q.; Choi, Y.; Yuan, W.; Feng, P.; Park, H.S.; Jung, J.U. Akt Kinase-Mediated Checkpoint of cGAS DNA Sensing Pathway. Cell Rep. 2015, 13, 440–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS–STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Shao, L.; Sampath, P.; Zhao, B.; Patel, N.V.; Zhu, J.; Behl, B.; Parise, R.A.; Beumer, J.H.; O’Sullivan, R.J.; et al. Oligoadenylate-Synthetase-Family Protein OASL Inhibits Activity of the DNA Sensor cGAS during DNA Virus Infection to Limit Interferon Production. Immunity 2019, 50, 51–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.S.; Cai, H.; Xue, W.; Wang, M.; Xia, T.; Li, W.J.; Xing, J.Q.; Zhao, M.; Huang, Y.J.; Chen, S.; et al. G3BP1 promotes DNA binding and activation of cGAS. Nat. Immunol. 2019, 20, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Okazawa, H. PQBP1, an intrinsically disordered/denatured protein at the crossroad of intellectual disability and neurodegenerative diseases. Neurochem. Int. 2018, 119, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Xiong, M.; Wang, S.; Wang, Y.-Y.; Ran, Y. The Regulation of cGAS. Virol. Sin. 2018, 33, 117–124. [Google Scholar] [CrossRef] [Green Version]
- König, R.; Zhou, Y.; Elleder, D.; Diamond, T.L.; Bonamy, G.M.; Irelan, J.T.; Chiang, C.Y.; Tu, B.P.; De Jesus, P.D.; Lilley, C.E.; et al. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 2008, 135, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; König, R.; Deng, M.; Riess, M.; Mo, J.; Zhang, L.; Petrucelli, A.; Yoh, S.M.; Barefoot, B.; Samo, M.; et al. NLRX1 Sequesters STING to Negatively Regulate the Interferon Response, Thereby Facilitating the Replication of HIV-1 and DNA Viruses. Cell Host Microbe 2016, 19, 515–528. [Google Scholar] [CrossRef] [Green Version]
- Barouch, D.H.; Ghneim, K.; Bosche, W.J.; Li, Y.; Berkemeier, B.; Hull, M.; Bhattacharyya, S.; Cameron, M.; Liu, J.; Smith, K.; et al. Rapid Inflammasome Activation following Mucosal SIV Infection of Rhesus Monkeys. Cell 2016, 165, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Deng, M.; Petrucelli, A.S.; Zhu, C.; Mo, J.; Zhang, L.; Tam, J.W.; Ariel, P.; Zhao, B.; Zhang, S.; et al. Viral DNA Binding to NLRC3, an Inhibitory Nucleic Acid Sensor, Unleashes STING, a Cyclic Dinucleotide Receptor that Activates Type I Interferon. Immunity 2019, 50, 591–599.e6. [Google Scholar] [CrossRef] [Green Version]
- Du, M.; Chen, Z.J. DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 2018, 361, 704–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ablasser, A.; Hemmerling, I.; Schmid-Burgk, J.L.; Behrendt, R.; Roers, A.; Hornung, V. TREX1 deficiency triggers cell-autonomous immunity in a cGAS-dependent manner. J. Immunol. 2014, 192, 5993–5997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Bonifati, S.; Qin, Z.; St Gelais, C.; Wu, L. SAMHD1 Suppression of Antiviral Immune Responses. Trends Microbiol. 2019, 27, 254–267. [Google Scholar] [CrossRef]
- Coquel, F.; Silva, M.J.; Techer, H.; Zadorozhny, K.; Sharma, S.; Nieminuszczy, J.; Mettling, C.; Dardillac, E.; Barthe, A.; Schmitz, A.L.; et al. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 2018, 557, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Bonifati, S.; Qin, Z.; St Gelais, C.; Kodigepalli, K.M.; Barrett, B.S.; Kim, S.H.; Antonucci, J.M.; Ladner, K.J.; Buzovetsky, O.; et al. SAMHD1 suppresses innate immune responses to viral infections and inflammatory stimuli by inhibiting the NF-kappaB and interferon pathways. Proc. Natl. Acad. Sci. USA 2018, 115, E3798–E3807. [Google Scholar] [CrossRef] [Green Version]
- Majer, C.; Schussler, J.M.; Konig, R. Intertwined: SAMHD1 cellular functions, restriction, and viral evasion strategies. Med. Microbiol. Immunol. 2019, 208, 513–529. [Google Scholar] [CrossRef]
- Maelfait, J.; Bridgeman, A.; Benlahrech, A.; Cursi, C.; Rehwinkel, J. Restriction by SAMHD1 Limits cGAS/STING-Dependent Innate and Adaptive Immune Responses to HIV-1. Cell Rep. 2016, 16, 1492–1501. [Google Scholar] [CrossRef] [Green Version]
- Ayinde, D.; Bruel, T.; Cardinaud, S.; Porrot, F.; Prado, J.G.; Moris, A.; Schwartz, O. SAMHD1 Limits HIV-1 Antigen Presentation by Monocyte-Derived Dendritic Cells. J. Virol. 2015, 89, 6994–7006. [Google Scholar] [CrossRef] [Green Version]
- Sultana, T.; Mamede, J.I.; Saito, A.; Ode, H.; Nohata, K.; Cohen, R.; Nakayama, E.E.; Iwatani, Y.; Yamashita, M.; Hope, T.J.; et al. Multiple Pathways to Avoid Beta Interferon Sensitivity of HIV-1 by Mutations in Capsid. J. Virol. 2019, 93, e00986-19. [Google Scholar] [CrossRef] [Green Version]
- Pertel, T.; Hausmann, S.; Morger, D.; Zuger, S.; Guerra, J.; Lascano, J.; Reinhard, C.; Santoni, F.A.; Uchil, P.D.; Chatel, L.; et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 2011, 472, 361–365. [Google Scholar] [CrossRef] [Green Version]
- Bejarano, D.A.; Peng, K.; Laketa, V.; Börner, K.; Jost, K.L.; Lucic, B.; Glass, B.; Lusic, M.; Müller, B.; Kräusslich, H.-G. HIV-1 nuclear import in macrophages is regulated by CPSF6-capsid interactions at the nuclear pore complex. ELife 2019, 8, e41800. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Pan, Q.; Ding, S.; Qian, J.; Xu, F.; Zhou, J.; Cen, S.; Guo, F.; Liang, C. The interferon-inducible MxB protein inhibits HIV-1 infection. Cell Host Microbe 2013, 14, 398–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilliams, M.; Ginhoux, F.; Jakubzick, C.; Naik, S.H.; Onai, N.; Schraml, B.U.; Segura, E.; Tussiwand, R.; Yona, S. Dendritic cells, monocytes and macrophages: A unified nomenclature based on ontogeny. Nat. Rev. Immunol. 2014, 14, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Noyce, R.S.; Taylor, K.; Ciechonska, M.; Collins, S.E.; Duncan, R.; Mossman, K.L. Membrane perturbation elicits an IRF3-dependent, interferon-independent antiviral response. J. Virol. 2011, 85, 10926–10931. [Google Scholar] [CrossRef] [Green Version]
- Swiecki, M.; Colonna, M. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 2015, 15, 471–485. [Google Scholar] [CrossRef]
- Vermeire, J.; Roesch, F.; Sauter, D.; Rua, R.; Hotter, D.; Van Nuffel, A.; Vanderstraeten, H.; Naessens, E.; Iannucci, V.; Landi, A.; et al. HIV Triggers a cGAS-Dependent, Vpu- and Vpr-Regulated Type I Interferon Response in CD4(+) T Cells. Cell Rep. 2016, 17, 413–424. [Google Scholar] [CrossRef] [Green Version]
- Aroh, C.; Wang, Z.; Dobbs, N.; Luo, M.; Chen, Z.; Gao, J.; Yan, N. Innate Immune Activation by cGMP-AMP Nanoparticles Leads to Potent and Long-Acting Antiretroviral Response against HIV-1. J. Immunol. 2017, 199, 3840–3848. [Google Scholar] [CrossRef] [Green Version]
- Doehle, B.P.; Chang, K.; Fleming, L.; McNevin, J.; Hladik, F.; McElrath, M.J.; Gale, M., Jr. Vpu-deficient HIV strains stimulate innate immune signaling responses in target cells. J. Virol. 2012, 86, 8499–8506. [Google Scholar] [CrossRef] [Green Version]
- Yan, N.; Regalado-Magdos, A.D.; Stiggelbout, B.; Lee-Kirsch, M.A.; Lieberman, J. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat. Immunol. 2010, 11, 1005–1013. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Ducroux, A.; Ponnurangam, A.; Vieyres, G.; Franz, S.; Musken, M.; Zillinger, T.; Malassa, A.; Ewald, E.; Hornung, V.; et al. cGAS-Mediated Innate Immunity Spreads Intercellularly through HIV-1 Env-Induced Membrane Fusion Sites. Cell Host Microbe 2016, 20, 443–457. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, D.J.; Miranda, D., Jr.; Marsden, M.D.; Dizon, T.M.; Bontemps, J.R.; Davila, S.J.; Del Mundo, L.E.; Ha, T.; Senaati, A.; Zack, J.A.; et al. Disruption of Type I Interferon Induction by HIV Infection of T Cells. PLoS ONE 2015, 10, e0137951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seissler, T.; Marquet, R.; Paillart, J.C. Hijacking of the Ubiquitin/Proteasome Pathway by the HIV Auxiliary Proteins. Viruses 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strebel, K. HIV accessory proteins versus host restriction factors. Curr. Opin. Virol. 2013, 3, 692–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rustagi, A.; Gale, M., Jr. Innate antiviral immune signaling, viral evasion and modulation by HIV-1. J. Mol. Biol. 2014, 426, 1161–1177. [Google Scholar] [CrossRef] [PubMed]
- Malim, M.H.; Emerman, M. HIV-1 accessory proteins--ensuring viral survival in a hostile environment. Cell Host Microbe 2008, 3, 388–398. [Google Scholar] [CrossRef] [Green Version]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef]
- Marin, M.; Rose, K.M.; Kozak, S.L.; Kabat, D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat. Med. 2003, 9, 1398–1403. [Google Scholar] [CrossRef]
- Kobayashi, M.; Takaori-Kondo, A.; Miyauchi, Y.; Iwai, K.; Uchiyama, T. Ubiquitination of APOBEC3G by an HIV-1 Vif-Cullin5-Elongin B-Elongin C complex is essential for Vif function. J. Biol. Chem. 2005, 280, 18573–18578. [Google Scholar] [CrossRef] [Green Version]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Willey, R.L.; Maldarelli, F.; Martin, M.A.; Strebel, K. Human immunodeficiency virus type 1 Vpu protein induces rapid degradation of CD4. J. Virol. 1992, 66, 7193–7200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, P.; Boso, G.; Langer, S.; Soonthornvacharin, S.; De Jesus, P.D.; Nguyen, Q.; Olivieri, K.C.; Portillo, A.J.; Yoh, S.M.; Pache, L.; et al. Large-Scale Arrayed Analysis of Protein Degradation Reveals Cellular Targets for HIV-1 Vpu. Cell Rep. 2018, 22, 2493–2503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, L.; Wang, Q.; Xu, Y.; Tsao, L.-C.; Nakagawa, T.; Guo, H.; Su, L.; Xiong, Y. Vpr Targets TET2 for Degradation by CRL4(VprBP) E3 Ligase to Sustain IL-6 Expression and Enhance HIV-1 Replication. Mol. Cell 2018, 70, 961–970.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Shun, M.-C.; Zhang, Y.; Hao, C.; Skowronski, J. HIV-1 Vpr counteracts HLTF-mediated restriction of HIV-1 infection in T cells. Proc. Natl. Acad. Sci. USA 2019, 116, 9568–9577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Descours, B.; Cribier, A.; Chable-Bessia, C.; Ayinde, D.; Rice, G.; Crow, Y.; Yatim, A.; Schwartz, O.; Laguette, N.; Benkirane, M. SAMHD1 restricts HIV-1 reverse transcription in quiescent CD4(+) T-cells. Retrovirology 2012, 9, 87. [Google Scholar] [CrossRef] [Green Version]
- Rosa, A.; Chande, A.; Ziglio, S.; De Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef] [Green Version]
- Usami, Y.; Wu, Y.; Göttlinger, H.G. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef] [Green Version]
- Faust, T.B.; Binning, J.M.; Gross, J.D.; Frankel, A.D. Making Sense of Multifunctional Proteins: Human Immunodeficiency Virus Type 1 Accessory and Regulatory Proteins and Connections to Transcription. Annu. Rev. Virol. 2017, 4, 241–260. [Google Scholar] [CrossRef] [Green Version]
- Bour, S.; Perrin, C.; Akari, H.; Strebel, K. The human immunodeficiency virus type 1 Vpu protein inhibits NF-kappa B activation by interfering with beta TrCP-mediated degradation of Ikappa, B. J. Biol. Chem. 2001, 276, 15920–15928. [Google Scholar] [CrossRef] [Green Version]
- Galão, R.P.; Le Tortorec, A.; Pickering, S.; Kueck, T.; Neil, S.J. Innate sensing of HIV-1 assembly by Tetherin induces NFκB-dependent proinflammatory responses. Cell Host Microbe 2012, 12, 633–644. [Google Scholar] [CrossRef] [Green Version]
- Manganaro, L.; de Castro, E.; Maestre, A.M.; Olivieri, K.; García-Sastre, A.; Fernandez-Sesma, A.; Simon, V. HIV Vpu Interferes with NF-κB Activity but Not with Interferon Regulatory Factor 3. J. Virol. 2015, 89, 9781–9790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doehle, B.P.; Chang, K.; Rustagi, A.; McNevin, J.; McElrath, M.J.; Gale, M., Jr. Vpu mediates depletion of interferon regulatory factor 3 during HIV infection by a lysosome-dependent mechanism. J. Virol. 2012, 86, 8367–8374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harman, A.N.; Nasr, N.; Feetham, A.; Galoyan, A.; Alshehri, A.A.; Rambukwelle, D.; Botting, R.A.; Hiener, B.M.; Diefenbach, E.; Diefenbach, R.J.; et al. HIV Blocks Interferon Induction in Human Dendritic Cells and Macrophages by Dysregulation of TBK1. J. Virol. 2015, 89, 6575–6584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Züst, R.; Cervantes-Barragan, L.; Habjan, M.; Maier, R.; Neuman, B.W.; Ziebuhr, J.; Szretter, K.J.; Baker, S.C.; Barchet, W.; Diamond, M.S.; et al. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 2011, 12, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dharan, A.; Campbell, E.M. Role of Microtubules and Microtubule-Associated Proteins in HIV-1 Infection. J. Virol. 2018, 92, e00085-18. [Google Scholar] [CrossRef] [Green Version]
- Dharan, A.; Opp, S.; Abdel-Rahim, O.; Keceli, S.K.; Imam, S.; Diaz-Griffero, F.; Campbell, E.M. Bicaudal D2 facilitates the cytoplasmic trafficking and nuclear import of HIV-1 genomes during infection. Proc. Natl. Acad. Sci. USA 2017, 114, E10707–E10716. [Google Scholar] [CrossRef] [Green Version]
- Corrales, L.; McWhirter, S.M.; Dubensky, T.W., Jr.; Gajewski, T.F. The host STING pathway at the interface of cancer and immunity. J. Clin. Investig. 2016, 126, 2404–2411. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Yi, M.; Qin, S.; Song, Y.; Chu, Q.; Wu, K. Activating cGAS-STING pathway for the optimal effect of cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 35. [Google Scholar] [CrossRef]
- Siddiqui, M.A.; Saito, A.; Halambage, U.D.; Ferhadian, D.; Fischer, D.K.; Francis, A.C.; Melikyan, G.B.; Ambrose, Z.; Aiken, C.; Yamashita, M. A Novel Phenotype Links HIV-1 Capsid Stability to cGAS-Mediated DNA Sensing. J. Virol. 2019, 93, e00706-19. [Google Scholar] [CrossRef] [Green Version]


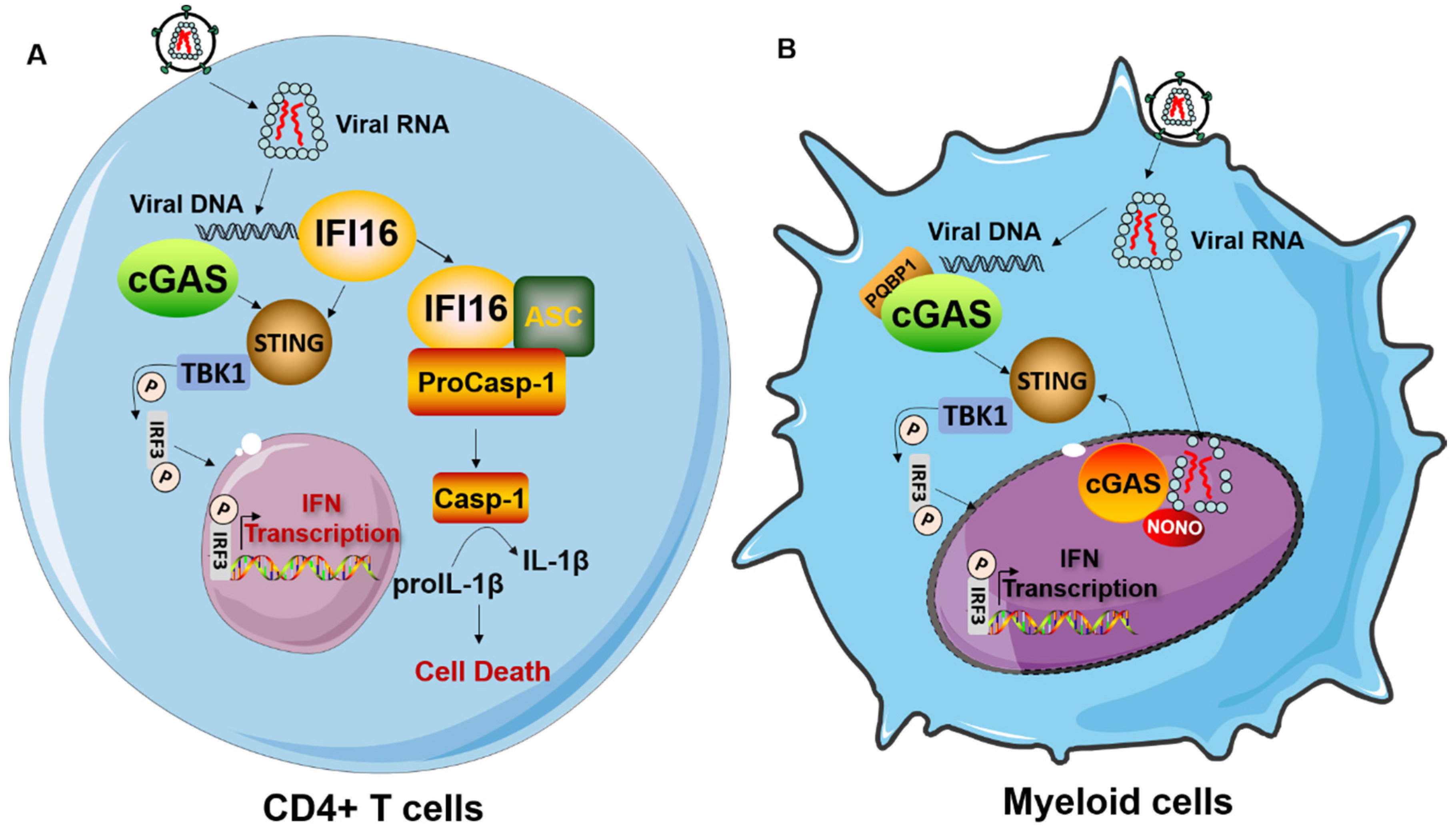{kind=link}
| Infection Course | PAMP | Sensor | Signal Axis | Immune Effector Response | Cell Type | Reference |
|---|---|---|---|---|---|---|
| Entry, Uncoating | Single-stranded RNA (ssRNA) | RIG-I/MDA5 | MAVS-TRAF3-TBK-IRFs | Inflammatory cytokines Type I IFNs | Macrophages | [23,27,35] |
| Single-stranded RNA (ssRNA) | TLR7/TLR8 | MyD88-TRAF6-NF-κB | Inflammatory cytokines Type I IFNs | Plasmacytoid DCs | [24,32] | |
| Viral Fusion | Unknown | Unknown | Type I IFNs | MDMs | [39] | |
| Uncoating, Reverse Transcription | RNA/DNA hybrids | DDX41 | STING-TBK1-IRFs | IFNs | BMDMs BMDCs | [40] |
| Double-stranded DNA (dsDNA) | cGAS | STING-TBK1-IRFs | Inflammatory cytokines and IFNs | DCs Macrophages CD4+ T cells | [16,41,42] | |
| Double-stranded DNA (dsDNA) | IFI16 | ASC-proCasp-1-IL1β | Caspase-1 activation Pyroptosis | Quiescent CD4+ T cells | [43] | |
| Single-stranded DNA (ssDNA) | IFI16 | STING-TBK1-IRFs | IFNs | Macrophages Activated CD4+ T cells | [22] | |
| Integration | Unknown | cGAS | STING-TBK1-IRFs | IFNs | MDDCs CD4+ T cells | [41,42] |
| Transcription Translation | Intron-containing RNA | Unknown | MAVS-TRAF3-TBK-IRFs | IFNs | MDMs CD4+ T cells | [36,37] |
| Abortive viral RNA | DDX3X | MAVS-TRAF3-TBK-IRFs | IFNs | DCs | [29] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, X.; Langer, S.; Zhang, Z.; Herbert, K.M.; Yoh, S.; König, R.; Chanda, S.K. Sensor Sensibility—HIV-1 and the Innate Immune Response. Cells 2020, 9, 254. https://doi.org/10.3390/cells9010254
Yin X, Langer S, Zhang Z, Herbert KM, Yoh S, König R, Chanda SK. Sensor Sensibility—HIV-1 and the Innate Immune Response. Cells. 2020; 9(1):254. https://doi.org/10.3390/cells9010254
Chicago/Turabian StyleYin, Xin, Simon Langer, Zeli Zhang, Kristina M. Herbert, Sunnie Yoh, Renate König, and Sumit K. Chanda. 2020. "Sensor Sensibility—HIV-1 and the Innate Immune Response" Cells 9, no. 1: 254. https://doi.org/10.3390/cells9010254
APA StyleYin, X., Langer, S., Zhang, Z., Herbert, K. M., Yoh, S., König, R., & Chanda, S. K. (2020). Sensor Sensibility—HIV-1 and the Innate Immune Response. Cells, 9(1), 254. https://doi.org/10.3390/cells9010254