1. Introduction
During chronic liver diseases (CLDs), liver fibrogenesis is sustained by an heterogeneous population of myofibroblast-like cells (MFs) that can originate mainly from hepatic stellate cells (HSC/MFs) and portal (myo)fibroblasts or, to a less extent, circulating and bone marrow-derived mesenchymal stem cell (MSCs) engrafting chronically injured liver [
1,
2,
3,
4,
5,
6]. At present, the involvement of epithelial to mesenchymal transition (EMT) as pro-fibrogenic mechanism leading to the origin of MFs from hepatocytes and/or cholangiocytes in progressive CLD is likely to be of minor relevance, as suggested by several elegant but mostly negative fate tracing studies [
7,
8,
9,
10,
11]. In the scenario of pro-fibrogenic phenotypic responses of hepatic MFs, their migration towards the site of injury and their ability to align with nascent and established fibrotic septa in response to several stimuli (polypeptide mediators, hypoxia, reactive oxygen species or ROS) is considered as a relevant issue [
4,
12,
13]. During CLD many peptide mediators, including platelet-derived growth factor (PDGF), monocyte chemoattractant protein-1 (MCP-1 or CCL2), angiotensin II (AT-II), vascular endothelial growth factor-A (VEGF-A) as well as ROS and/or hypoxic conditions, have been reported to stimulate HSC/MFs migration, then contributing to fibrosis progression [
12,
13,
14,
15,
16,
17]. Along these lines, OSM, a pleiotropic cytokine structurally and functionally related to the interleukin-6 (IL-6) cytokine family, has recently emerged as a mediator involved in CLDs progression [
18,
19,
20,
21,
22,
23,
24]. OSM acts through the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway and mitogen-activated protein kinases (MAPK) in order to critically regulate processes such as liver development and regeneration, hematopoiesis, and angiogenesis [
23]. Remarkably, these processes are also regulated by hypoxia through the involvement of hypoxia-inducible factor 1 alpha (HIF1α). A previous study by Vollmer and colleagues [
19] showed that HIF1α levels increased significantly after treatment of hepatocytes and hepatoma cells with OSM. These Authors also showed that HIF1α contributed to OSM-downstream signaling events, suggesting the existence of a cross-talk between OSM and hypoxia signaling in liver development and regeneration. Moreover, OSM is known to affect and regulate inflammatory response in several diseases affecting different organs and tissues [
20]. Concerning the liver, in addition to the role in development and regeneration [
23] as well as inflammatory response [
20] OSM has been suggested to be involved also in the pathogenesis of steatosis and hepatic insulin resistance [
21]. OSM is expressed in Kupffer cells even in normal liver but it is consistently over-expressed in cirrhotic livers [
18]. OSM can signal through 2 different heterodimeric receptors that share gp130 (a common subunit receptor for ligands of IL-6 family) and involve either leukemia inhibitor factor receptor β (LIFRβ) or OSM receptor β (OSMRβ). In particular, LIFRβ is weakly expressed in normal livers, but more intensely in cirrhotic liver, in reactive ductules, bile ducts, colangiocytes, and perisinusoidal areas; OSMRβ is expressed at low level in hepatocytes of normal livers, but is was reported to be unchanged in cirrhotic samples. Moreover, in a study from the same research group [
22] it was reported that OSM was able to modulate response of HSC/MFs by increasing collagen I and tissue inhibitor of metalloproteinase-1 (TIMP-1) secretion, suggesting a putative pro-fibrogenic role for OSM in CLD. More recently, a mechanistic experimental study has confirmed the ability of OSM to act as a pro-fibrogenic mediator by regulating both macrophage (resident and bone marrow-derived) and, as a consequence, MFs activation during the course of thioacetamide (TAA)-induced model of CLD [
24]. These authors showed that OSM up-regulated the expression of pro-fibrogenic mediators such as TGF-β and PDGF in macrophages, suggesting that OSM may be indirectly pro-fibrogenic by up-regulating through TGF-β and PDGF, collagen and TIMP1 synthesis in HSC/MFs. In the mentioned study, liver fibrosis was significantly prevented in OSM knockout mice submitted to the TAA protocol vs related control littermates. Moreover, continuous expression of OSM in normal mouse liver, as forced by means of hydrodynamic tail vein injection (HTVi), resulted in severe fibrosis [
24]. In the present study, we investigated the involvement of OSM in experimental and human NAFLD/NASH and we performed experiments in order to elucidate whether this cytokine may act directly on LX2 cells and primary culture of human HSC/MFs by regulating selected pro-fibrogenic phenotypic responses.
2. Materials and Methods
Materials: The methionine and choline-deficient (MCD) diet, the choline-deficient amino acid-refined (CDAA) diet, the high fat–high fructose diet (HFHF) as well as the related control diets, methionine choline supplemented (MCS) diet and choline-supplemented and amino acid-refined (CSAA) diet and normal (not supplemented with fat) diet were provided by Laboratorio Dottori Piccioni srl (Gessate, Milano, Italy). The kit for RNA retro-trascription and for real time PCR were purchased from Biorad (Berkely, CA, USA). Enhanced chemiluminescence (ECL) reagents, nitrocellulose membranes (Hybond-C extra) were from Amersham Pharmacia Biotech Inc. (Piscataway, NJ, USA). Human recombinant Oncostatin M (OSM) was from Petrotech (Rocky Hill, CT, USA). Monoclonal antibody against p-ERK1/2 (sc-7383), polyclonal antibody for p-STAT1 (sc-8394), STAT1 (sc-346), p-STAT3 (sc-8059), STAT3 (sc-7179), ERK1/2 (sc-292838), p-Akt1/2/3 (sc-7985-R), Akt1/2/3 (sc-8312), VEGF (sc-152), and HIF-1α (sc-10790) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Polyclonal antibody for p-JNK and JNK1/2 were from Cell Signaling Technology (Danvers, MA, USA). PD98059, SP600125, LY294002 and apocynin were from Calbiochem (La Jolla, CA, USA). Monoclonal antibodies for β-actin, crystal violet, DCFH-DA and all other reagents of analytical grade as well as primers for RT-PCR were from Sigma Chemical Co (Sigma Aldrich Spa, Milan, Italy), Boyden’s chambers were from Neuro Probe, Inc. (Gaithersburg, MD, USA). The monoclonal neutralizing antibody against Flk-1 was obtained from ImClone (New York, NY, USA).
Methods: Animal experimentation. In this study we used three different dietary protocols for induction of progressive NAFLD that were carried out in 8 week old C57Bl/6 mice (Harlan Laboratories, Indianapolis, IN, USA) (
n = 8 for any experimental group). Mice were fed as previously described [
25] on the following dietary regimens: (i) Methionine and choline-deficient (MCD) diet or methionine and choline sufficient (MCS) control diet, (ii) choline-devoided and L-amino acid-defined (CDAA) diet or choline-sufficient L-amino acid-defined (CSSA), (iii) high fat–high fructose (HFHF) diet. Mice were then sacrificed at different experimental time points (4 days, 2, 4, and 8 weeks for MCD or MCS protocol, 12 and 24 weeks for CDAA or CSAA protocol, 24 weeks for HFHF and standard control diet). Mice were kept under specific pathogen-free conditions and maintained with free access to pellet food and water. Liver samples were obtained and immediately used/processed for morphological or molecular biology analyses or frozen and thereafter maintained at −80 °C for further analysis. The experiments complied with EU and national ethical guidelines for animal experimentation and all experimental protocols were approved by the Animal Ethic Committee of University of Oriental Piedmont, Novara, Italy and Italian Ministry of Health.
Human patients: The study on NASH patients was approved by the Ethics Committee of the Azienda Ospedaliera Universitaria Città della Salute (Turin, Italy). For this study we analyzed liver biopsies from NASH patients (n = 20) or from patients with simple steatosis (n = 10), referring to the Division of Gastroenterology and Hepatology of the University of Turin. All samples were collected at the time of first diagnosis; all subjects gave informed consent to the analysis, and the study protocol, which conformed to the ethical guidelines of the 1975 Declaration of Helsinki, was planned according to the guidelines of the local ethics committee.
Immunohistochemistry analysis: Liver sections from human patients with NASH or with simple steatosis were employed. Immunostaining procedure was as previously described [
25]. Briefly, paraffin sections (2 μm thick), mounted on poli-
l-lysine coated slides, were incubated with (i) the monoclonal antibody against OSM (Santa Cruz Biotechnology, Dallas, TX, USA; dilution 1:200) or (ii) the monoclonal antibody against human CD68 (Biorad, Hercules, CA, USA; dilution 1:80) or (iii) the secondary monoclonal antibody alone, as negative control. After blocking endogenous peroxidase activity with 3% hydrogen peroxide and performing microwave antigen retrieval in sodium citrate buffer pH6, primary antibodies were labeled by using EnVision, HRP-labeled System (DAKO) and visualized by 3′-diaminobenzidine substrate.
LX2 cells culture: Human LX2 cells, a model of immortalized and activated, MF-like, human HSC, originally kindly provided by Prof. Scott L. Friedman (Icahn School of Medicine, MS, USA), were cultured in Dulbecco’s modified Eagle’s medium (Sigma Aldrich Spa, Milan, Italy), supplemented with 10% fetal calf serum and 1% antibiotics. In most experiments we also used human HSCs (Clinisciences, Nanterre, France), were used between passages 4 and 7 when showing a phenotype of fully activated, MF-like HSCs (HSC/MFs), plated to obtain the desired sub-confluence level and then left for 24 h in serum-free Iscove’s medium to have cells at the lowest level of spontaneous proliferation [
13]. LX2 cells or HSC/MFs were then exposed in culture conditions to human recombinant OSM 10 ng/mL for different times.
Cell migration and Chemotaxis: Non-oriented migration (chemokinesis) and chemotaxis of human LX2 (and HSC/MFs) were evaluated after exposure to PDGF-BB 10ng/mL, used as positive control, or to OSM 10 ng/mL, by performing the wound healing assay (20 h of incubation) or the modified Boyden’s chamber assay (6 h of incubation), as previously described [
7,
13]. For the wound healing assay LX2 or HSC/MFs cells were plated on collagen coated 24 wells (Falcon, Corning, NY, USA) and, were confluent, left for 24 h in their medium without serum to have cells at the lowest level of spontaneous proliferation. Then, a scratch on the cell monolayer was performed and the cells were exposed to medium with hrOSM (or where indicated with specific inhibitors) for 20 h, stained with crystal violet and finally observed at contrast phase microscope.
For the Boyden’s chamber assay, filter of 8 µm (Whatman Nuclepore™ track-etched polycarbonate membranes) were first coated with type I collagen (20 µg/mL) for 30 min at 37 °C.
2 × 104 cells were placed in the upper compartment and were allowed to migrate through the pores of the filter into the lower compartment, in which chemotactic agents are present. At the end of incubation time (6 h), filters were removed from the Boyden’s chamber and stained with crystal violet. In some experiments, cells were pre-treated with specific inhibitors to check the relevance of specific proteins in process described.
Proliferation: Proliferation of human LX2 cells (and HSC/MFs) was evaluated by crystal violet proliferation assay by seeding cells in a 96-well plate at a density of 1.5 × 103 cells per well. The cells were incubated in serum-free medium (SFM) for 24 h and then exposed to OSM 10 ng/mL. At the desired time, the medium was removed, and the cells were washed twice with phosphate-buffered saline, fixed with 11% glutaraldehyde; after fixation, cells were washed and then stained with 0.1% (w/v) crystal violet solution for 10 min. After washing with water, the crystal violet was solubilized with 50 μL of 10% acetic acid solution, and absorbance was measured at 595–650 nm using a microplate reader (SpectraMAX M3; Molecular Devices, Sunnyvale, CA, USA). In some experiments, BrdU incorporation assay was performed. LX2 cells or HSC/MFs were seeded in culture plates (1 × 104 cells per well, 96 multiwell), for 24 h up to 72 h. The cell proliferation rate was evaluated by means of a colorimetric assay kit supplied by Roche Diagnostic (11647229001) according to manufacturer’s instructions.
Detection of intracellular generation of ROS. (A) DCFH-DA technique: Detection of ROS generation in cultured cells was performed by using the semi-quantitative DCFH-DA technique as previously described [
7]. Cultured cells, seeded in 12-well culture plates (10
5 cells/well), were exposed to OSM 10 ng/mL for 15 min or pre-treated with apocynin for 1h and then exposed with OSM 10 ng/mL. ROS generation was detected as the conversion of 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA, 1 μM) into the corresponding fluorescent derivative. Cells were observed and photographed under a Leica fluorescence microscope (DMI 4000 B model). (B) combination of DCFH-DA technique and flow cytometric analysis: cells were seeded in P35 dishes (5 × 10
5 cells/dish), cultured for 24 h and exposed to OSM alone or OSM plus 250 nM apocynin for 1 h before addition of 5 μM DCFH-DA (15 min of incubation in the dark). Cells were rapidly washed with PBS, collected by trypsinization, briefly centrifuged (1600 rpm for 5 min) and re-suspended in PBS for analysis. Detection of DCF green fluorescence (FL1) was performed on at least 5000 cells per sample with a FACScan equipped with a 488 nm argon laser using the CellQuest software (version 1.0.264.21, Becton-Dickinson, Milano, Italy). The peak of FL1 intensity of DCFH-DA-stained control cells grown without OSM was set to channel 101 and retained for all measurements [
24]. As a positive control, cells were treated with 100 μM H
2O
2 for 15 min, stained with DCFH-DA as above and assayed during the same analytical session.
Western blot analysis: Total cell lysates, obtained as described [
7,
11,
24], were subjected to sodium dodecyl sulfate-polyacrylamide gel-electrophoresis on 13.5%, 10%, or 7.5% acrylamide gels, incubated with desired primary antibodies, then with peroxidase-conjugated anti-mouse or anti-rabbit immunoglobulins in Tris-buffered saline-Tween containing 2% (
w/
v) non-fat dry milk and finally developed with the ECL reagents according to manufacturer’s instructions. Sample loading was evaluated by reblotting the same membrane with the un-phosphorylated form of protein or with β-actin antibody. In some experiments, protein levels in culture medium obtained from LX2 or HSC/MFs exposed to OSM for the desired time were evaluated by an immunoprecipitation procedure, as described previously [
13].
Quantitative real-time PCR (Q-PCR): RNA extraction, complementary DNA synthesis, quantitative real-time PCR (Q-PCR) reactions were performed as previously described [
6,
10]. mRNA levels were measured by Q-PCR, using the SYBR
® green method as described [
25]. The amplification mix was prepared using iTaq Universal Syber Green SuperMix (Biorad Laboratories, Berkeley, CA, USA) following manufacturer’s instructions and realtime PCR was performed using Miniopticon ThermoCycler Instrument (Biorad Laboratories, Berkeley, CA, USA). Oligonucleotide sequence of primers used for RT-PCR were:
| Primer | Sense | Reverse |
| murine OSM | TTTCTCTGGGGATACCATCG | GGAGACACGATGGGCTATGT |
| murine OSMβR | GGAGACACGATGGGCTATGT | CATCTGAGGTGATGGTGGTG |
| murine TBP | CACATCACAGCTCCCCACCA | AGCGGAGAAGATGCTGGAAAC |
| human OSM | TACTGCTCACACAGAGGACGC | CTATAGCCGCCATGCTCGC |
| human OSMR | CGCGTCAGGTTTGCACTTA | GTGTGTGGCACATTCCAAG |
| human LIFR | GGCTCATCACCACCTTCCAA | CCCCTTTCCCATCCCAACAA |
| human HIF1α | CCACCTATGACCTGCTTGGT | TATCCAGGCTGTGTCGACTG |
| human VEGF | CCCACTGAGGAGTCCAACAT | TTTCTTGCGCTTTCGTTTTT |
| human CCL2 | CCCCAGTCACCTGCTGTTAT | AGATCTCCTTGGCCACAATG |
| human IL6 | AGGAGACTTGCCTGGTGAAA | CAGGGGTGGTTATTGCATCT |
| human TNF-α | AACCTCCTCTCTGCCATCAA | GGAAGACCCCTCCCAGATAG |
| human GAPDH | TGGTATCGTGGAGGACTCATGGAC | ATGCCAGTGAGCTTCCCGTTCAGC |
TATA binding protein (TBP) and Gliceraldehyde-3-phosphate dehydrogenase (GAPDH) were used as internal reference for murine and human sample respectively, and co-amplified with target samples using identical Q-PCR conditions. Samples were run in triplicate and mRNA expression was generated for each sample. Specificity of the amplified PCR products was determined by melting curve analysis and confirmed by agarose gel electrophoresis.
Statistical analysis: Data in bar graphs represent means ± SEM, and were obtained from average data of at least three independent experiments. Luminograms and morphological images are representative of at least three experiments with similar results. Statistical analysis for these experiments was performed by Student’s t-test or ANOVA for analysis of variance when appropriate (p < 0.05 was considered significant).
4. Discussion
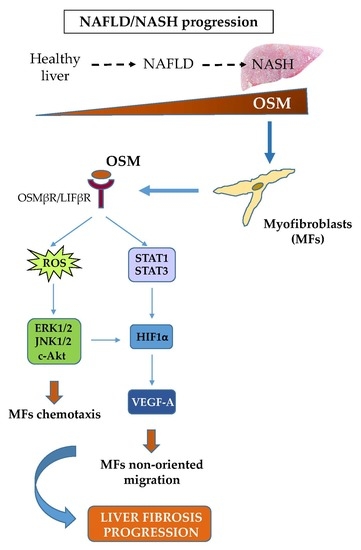
Migration of hepatic MFs is a typical and distinctive pro-fibrogenic feature that allows these cells to align with inflammatory cells along fibrotic septa during liver fibrosis progression. This event occurs during CLDs in response to different stimuli and mediators, including polypeptide mediators, ROS as well as hypoxia [
6,
7,
11]. In this scenario, literature data have already proposed a role for OSM in modulating CLD progression, likely by modulating defined processes like development, regeneration, hematopoiesis, and angiogenesis [
20,
22]. OSM is normally expressed in Kupffer cells and its expression, variable in normal livers, increases in cirrhotic livers [
21]; it was proposed that this cytokine may up-regulate selected pro-fibrogenic responses of HSCs, including synthesis of collagen I and TIMP1 [
22]. Along these lines, the present study was designed to investigate whether OSM may be involved also in NAFLD progression and may directly modulate other relevant pro-fibrogenic responses of MFs like proliferation and migration. Data in this study provide evidence that OSM is potentially involved in NAFLD progression as shown by data obtained by analyzing either the livers of mice fed on different dietary protocols (i.e., mice fed on either MCD, CDAA or HFHF diets) or in human liver specimens from NAFLD/NASH patients. In particular, human data indicate that OSM is up-regulated in NASH patients but not in patients that have only histological evidence of simple steatosis. Similarly, OSM expression increases in murine livers in parallel with the development of NASH-associated fibrosis. In our knowledge, this is the first report involving up-regulation of OSM expression in experimental and clinical progressive NAFLD, and further experiments and analyses are currently in progress in order to verify whether this mediator (or its related receptor subunits) may be validated as a marker of NAFLD progression and/or may have prognostic significance. This is a potentially relevant issue for a disease that is rapidly emerging as the most relevant CLD worldwide and that affects a large number of individuals in the general population.
Concerning the putative pro-fibrogenic role of OSM, already proposed by two earlier studies [
18,
22] and by a more mechanistic and recent one [
24], we here propose that OSM is indeed able to directly target hepatic MFs. In particular, the present study shows that OSM can induce migration of both LX2 and HSC/MFs and that OSM dependent migration occurs as a biphasic process involving distinct but interrelated early and late events. These data are fully in agreement with previously published results investigating the pro-migratory action of chemotactic polypeptides and controlled conditions of hypoxia [
12,
13]. This conclusion is supported by several lines of evidence. First, OSM-induced early migration is triggered by ROS released within few minutes, likely as a consequence of the parallel activation of the ROS-generating membrane complex NADPH-oxidase, similarly to what described for HSC/MFs in response to other chemoattractant polypeptides [
13]. The intracellular generation of ROS after OSM treatment was demonstrated by performing the DCFH-DA technique in presence or absence of the NADPH pharmacological inhibitor apocynin. This inhibitor, although not specific/selective, has the advantage to block all NADPH-oxidase (NOX) isoforms expressed by hepatic MFs (i.e., NOX1, NOX2, and NOX4). Apocynin pretreatment reduced early LX2 and HSC/MFs chemotaxis, whereas it was ineffective on late OSM-dependent migration. Our data also suggest, as previously demonstrated for other stimuli [
13,
25], that the rise of intracellular ROS generation is related to an early redox-dependent phosphorylation (i.e., activation) of ERK1/2 and JNK1/2 isoforms with a kinetic compatible with that of ROS release. This notion is supported by data demonstrating a reduction of chemotaxis in both cell types obtained by pre-treating cells with specific pharmacological inhibitors of ERK1/2 and JNK1/2 (PD98059 and SP600125, respectively). A direct link between the increase on intracellular levels of ROS and redox-related activation of both ERK1/2 and JNK1/2 it is now well established in the redox signaling scenario. It has been suggested that the increased kinase activity is likely to be the result of direct activation of defined signaling components or of inhibition of phosphatases responsible for the negative feed-back control of activated signaling pathways (like JNK specific phosphatases or, more generally, protein tyrosine phosphatases) [
26,
27,
28]. The involvement of these different signaling pathways is in line with that observed for all the cytokine belonging to IL-6 type family, even if the signal transduction pathways of these cytokines, still currently investigated, are sometimes suggested to be cell-type specific.
A second role for OSM-related increase in intracellular ROS levels is related to the modulation of JAK/STAT signaling pathway. This event has been already described by Madamanchi et al. in vascular smooth muscle cells in which JAK2 was rapidly activated after treatment with hydrogen peroxide and both STAT1 and STAT3 were phosphorylated on tyrosine residues to then translocate to the nucleus in a JAK2-dependent manner [
29]. Moreover, Authors showed that the inhibition of JAK2 activity by means of AG-490 partially inhibited hydrogen peroxide-induced ERK2 activity, suggesting that JAK protein is upstream of the Ras/ERK pathway and have a key role in the adaptive response to oxidative stress [
29]. Along these lines, also in LX2 and HSC/MFs the early intracellular generation of ROS is able to sustain late migration by the involvement of JAK/STAT3 signaling pathway; this notion is supported by results showing AG490-dependent inhibition of JAK pathway and apocynin-dependent inhibition of STAT3 pathway. These results suggest that OSM can affect cell migration and are in agreement with a study in which the inhibition of JAK family members, in particular of JAK1, significantly reduced activation, proliferation, and migration of rat hepatic stellate cells [
29]. Our results are also in agreement with literature data showing a role for JAK/STAT pathway in the modulation of liver fibrosis, as in the case of STAT3 activation in hepatic stellate cells treated with IL-6 and leptin [
30,
31].
Results obtained in the wound healing assay also provided evidence that OSM-dependent non oriented migration required late HIF1-dependent expression and release of VEGF-A. These results are homologous to those reported for exposure to hypoxic conditions and are potentially in agreement with literature data showing a role for OSM in modulating hypoxia-regulated processes (including hematopoiesis, angiogenesis, liver development, and regeneration), then suggesting a possible cross-talk between OSM and hypoxia signaling pathways [
23]. According to literature data obtained in different cell types [
19], we report that in both LX2 cells and HSC/MFs OSM can induce a significant up-regulation of HIF1α protein levels under normoxic conditions. Interestingly, as previously reported for HSC/MFs exposed to hypoxia [
26], HIF1α up-regulation was associated with increased production and release of VEGF-A that, in turn, was the real responsible for late OSM-dependent migration. This notion was supported by the following observations: (i) OSM-related increase in HIF1α stabilization was rapidly detected (starting from 3 h and persisting for the entire experimental protocol; (ii) VEGF-A was released in the extracellular medium of cells exposed to OSM only after 24 h in LX2 cells; (iii) conditioned medium obtained from cells treated with OSM efficiently stimulated only late non-oriented migration; and (iv) both pre-treatment of cells with SU1498, a pharmacological inhibitor of the tyrosine-kinase receptor subunit of VEGFR-2 and with a neutralizing antibody for VEGFR-2 inhibited non-oriented OSM-dependent migration. These results obtained in both LX2 cells and HSC/MFs are in agreement with data obtained in previous studies related to the pro-migratory action of VEGF-A [
26,
32], and suggest a potential role for OSM-dependent expression and release of VEGF-A.
Finally, although in our experimental conditions OSM was found ineffective on the synthesis of collagen I and of TIMP1 (data not shown), differently from what reported by others [
33], we additionally observed that hrOSM up-regulated in LX2 cells transcript levels of the chemokine CCL2, the most relevant chemokine during both experimental and human conditions of progressive NAFLD [
34], as well as of the pro-inflammatory cytokines TNF e IL-6. However, these data are preliminary and further experiments will be necessary to mechanistically characterize this issue.
In conclusion, data reported in the present study provide novel evidence adding further knowledge on the proposed pro-fibrogenic role of OSM in the progression of CLD and suggest for the first time a possible involvement of this cytokine in the progression of NAFLD.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

















