Understanding IGF-II Action through Insights into Receptor Binding and Activation


Abstract
:1. Introduction
2. IGF-II and Cancer
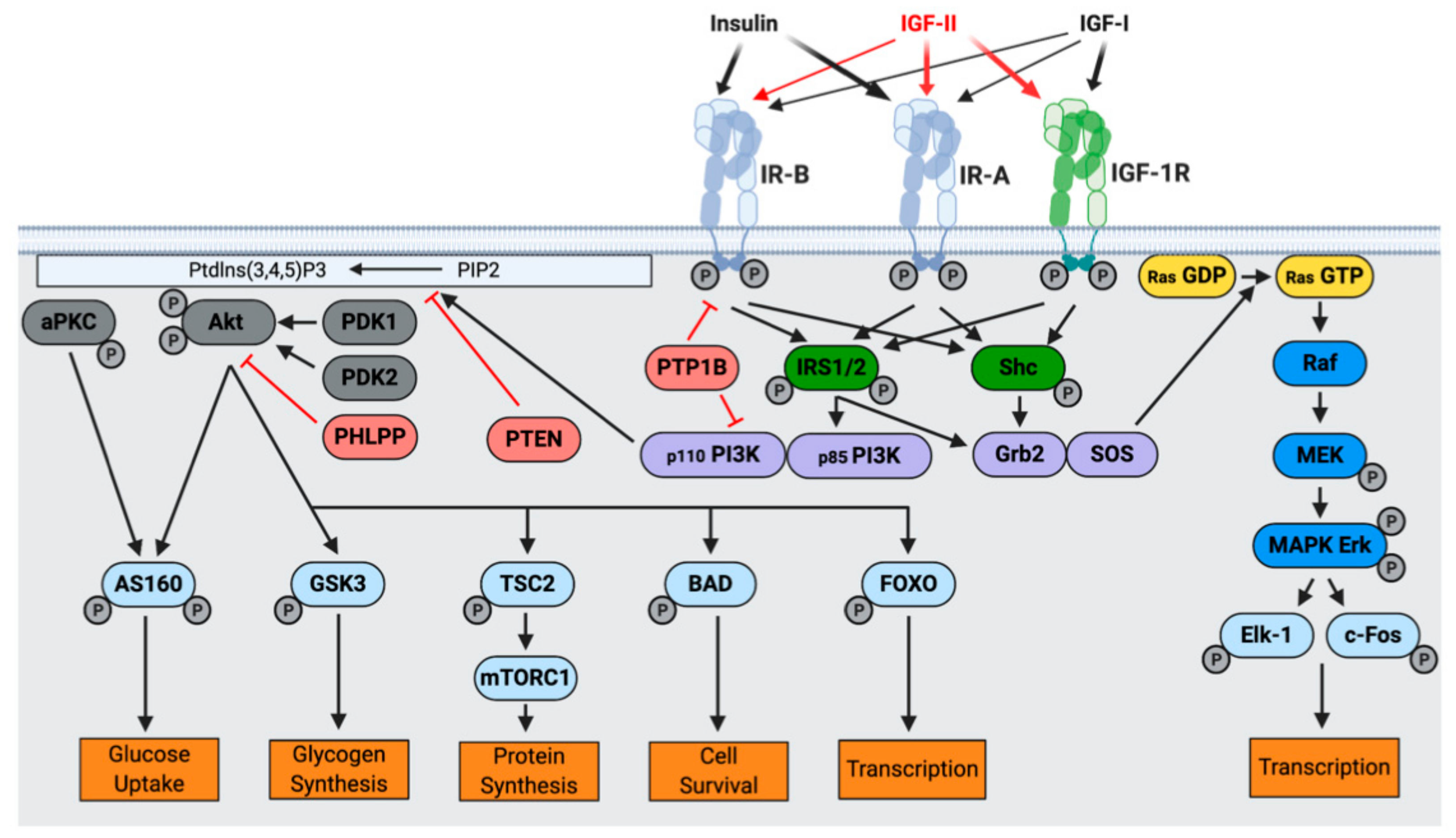
3. IGF-II Signaling
4. How does IGF-II Bind and Activate IGF-1R and IR-A?
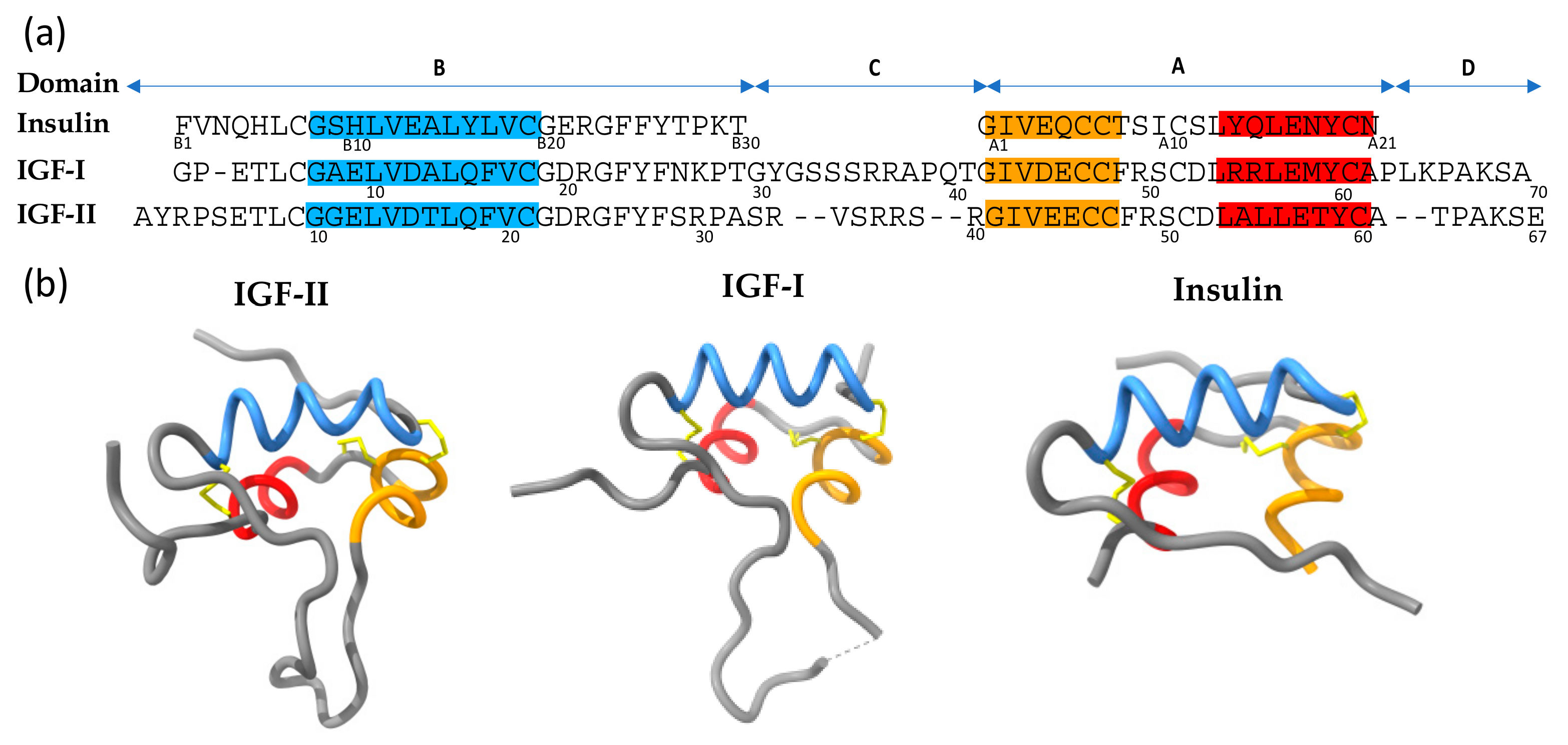
4.1. IGF-II Structure
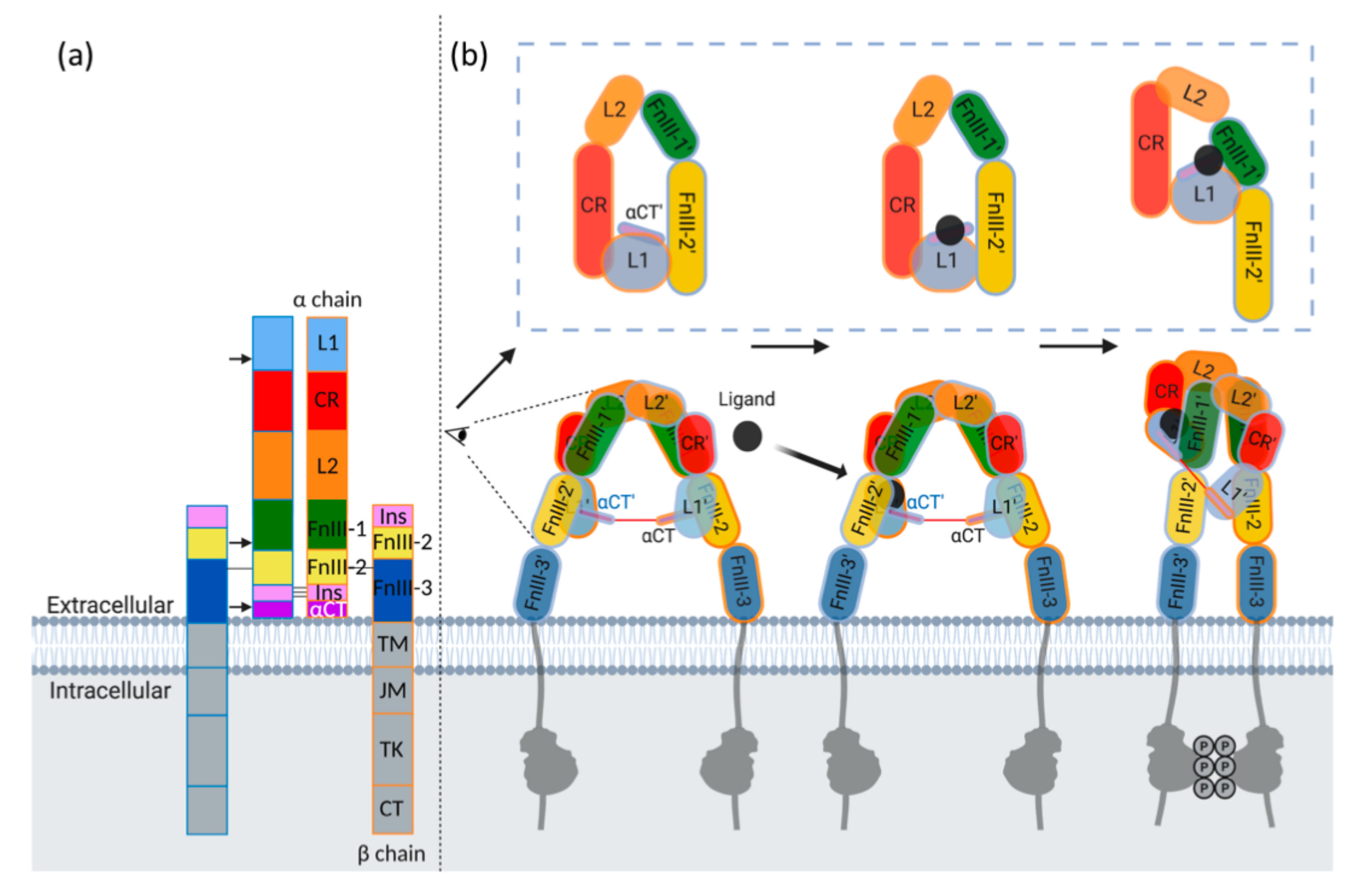
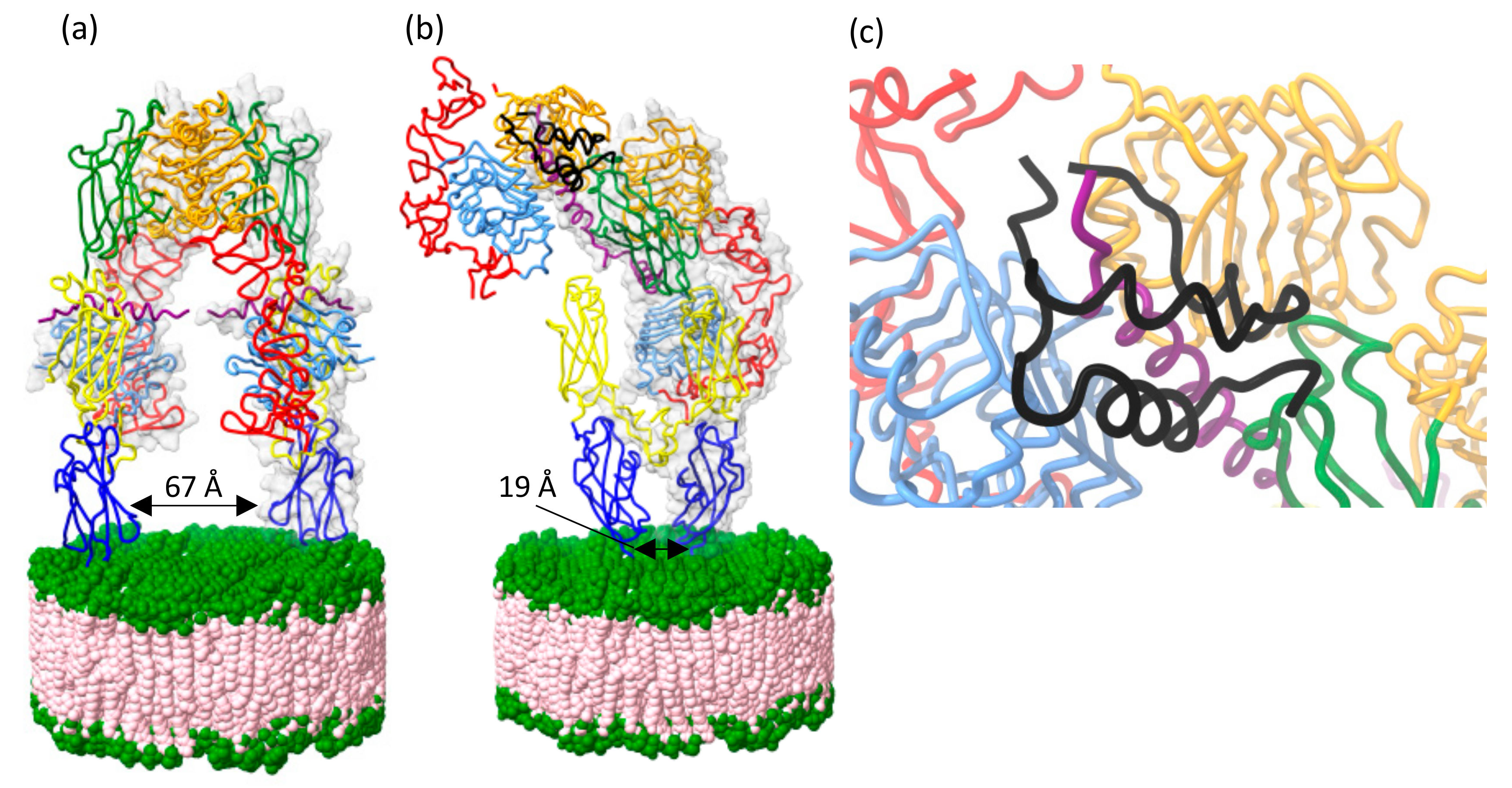
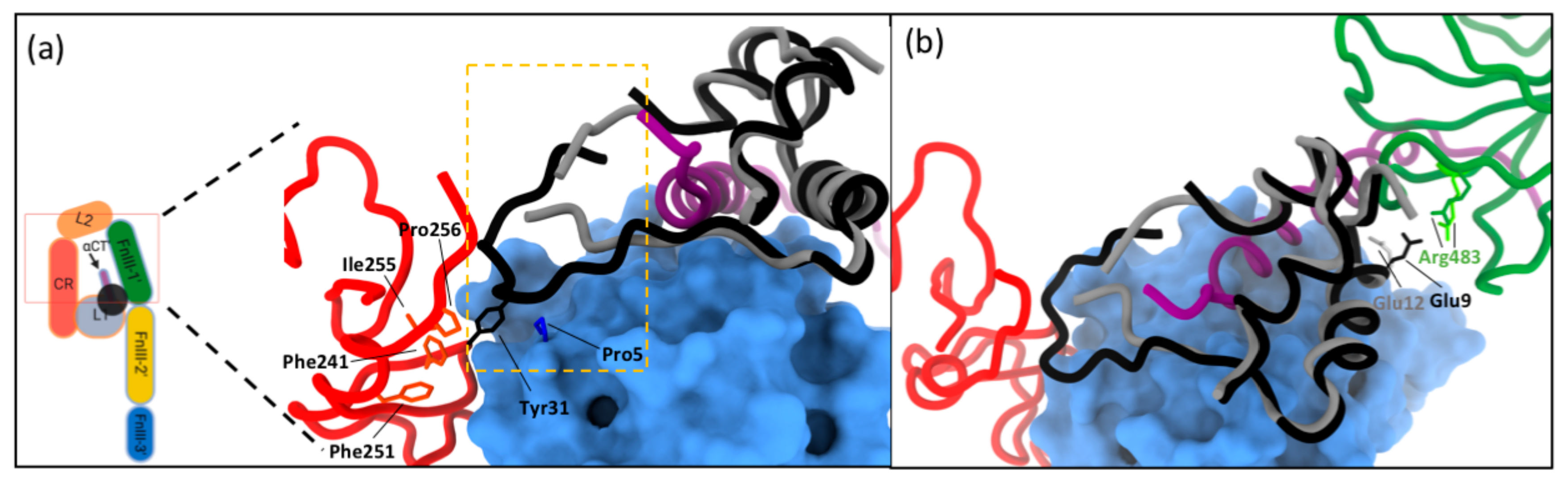
4.2. Receptor Structure, Mechanism of Binding and Activation
5. Conclusions and Implications of Structural Information for Developing Treatments for Disease
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Denley, A.; Cosgrove, L.J.; Booker, G.W.; Wallace, J.C.; Forbes, B.E. Molecular interactions of the IGF system. Cytokine Growth Factor Rev. 2005, 16, 421–439. [Google Scholar] [CrossRef] [PubMed]
- Versteyhe, S.; Klaproth, B.; Borup, R.; Palsgaard, J.; Jensen, M.; Gray, S.G.; De Meyts, P. IGF-I, IGF-II, and Insulin Stimulate Different Gene Expression Responses through Binding to the IGF-I Receptor. Front. Endocrinol. 2013, 4, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morcavallo, A.; Gaspari, M.; Pandini, G.; Palummo, A.; Cuda, G.; Larsen, M.R.; Vigneri, R.; Belfiore, A. Research resource: New and diverse substrates for the insulin receptor isoform A revealed by quantitative proteomics after stimulation with IGF-II or insulin. Mol. Endocrinol. 2011, 25, 1456–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, V.; Jawerbaum, A.; Mazzucco, M.B.; Gauster, M.; Desoye, G.; Hiden, U. IGF2 stimulates fetal growth in a sex- and organ-dependent manner. Pediatric Res. 2018, 83, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Randhawa, R.; Cohen, P. The role of the insulin-like growth factor system in prenatal growth. Mol. Genet. Metab. 2005, 86, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Agrogiannis, G.D.; Sifakis, S.; Patsouris, E.S.; Konstantinidou, A.E. Insulin-like growth factors in embryonic and fetal growth and skeletal development (Review). Mol. Med. Rep. 2014, 10, 579–584. [Google Scholar] [CrossRef] [Green Version]
- DeChiara, T.M.; Efstratiadis, A.; Robertson, E.J. A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature 1990, 345, 78–80. [Google Scholar] [CrossRef]
- Stylianopoulou, F.; Efstratiadis, A.; Herbert, J.; Pintar, J. Pattern of the insulin-like growth factor II gene expression during rat embryogenesis. Development 1988, 103, 497–506. [Google Scholar]
- Soares, M.B.; Turken, A.; Ishii, D.; Mills, L.; Episkopou, V.; Cotter, S.; Zeitlin, S.; Efstratiadis, A. Rat insulin-like growth factor II gene: A single gene with two promoters expressing a multitranscript family. J. Mol. Biol. 1986, 192, 737–752. [Google Scholar] [CrossRef]
- Gluckman, P.D.; Butler, J.H. Parturition-related changes in insulin-like growth factors-I and -II in the perinatal lamb. J. Endocrinol. 1983, 99, 223–232. [Google Scholar] [CrossRef]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr. Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.C.; Choufani, S.; Ferreira, J.C.; Weksberg, R. Growth regulation, imprinted genes, and chromosome 11p15. 5. Pediatric Res. 2007, 61, 43–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gicquel, C.; Le Bouc, Y. Hormonal regulation of fetal growth. Horm. Res. 2006, 65 (Suppl. 3), 28–33. [Google Scholar] [CrossRef]
- Holly, J.M.P.; Biernacka, K.; Perks, C.M. The Neglected Insulin: IGF-II, a Metabolic Regulator with Implications for Diabetes, Obesity, and Cancer. Cells 2019, 8, 1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakar, S.; Werner, H.; Rosen, C.J. Insulin-like growth factors: Actions on the skeleton. J. Mol. Endocrinol. 2018, 61, T115–T137. [Google Scholar] [CrossRef]
- Devedjian, J.C.; George, M.; Casellas, A.; Pujol, A.; Visa, J.; Pelegrín, M.; Gros, L.; Bosch, F. Transgenic mice overexpressing insulin-like growth factor-II in beta cells develop type 2 diabetes. J. Clin. Investig. 2000, 105, 731–740. [Google Scholar] [CrossRef] [Green Version]
- Zaina, S.; Pettersson, L.; Thomsen, A.B.; Chai, C.-M.; Qi, Z.; Thyberg, J.; Nilsson, J. Shortened life span, bradycardia, and hypotension in mice with targeted expression of an Igf2 transgene in smooth muscle cells. Endocrinology 2003, 144, 2695–2703. [Google Scholar] [CrossRef] [Green Version]
- Constância, M.; Hemberger, M.; Hughes, J.; Dean, W.; Ferguson-Smith, A.; Fundele, R.; Stewart, F.; Kelsey, G.; Fowden, A.; Sibley, C.; et al. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature 2002, 417, 945–948. [Google Scholar] [CrossRef]
- Ferron, S.; Radford, E.; Domingo-Muelas, A.; Kleine, I.; Ramme, A.; Gray, D.; Sandovici, I.; Constancia, M.; Ward, A.; Menheniott, T. Differential genomic imprinting regulates paracrine and autocrine roles of IGF2 in mouse adult neurogenesis. Nat. Commun. 2015, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Charalambous, M.; Menheniott, T.R.; Bennett, W.R.; Kelly, S.M.; Dell, G.; Dandolo, L.; Ward, A. An enhancer element at the Igf2/H19 locus drives gene expression in both imprinted and non-imprinted tissues. Dev. Biol. 2004, 271, 488–497. [Google Scholar] [CrossRef]
- Feil, R.; Walter, J.; Allen, N.D.; Reik, W. Developmental control of allelic methylation in the imprinted mouse Igf2 and H19 genes. Development 1994, 120, 2933–2943. [Google Scholar] [PubMed]
- DeChiara, T.M.; Robertson, E.J.; Efstratiadis, A. Parental imprinting of the mouse insulin-like growth factor II gene. Cell 1991, 64, 849–859. [Google Scholar] [CrossRef]
- Stylianopoulou, F.; Herbert, J.; Soares, M.B.; Efstratiadis, A. Expression of the insulin-like growth factor II gene in the choroid plexus and the leptomeninges of the adult rat central nervous system. Proc. Natl. Acad. Sci. USA 1988, 85, 141–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegler, A.N.; Schneider, J.S.; Qin, M.; Tyler, W.A.; Pintar, J.E.; Fraidenraich, D.; Wood, T.L.; Levison, S.W. IGF-II promotes stemness of neural restricted precursors. Stem Cells 2012, 30, 1265–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracko, O.; Singer, T.; Aigner, S.; Knobloch, M.; Winner, B.; Ray, J.; Clemenson, G.D.; Suh, H.; Couillard-Despres, S.; Aigner, L. Gene expression profiling of neural stem cells and their neuronal progeny reveals IGF2 as a regulator of adult hippocampal neurogenesis. J. Neurosci. 2012, 32, 3376–3387. [Google Scholar] [CrossRef] [Green Version]
- Lehtinen, M.K.; Zappaterra, M.W.; Chen, X.; Yang, Y.J.; Hill, A.D.; Lun, M.; Maynard, T.; Gonzalez, D.; Kim, S.; Ye, P.; et al. The cerebrospinal fluid provides a proliferative niche for neural progenitor cells. Neuron 2011, 69, 893–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegler, A.N.; Levison, S.W.; Wood, T.L. Insulin and IGF receptor signalling in neural-stem-cell homeostasis. Nat. Rev. Endocrinol. 2015, 11, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, A.N.; Feng, Q.; Chidambaram, S.; Testai, J.M.; Kumari, E.; Rothbard, D.E.; Constancia, M.; Sandovici, I.; Cominski, T.; Pang, K.; et al. Insulin-like Growth Factor II: An Essential Adult Stem Cell Niche Constituent in Brain and Intestine. Stem Cell Rep. 2019, 12, 816–830. [Google Scholar] [CrossRef] [Green Version]
- Forbes, B.E.; McCarthy, P.; Norton, R.S. Insulin-like growth factor binding proteins: A structural perspective. Front. Endocrinol. 2012, 3, 38. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.; Jones, E.Y.; Forbes, B.E. Interactions of IGF-II with the IGF2R/cation-independent mannose-6-phosphate receptor mechanism and biological outcomes. Vitam. Horm. 2009, 80, 699–719. [Google Scholar] [CrossRef]
- Brown, J.; Delaine, C.; Zaccheo, O.J.; Siebold, C.; Gilbert, R.J.; Van Boxel, G.; Denley, A.; Wallace, J.C.; Hassan, A.B.; Forbes, B.E. Structure and functional analysis of the IGF-II/IGF2R interaction. EMBO J. 2008, 27, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Zha, J.; Lackner, M.R. Targeting the insulin-like growth factor receptor-1R pathway for cancer therapy. Clin. Cancer Res. 2010, 16, 2512–2517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belfiore, A.; Malaguarnera, R. Insulin receptor and cancer. Endocr. Relat. Cancer 2011, 18, R125–R147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livingstone, C. IGF2 and cancer. Endocr. Relat. Cancer 2013, 20, R321–R339. [Google Scholar] [CrossRef] [Green Version]
- Pollak, M. Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 2008, 8, 915. [Google Scholar] [CrossRef] [PubMed]
- Samani, A.A.; Yakar, S.; LeRoith, D.; Brodt, P. The role of the IGF system in cancer growth and metastasis: Overview and recent insights. Endocr. Rev. 2007, 28, 20–47. [Google Scholar] [CrossRef]
- Sciacca, L.; Costantino, A.; Pandini, G.; Mineo, R.; Frasca, F.; Scalia, P.; Sbraccia, P.; Goldfine, I.D.; Vigneri, R.; Belfiore, A. Insulin receptor activation by IGF-II in breast cancers: Evidence for a new autocrine/paracrine mechanism. Oncogene 1999, 18, 2471–2479. [Google Scholar] [CrossRef]
- Chao, W.; D’Amore, P.A. IGF2: Epigenetic regulation and role in development and disease. Cytokine Growth Factor Rev. 2008, 19, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Harris, L.K.; Westwood, M. Biology and significance of signalling pathways activated by IGF-II. Growth Factors 2012, 30, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Lui, J.C.; Baron, J. Evidence that Igf2 down-regulation in postnatal tissues and up-regulation in malignancies is driven by transcription factor E2f3. Proc. Natl. Acad. Sci. USA 2013, 110, 6181–6186. [Google Scholar] [CrossRef] [Green Version]
- Arcaro, A. Targeting the insulin-like growth factor-1 receptor in human cancer. Front. Pharmacol. 2013, 4, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallagher, E.J.; LeRoith, D. The proliferating role of insulin and insulin-like growth factors in cancer. Trends Endocrinol. Metab. TEM 2010, 21, 610–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denley, A.; Wallace, J.C.; Cosgrove, L.J.; Forbes, B.E. The insulin receptor isoform exon 11- (IR-A) in cancer and other diseases: A review. Horm. Metab. Res. 2003, 35, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Giorgino, F.; Belfiore, A.; Milazzo, G.; Costantino, A.; Maddux, B.; Whittaker, J.; Goldfine, I.D.; Vigneri, R. Overexpression of insulin receptors in fibroblast and ovary cells induces a ligand-mediated transformed phenotype. Mol. Endocrinol. 1991, 5, 452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vella, V.; Nicolosi, M.L.; Cantafio, P.; Massimino, M.; Lappano, R.; Vigneri, P.; Ciuni, R.; Gangemi, P.; Morrione, A.; Malaguarnera, R.; et al. DDR1 regulates thyroid cancer cell differentiation via IGF-2/IR-A autocrine signaling loop. Endocr. Relat. Cancer 2019, 26, 197–214. [Google Scholar] [CrossRef]
- Tominaga, K.; Shimamura, T.; Kimura, N.; Murayama, T.; Matsubara, D.; Kanauchi, H.; Niida, A.; Shimizu, S.; Nishioka, K.; Tsuji, E.I.; et al. Addiction to the IGF2-ID1-IGF2 circuit for maintenance of the breast cancer stem-like cells. Oncogene 2017, 36, 1276–1286. [Google Scholar] [CrossRef] [Green Version]
- Laviola, L.; Natalicchio, A.; Giorgino, F. The IGF-I signaling pathway. Curr. Pharm. Des. 2007, 13, 663–669. [Google Scholar] [CrossRef]
- Siddle, K. Signalling by insulin and IGF receptors: Supporting acts and new players. J. Mol. Endocrinol. 2011, 47, R1–R10. [Google Scholar] [CrossRef] [Green Version]
- Cabail, M.Z.; Li, S.; Lemmon, E.; Bowen, M.E.; Hubbard, S.R.; Miller, W.T. The insulin and IGF1 receptor kinase domains are functional dimers in the activated state. Nat. Commun. 2015, 6, 6406. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, S.R.; Wei, L.; Ellis, L.; Hendrickson, W.A. Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature 1994, 372, 746–754. [Google Scholar] [CrossRef]
- De Meyts, P.; Whittaker, J. Structural biology of insulin and IGF1 receptors: Implications for drug design. Nat. Rev. Drug Discov. 2002, 1, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Hanke, S.; Mann, M. The phosphotyrosine interactome of the insulin receptor family and its substrates IRS-1 and IRS-2. Mol. Cell. Proteom. 2009, 8, 519–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Hakuno, F.; Takahashi, S.I. IGF1 receptor signaling pathways. J. Mol. Endocrinol. 2018, 61, T69–T86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, A.M.; Forbes, B.E.; Aplin, S.E.; Wallace, J.C.; Francise, G.L.; Norton, R.S. Solution structure of human insulin-like growthfactor II. Relationship to receptor and binding protein interactions. J. Mol. Biol. 1995, 248, 385–401. [Google Scholar] [CrossRef]
- Smith, G.D.; Pangborn, W.A.; Blessing, R.H. The structure of T6 human insulin at 1.0 Å resolution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2003, 59, 474–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKern, N.M.; Lawrence, M.C.; Streltsov, V.A.; Lou, M.Z.; Adams, T.E.; Lovrecz, G.O.; Elleman, T.C.; Richards, K.M.; Bentley, J.D.; Pilling, P.A.; et al. Structure of the insulin receptor ectodomain reveals a folded-over conformation. Nature 2006, 443, 218–221. [Google Scholar] [CrossRef]
- Xu, Y.; Kong, G.K.; Menting, J.G.; Margetts, M.B.; Delaine, C.A.; Jenkin, L.M.; Kiselyov, V.V.; De Meyts, P.; Forbes, B.E.; Lawrence, M.C. How ligand binds to the type 1 insulin-like growth factor receptor. Nat. Commun. 2018, 9, 821. [Google Scholar] [CrossRef] [Green Version]
- Kavran, J.M.; McCabe, J.M.; Byrne, P.O.; Connacher, M.K.; Wang, Z.; Ramek, A.; Sarabipour, S.; Shan, Y.; Shaw, D.E.; Hristova, K. How IGF-1 activates its receptor. Elife 2014, 3, e03772. [Google Scholar] [CrossRef]
- Lou, M.; Garrett, T.P.; McKern, N.M.; Hoyne, P.A.; Epa, V.C.; Bentley, J.D.; Lovrecz, G.O.; Cosgrove, L.J.; Frenkel, M.J.; Ward, C.W. The first three domains of the insulin receptor differ structurally from the insulin-like growth factor 1 receptor in the regions governing ligand specificity. Proc. Natl. Acad. Sci. USA 2006, 103, 12429–12434. [Google Scholar] [CrossRef] [Green Version]
- Garrett, T.P.; McKern, N.M.; Lou, M.; Frenkel, M.J.; Bentley, J.D.; Lovrecz, G.O.; Elleman, T.C.; Cosgrove, L.J.; Ward, C.W. Crystal structure of the first three domains of the type-1 insulin-like growth factor receptor. Nature 1998, 394, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Weis, F.; Menting, J.G.; Margetts, M.B.; Chan, S.J.; Xu, Y.; Tennagels, N.; Wohlfart, P.; Langer, T.; Muller, C.W.; Dreyer, M.K.; et al. The signalling conformation of the insulin receptor ectodomain. Nat. Commun. 2018, 9, 4420. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Kirk, N.S.; Venugopal, H.; Margetts, M.B.; Croll, T.I.; Sandow, J.J.; Webb, A.I.; Delaine, C.A.; Forbes, B.E.; Lawrence, M.C. How IGF-II Binds to the Human Type 1 Insulin-like Growth Factor Receptor. Structure 2020. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Choi, E.; Yu, H.; Bai, X.-c. Structural basis of the activation of type 1 insulin-like growth factor receptor. Nat. Commun. 2019, 10, 4567. [Google Scholar] [CrossRef] [PubMed]
- De Meyts, P. Insulin/receptor binding: The last piece of the puzzle. BioEssays 2015, 37, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Alvino, C.L.; McNeil, K.A.; Ong, S.C.; Delaine, C.; Booker, G.W.; Wallace, J.C.; Whittaker, J.; Forbes, B.E. A novel approach to identify two distinct receptor binding surfaces of insulin-like growth factor II. J. Biol. Chem. 2009, 284, 7656–7664. [Google Scholar] [CrossRef] [Green Version]
- Gauguin, L.; Delaine, C.; Alvino, C.L.; McNeil, K.A.; Wallace, J.C.; Forbes, B.E.; De Meyts, P. Alanine scanning of a putative receptor binding surface of insulin-like growth factor-I. J. Biol. Chem. 2008, 283, 20821–20829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristensen, C.; Kjeldsen, T.; Wiberg, F.C.; Schäffer, L.; Hach, M.; Havelund, S.; Bass, J.; Steiner, D.F.; Andersen, A.S. Alanine scanning mutagenesis of insulin. J. Biol. Chem. 1997, 272, 12978–12983. [Google Scholar] [CrossRef] [Green Version]
- Gutmann, T.; Schafer, I.B.; Poojari, C.; Brankatschk, B.; Vattulainen, I.; Strauss, M.; Coskun, U. Cryo-EM structure of the complete and ligand-saturated insulin receptor ectodomain. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef] [Green Version]
- Uchikawa, E.; Choi, E.; Shang, G.; Yu, H.; Bai, X.C. Activation mechanism of the insulin receptor revealed by cryo-EM structure of the fully liganded receptor-ligand complex. Elife 2019, 8. [Google Scholar] [CrossRef]
- Kiselyov, V.V.; Versteyhe, S.; Gauguin, L.; De Meyts, P. Harmonic oscillator model of the insulin and IGF1 receptors’ allosteric binding and activation. Mol. Syst. Biol. 2009, 5, 243. [Google Scholar] [CrossRef] [PubMed]
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr. Rev. 2009, 30, 586–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gualberto, A.; Pollak, M. Emerging role of insulin-like growth factor receptor inhibitors in oncology: Early clinical trial results and future directions. Oncogene 2009, 28, 3009–3021. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IGF-II | IGF-I [57,65] | Insulin [64] | ||||
|---|---|---|---|---|---|---|
| Site 1 | Cys9 | [63] | Cys6 | [64] | CysB7 | [62] |
| Leu13 | Leu10 | LeuB11 | ||||
| Leu17 | Leu14 | LeuB15 | ||||
| Asp23 | Asp20 | GluB21 | ||||
| Ser29 | Asn26 | ProB28 | ||||
| Arg30 | Lys27 | |||||
| Thr58 | Met59 | AsnA18 | ||||
| Thr62 | Lys65 | |||||
| Val14 | [1,63] | Val11 | [1,64] | ValB12 | [62,65] | |
| Gln18 | Gln15 | TyrB16 | ||||
| Gly25 | Gly22 | GlyB23 | ||||
| Phe26 | Phe23 | PheB24 | ||||
| Tyr27 | Tyr24 | PheB25 | ||||
| Phe28 | Phe25 | TyrB26 | ||||
| Tyr31 | ||||||
| Arg36 | ||||||
| Arg37 | ||||||
| Gly41 | Gly42 | GlyA1 | ||||
| Ile42 | Ile43 | IleA2 | ||||
| Val43 | Val44 | ValA3 | ||||
| - | Asp45 a | GluA4 | ||||
| Glu45 | Glu46 | GluA5 | ||||
| Phe48 | Phe49 | ThrA8 | ||||
| Tyr59 | Tyr60 | TyrA19 | ||||
| Ala61 | Ala62 | AsnA21 | ||||
| Site 2 | Glu6 | [63] | Glu3 | [1] | GlnB4 | [62] |
| Thr7 | [63] | Thr4 | [64] | HisB5 | [62] | |
| Cys9 | [63] | Cys6 | [64] | CysB7 | [62] | |
| Glu12 | [63,66] | Glu9 # | [64,67] | HisB10 * | [62,68] | |
| Asp15 | [66] | Asp12 | [64,67] | GluB13 * | [62,68] | |
| Phe19 | [66] | Phe16# | [64,67] | LeuB17 * | [62,68] | |
| Cys47 | [63] | Cys48 | [64] | CysA7 | [62] | |
| - | - | SerA12 | [62,68] | |||
| Leu53 | [66] | Leu54# | [67] | LeuA13 * | [62,68] | |
| Glu57 | [66] | Glu58# | [67] | GluA17 * | [62,68] | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blyth, A.J.; Kirk, N.S.; Forbes, B.E. Understanding IGF-II Action through Insights into Receptor Binding and Activation. Cells 2020, 9, 2276. https://doi.org/10.3390/cells9102276
Blyth AJ, Kirk NS, Forbes BE. Understanding IGF-II Action through Insights into Receptor Binding and Activation. Cells. 2020; 9(10):2276. https://doi.org/10.3390/cells9102276
Chicago/Turabian StyleBlyth, Andrew J., Nicholas S. Kirk, and Briony E. Forbes. 2020. "Understanding IGF-II Action through Insights into Receptor Binding and Activation" Cells 9, no. 10: 2276. https://doi.org/10.3390/cells9102276
APA StyleBlyth, A. J., Kirk, N. S., & Forbes, B. E. (2020). Understanding IGF-II Action through Insights into Receptor Binding and Activation. Cells, 9(10), 2276. https://doi.org/10.3390/cells9102276