1. Introduction
Nowadays, it is well known that the circadian clock plays a paramount role in living organisms. It enables adaptation of behavior and physiology, including metabolism, body temperature, sleep–wake cycle and hormone secretion to daily environmental changes like the light/dark cycle [
1,
2]. The circadian clock shows a hierarchical structure to maintain the rhythm throughout an organism, with the central circadian pacemaker (located in the suprachiasmatic nucleus (SCN)) on top of the hierarchy, controlling the rhythmicity of downstream oscillators in the periphery [
2]. The maintenance of a rhythm with a period length of about 24 h is ensured by self-sustained transcriptional–translational feedback loops [
3]. This involves a complex molecular machinery resulting in the rhythmic expression of so-called clock genes, including the trans-activating components CLOCK and BMAL1 (brain and muscle ARNT-Like 1), and the trans-inhibiting components period (isoforms PER1-3) and cryptochrome (isoforms CRY1 and 2).
Neurosteroids, a specific class of steroids characterized by de novo synthesis in the nervous system, were discovered decades ago [
4], and are known to exert brain-specific functions [
5]. The first step of neurosteroid production takes place in mitochondria with the transfer of cholesterol from the outer to the inner mitochondrial membrane. The translocator protein TSPO is a component of the mitochondrial cholesterol import machinery. Cholesterol is then converted to pregnenolone (P5), the precursor of all steroids [
6]. P5 is further metabolized into subsequent neurosteroids either in mitochondria or in the endoplasmic reticulum (ER) major pathways. In the brain, neurosteroids are synthesized by both neurons and glial cells [
5]. These molecules play an important role in the nervous system as they can bind either on nuclear receptors to regulate gene expression or act through the activation of membrane-associated signaling pathways to modulate neuronal function [
7]. Indeed, neurosteroids—such as pregnenolone, allopregnanolone, or dehydroepiandrosterone—modulate neurotransmission by acting as allosteric modulators of neurotransmitter receptors (e.g., NMDA or GABA receptors) [
8]. As they regulate important processes, such as learning and memory, they represent an attractive therapeutic target in brain disorders, including Alzheimer’s disease (AD), epilepsy, and mood disorders [
8,
9]. Strikingly, the link between neurosteroid synthesis and the circadian clock remains elusive until today.
By anticipating the time of day, the circadian clock orchestrates numerous processes in the body, especially metabolic reactions, including glucose and fatty acid metabolism [
10]. Since mitochondria are known as metabolic hubs, orchestrating a large variety of metabolic processes, it is not surprising that a lot of these processes are under the control of the circadian clock, including mitochondrial dynamics. Mitochondria are remarkably dynamic organelles that fuse and divide in order to maintain a homogenous population of the organelles [
11]. On the one hand, mitochondrial fusion allows mitochondria to interact and communicate with each other, and requires the action of two evolutionarily distinct dynamin-related GTPases, mitofusin 1 and 2 (MFN1/2, for fusion of outer membranes), and optic atrophy 1 (OPA1, for fusion of inner membranes). On the other hand, mitochondrial fission allows the renewal, redistribution, and proliferation of organelles involving the dynamin-related protein 1 (DRP1), master fission mediator, as well as fission protein 1 (FIS1).
We recently provided the first evidence that mitochondrial dynamics are controlled by the clock with a direct control mechanism via phosphorylation of DRP1 at serine 637 [
12]. In particular, we showed that mitochondrial energy metabolism (oxidative phosphorylation, adenosine triphosphate (ATP) production) follows a circadian rhythm, and that suppression of DRP1 activity eliminates circadian ATP production. In addition, blocking DRP1 function impaired the core circadian clock, suggesting feedback regulation of mitochondrial metabolism back to the molecular clock.
Mitochondria are not only the powerhouse of cells, but they are also involved in numerous of cellular functions, including steroidogenesis. Hints for a link between steroid synthesis and mitochondrial architecture were shown in peripheral steroidogenic tissues by Duarte and collaborators who demonstrated that mitochondrial fusion directly correlates with increased steroid production, particularly progesterone, in Leydig cells [
13,
14]. They showed that MFN2 knockdown significantly decreased mitochondrial fusion and steroid production, suggesting that mitochondrial fusion is required for steroidogenesis.
Thus, mitochondrial dynamics seem to play a role in steroid production, while being under control of the circadian clock. However, nothing is known about the implication of the circadian clock in mitochondrial neurosteroidogenesis. Therefore, we decided to first investigate whether mitochondrial neurosteroid production follows a circadian rhythm, and then to explore potential links with the clock-controlled mitochondrial architecture. For this purpose, we used synchronized human A172 glioma cells, which are steroidogenic cells with a functional core molecular clock [
15]. Key findings were recapitulated in mouse brain lysates.
2. Materials and Methods
2.1. Cell Culture
Human A172 glioma cells, A172 glioma cells transfected with luciferase under control of the BMAL1 promoter (generously provided by Steven Brown from the University of Zurich), wild-type Hela cells, and DRP1 KO HeLa cells (generously provided by Stephan Frank from the University of Basel) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma, Buchs SG, Switzerland, no. D6429) supplemented with 1% penicillin and streptomycin (Sigma, no. P4333), 1% GlutaMax (Thermo Fisher Scientific, Allschwil, Switzerland, no. 35050061), and 10% foetal bovine serum (FBS) (Sigma, no. F9665) at 37 °C in a humidified atmosphere containing 7.5% CO2. Cell passaging was performed twice a week. Human A172 glioma cells transfected with mitochondria-targeted GFP were maintained under the same conditions.
2.2. Mouse Brain Homogenates
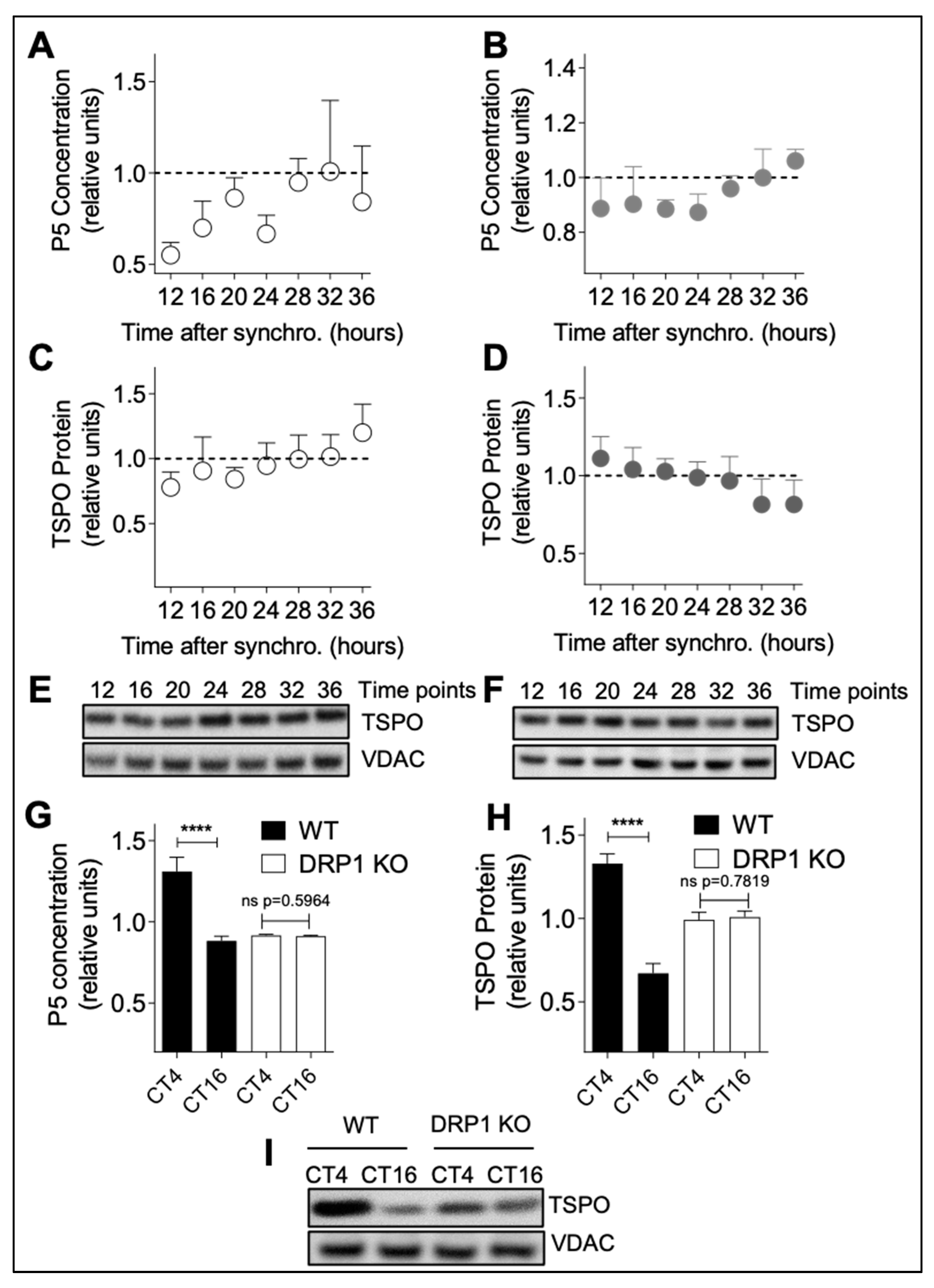
Drp1
flx/flx CreERT2 (DRP1 KO) mice were obtained by crossing Drp1
flx/flx mice with mice expressing an inducible Cre recombinase transgene under the control of the CamKIIa promoter (Cre+), which is active in the hippocampus and the cortex of adult mice (from the European Mouse Mutant Archive EMMA strain 02125) [
16]. At 8 weeks of age, mice were injected intraperitoneally with 1 mg tamoxifen twice daily for five consecutive days to induce recombination of the Drp1 locus [
17]. mPer1/mPer2 double-mutant (PER1/PER2) mice were generated as described previously [
18]. Mouse brain samples were kindly provided by Jürgen A. Ripperger and Urs Albrecht (University of Fribourg, Switzerland).
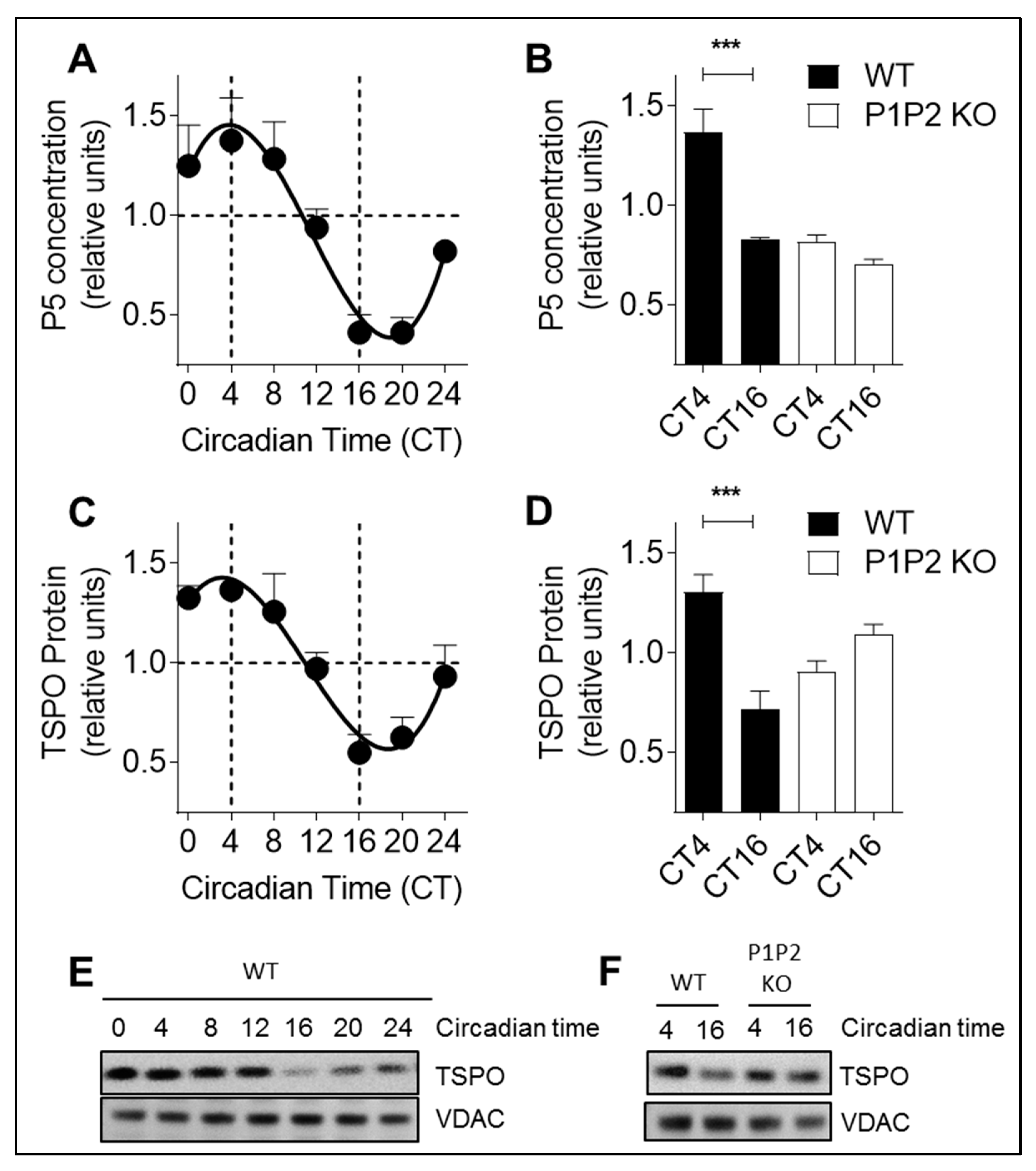
Animal experiments were performed according to standardized experimental designs assessing circadian rhythms in mice [
19]. Mice were housed at 24 °C on a conventional 12:12 light/dark cycle until the age of 10–12 weeks. Then, they were placed in constant darkness 5 days before the beginning of the experiment with free access to ordinary food (normal chow) and water. Indeed, organisms kept under constant condition (12:12 dark/dark condition) display persisting and intrinsic circadian rhythms (of about 24 h) that may differ from daily rhythms (rhythms synchronized by external clues, such as light/dark cycles). Under constant dark conditions, the time is expressed in circadian time (CT) units, where CT0 is the start of subjective daytime and CT12 is the start of subjective night-time (corresponding to the onset of activity for nocturnal animals like mice) [
19]. Animals were sacrificed every 4 h over 24 h and brains were harvested. All experiments were performed in accordance with Swiss animal protection legislation and with approval of the Basel Veterinary Committee for Animal Care: permits 2393 and 23288. The date when the ethical approval was obtained is 22 February 2012 for both permits.
Brain homogenates of wild-type mice, PER1/PER2 and DRP1 knock-out mice were prepared as previously described [
12]. Brains were dissected on ice and washed in ice-cold buffer (210 mM mannitol, 70 mM sucrose, 10 mM Hepes, 1 mM EDTA, 0.45% BSA, 0.5 mM DTT, and Complete Protease Inhibitor mixture tablets (Roche Diagnostics, Sigma, no. 11836153001)). After removing the cerebellum, tissue samples were homogenized in 2 mL of buffer with a glass homogenizer (10–15 strokes, 400 rpm), and protein concentration was determined before pregnenolone and western blot analysis.
2.3. Transfection with Mitochondria-Targeted GFP
To visualize mitochondrial structures, A172 glioma cells were transfected with plasmid DNA of a mitochondria-targeted GFP (mito-GFP, mitochondria targeting sequence in pEGFP-N1 plasmid for mammalian expression) [
20] using transfection reagent Lipofectamine 2000 (Thermo Fisher Scientific, no. 11668027). Cells were seeded 24 h before transfection at a density of 5 × 10
5 cells in 24-wells plates (Falcon, Amsterdam, The Netherlands, no. 353047) to reach a confluency of 70–90%. At the day of transfection, medium was exchanged with OptiMEM I reduced serum medium (Thermo Fisher Scientific, no. 31985070) containing 5% FBS and no antibiotics. Plasmid DNA was diluted in OptiMEM to a final concentration of 5 μg. Then, OptiMEM containing 4 μL Lipofectamine 2000 and diluted plasmid DNA were mixed 1:1, incubated for 5 min at room temperature and added to cells. After 6 h incubation at 37 °C, medium was changed to OptiMEM containing 5% FBS. After 48 h medium was changed to selection medium (DMEM with 1% Penicillin Streptomycin and GlutaMax, 10% FBS and 300 μg/mL of antibiotic Geneticin (Thermo Fisher Scientific, no. 11811031). Stably transfected cells were selected for two months.
2.4. Circadian Clock Synchronization
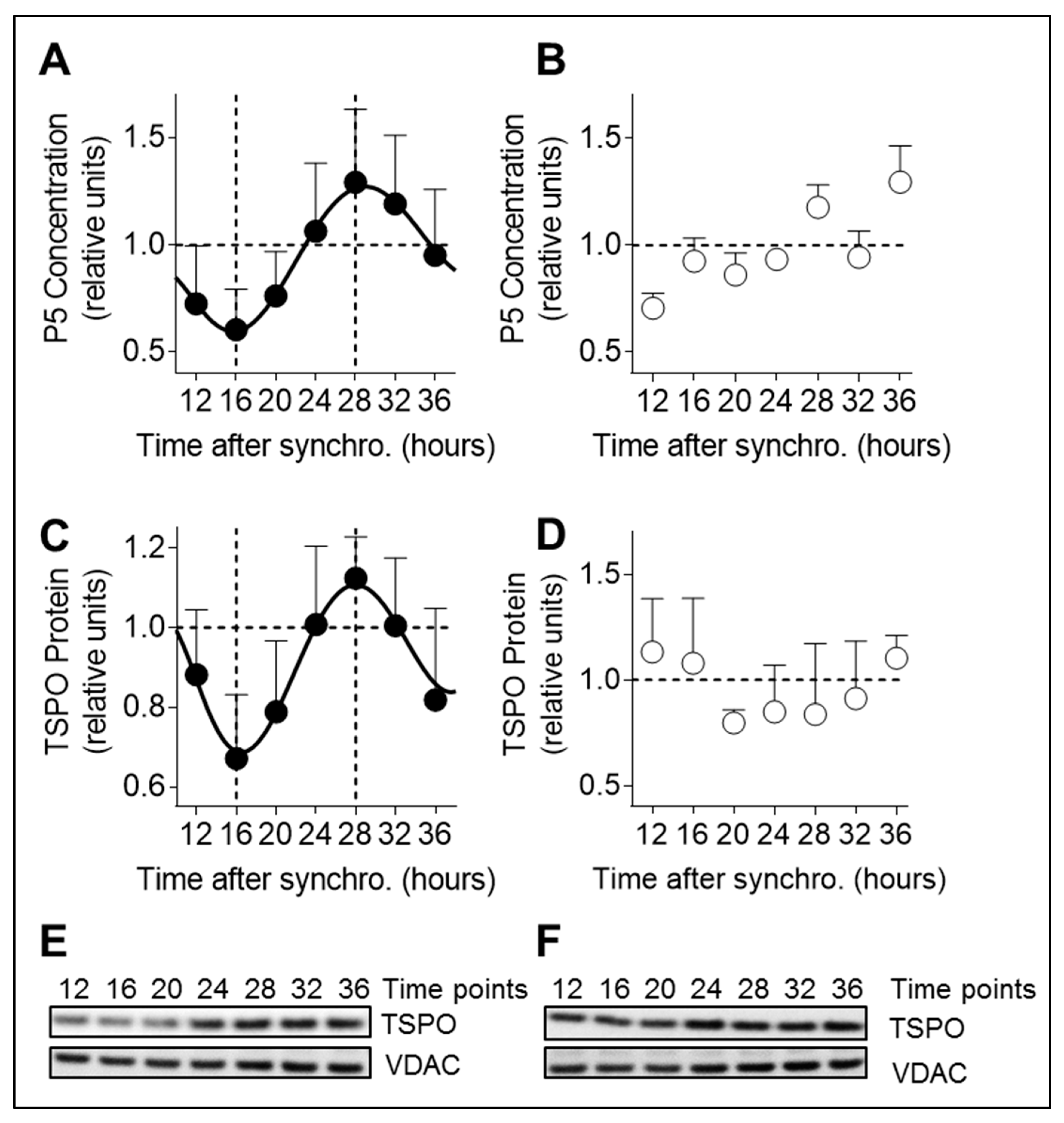
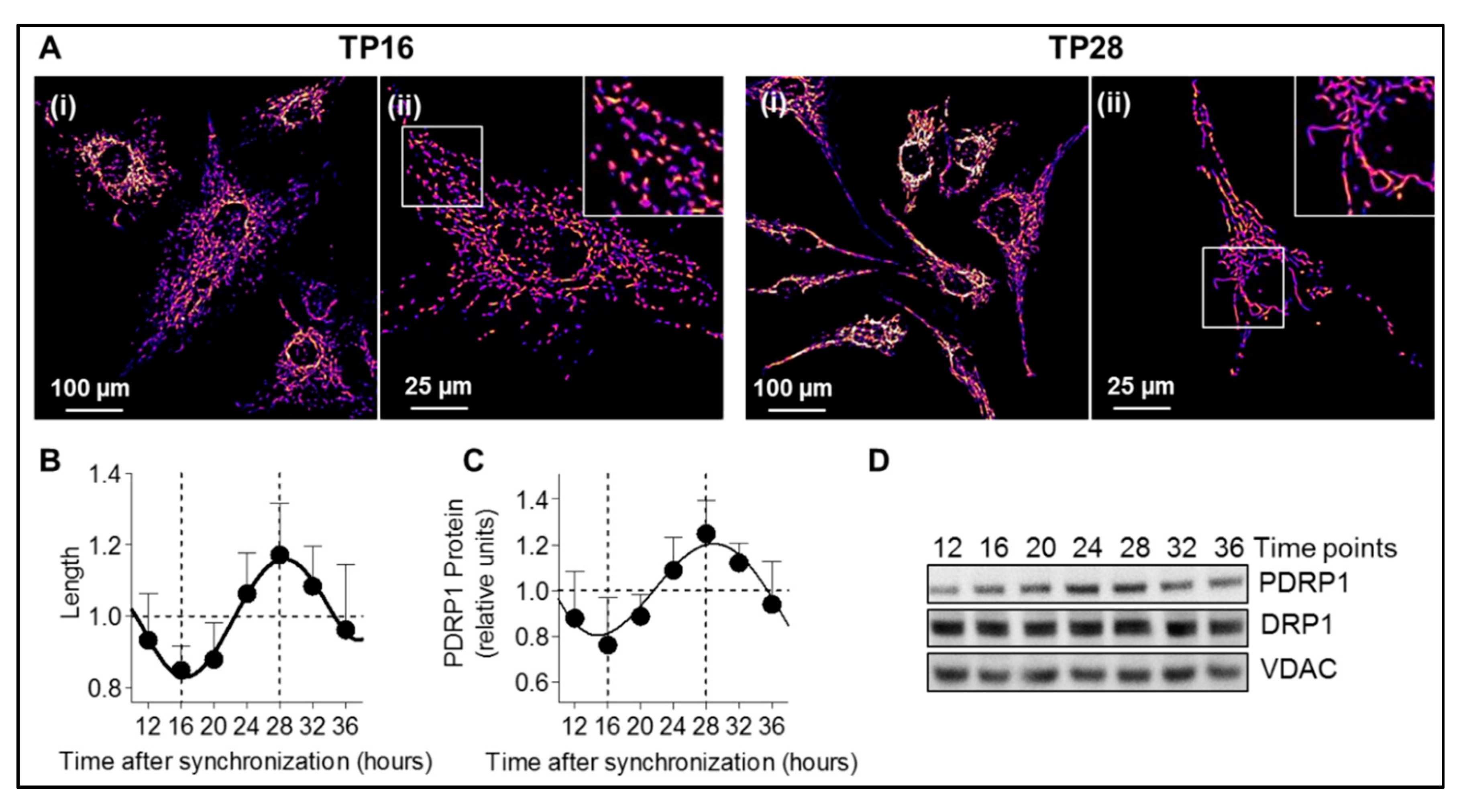
To analyze all parameters (mitochondrial morphology, phosphorylation of DRP1 at serine 637, pregnenolone production, and TSPO protein level) in a circadian time dependent manner, circadian clocks of A172 glioma cells were synchronized using a horse serum shock [
21]. Cells were incubated 2 h at 37 °C with 50% horse serum in DMEM, then washed three times with PBS and placed in DMEM medium supplemented with 1% penicillin/streptomycin, 1% GlutaMax, and 5% FBS at 37 °C in a humidified atmosphere containing 7.5% CO2. Corresponding experiments were then performed from 8 h to 48 h post-synchronization in 4 h intervals.
2.5. MDIVI-1 Treatment Paradigm
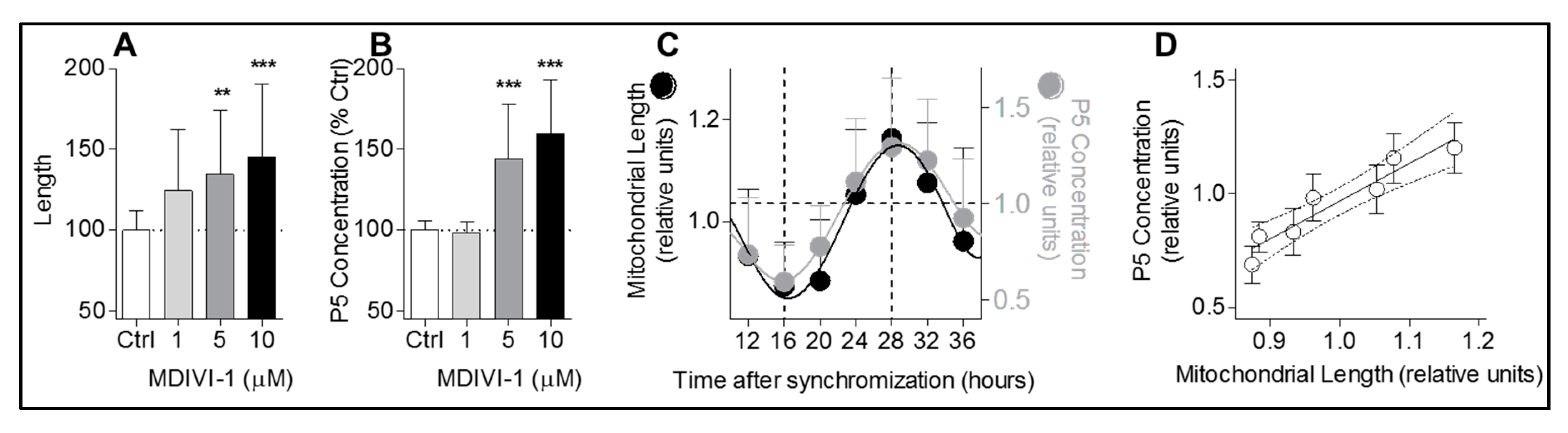
For the purpose of this study, the small molecule inhibitor MDIVI-1 (Sigma-Aldrich, Buchs SG, Switzerland, no. MO199) was used to disturb mitochondrial dynamics in two experimental approaches. First, in a non-circadian approach, pregnenolone (P5) was quantified after MDIVI-1 administration. Cells were treated with different concentration of MDIVI-1 and two inhibitors of downstream enzymes of the neurosteroid synthesis pathway to ensure pregnenolone accumulation: abiraterone (MedChem Express, Bromma, Sweden, no. HY-75054) which inhibits 17α-hydroxylase, and trilostane (Sigma-Aldrich, no. SML0141), an inhibitor of 3β-hydroxysteroid dehydrogenase. Treatment was performed in a salt buffer (140 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 1 mM MgSO4 × 7H2O, 10 mM HEPES, 10 mM D-glucose, and 0.1% BSA, pH adjusted to 7.4) with 1, 5, and 10 μM MDIVI-1, 100 nM abiraterone, and 25 μM trilostane for 2 h at 37 °C. Directly after, pregnenolone concentration was quantified in supernatants (see pregnenolone quantification) and mitochondrial morphology was investigated by confocal microscopy. Secondly, in a circadian approach, MDIVI-1 was applied to chronically disturb mitochondrial network morphology. After synchronization, medium was supplemented with 5 μM MDIVI-1 and after 24 h cells were retreated with 5 μM MDIVI-1 to ensure a chronically disturbed mitochondrial network. During this chronic treatment, cells were harvested from 8 to 48 h after synchronization with 4 h intervals for pregnenolone and TSPO protein quantification.
2.6. Confocal Microscopy
Mitochondrial morphology was assessed in human A172 glioma cells transfected with mito-GFP plasmid. Coverslips were coated with 0.05 mg/mL collagen (BD Biosciences Discovery Labware, Allschwil, Switzerland, no. 354236) for 3 h at room temperature. Coverslips were washed with ultrapure water and cells were added on pre-coated coverslips. Cells were synchronized 24 h later and were fixed 15 min with 1 mL of 2% paraformaldehyde (Sigma-Aldrich, no. P6148) from 8 h to 48 h with 4 h intervals. Cells were mounted on glass slides using mounting medium (Thermo Fisher Scientific, no. P36930) and analyzed with a confocal microscope (Leica DMI4000B confocal microscope, Leica Microsystems (Heerbrugg, Switzerland) with Leica Application Suite Advanced Fluorescence (Leica LAS AF) software version 2.51.6757). Images were acquired using a HCX PL APO objective with a magnification of 63x/1.10-0.60 and oil immersion (Leica Microsystems SPE) with Type F immersion oil (Leica Microsystems, no. 11513859). For mito-GFP detection an excitation beam splitter DD 488/365 was used, and emission was detected in a bandwidth of 508–526 nm.
2.7. Mitochondrial Morphology Quantification
Mitochondrial shape parameters were quantified using the open-source software package ImageJ and as previously described [
22]. Briefly, images were background-subtracted (rolling ball radius = 50 pixels) and uneven labeling of mitochondria was improved through local contrast enhancement using contrast-limited adaptive histogram equalization (“CLAHE”). To segment mitochondria, the “Tubeness” filter was applied. After setting an automated threshold, the “Analyze Particles” plugin was used to determine the area and perimeter of individual mitochondria and the “Skeletonize” function was used to measure mitochondrial length.
Three parameters were assessed, namely:
- -
Mitochondrial length: the length reports the mitochondrial length or elongation in pixel, after the mitochondria are reduced to a single-pixel-wide shape (“Skeletonize” function on ImageJ).
- -
Form factor (FF): The form factor value describes the particle’s shape complexity of the mitochondria, as the inverse of the circularity.
- -
Aspect ratio: this parameter is independent of area and perimeter and is defined as the ratio of the major on the minor axis.
2.8. Gel Electrophoresis and Immunoblotting
A172 glioma cells were seeded one day before synchronization at a density of 0.15 × 106 cells/mL in DMEM + 1% penicillin/streptomycin + 1% GlutaMax + 10% FBS in 60 mm dishes (Falcon, no. 353004) and were harvested in 100 μL ice-cold protein lysis buffer (150 mM Tris Ultrapure, 150 mM NaCl, 1% Nonidet-P40, 0.1% SDS, 2 mM EDTA) after a washing step with cold PBS- from 8 to 48 h post-synchronization in 4 h intervals. Shortly before harvesting, proteases and phosphatases inhibitors were added to the lysis buffer (complete Mini tablet, Roche Diagnostics, Sigma, no. 11836153001; 1 mM Na3VO4 and 5 mM NaF). Equal protein amounts of each time points were then used. Cell lysates were added to 12.5 μL NUPAGE LDS sample buffer (4×) (Thermo Fisher Scientific, no. NP0007), 2.5 μL DTT (50 mM) (Sigma-Aldrich, no. 233155) and ultrapure water (final volume 50 μl). Samples were heated for 5 min at 95 °C and centrifuged at 10,000 rpm for 5 min at 4 °C. Gel electrophoresis was performed on the NUPAGE Novex 4–12% Bis-Tris Protein gel, 1.0 mm, 15 well (Thermo Fisher Scientific, no. NP0323BOX) according to the manufacturer’s protocol
Before performing immunoblotting, membranes were blocked for 1 h at room temperature with blocking buffer consisting of 5% BSA (Sigma-Aldrich, no. A7906) diluted in TBS-0.1% Tween 20 (Sigma-Aldrich, no. 93773). Primary antibodies were used for immunoblotting: anti-PBR (TSPO) antibody EPR5384 (Abcam, Cambridge, UK, no. ab109497), phospho-DRP1 Ser637 (Cell Signaling Technology, Beverly, MA, United States, no. 4867), DRP1 (D6C7) Rabbit mAb (Cell Signaling Technology, no. 8570), and VDAC (Cell Signaling Technology, no. 4866). Membranes were incubated with primary antibody diluted 1:1000 (except for Anti-PBR 1:10000) in antibody incubation buffer consisting of 0.6% BSA in TBS-0.1% Tween 20 and incubated at 4 °C overnight. Then, membranes were incubated with the second antibody against rabbit IgG HRP-linked (Cell Signaling Technology, no. 7074) diluted 1:1000 in antibody incubation buffer for 1 h at room temperature. Protein bands were detected by enhanced chemiluminescent reaction using SuperSignal West Dura Extended Duration Substrate (Thermo Fisher Scientific, no. 34075). For detection, a Gene Gnome Chemiluminescent Imaging System (Syngene) was used. Image analysis was performed using ImageJ (
https://imagej.nih.gov/ij/ ImageJ with Java 1.8.0_172).
2.9. Pregnenolone Quantification
A172 glioma cells were seeded 24 h before synchronization at a density of 0.15 × 106 cells/mL in DMEM + 1% penicillin/streptomycin + 1% GlutaMax + 10% FBS in 60 mm dishes and were harvested in 250 μL PBS+ (Thermo Fisher Scientific, no. 21300-058) and lysed with ultrasound from 8 to 48 h post-synchronization in 4 h intervals. Pregnenolone concentration was quantified in cell lysates and mouse brain lysates with a pregnenolone direct ELISA (DRG, Marburg, Germany no. EIA-4170) according to instructions of the manufacturer. Measurements were then performed with a Cytation 3 Cell Imaging Multi-Mode reader (BioTek, Luzern, Switzerland).
2.10. siRNA Transfection
A172 glioma cells were seeded 24 h prior to transfection at 0.15 × 10
6 cells/mL in 60 mm dishes (Falcon, no. 353004) in DMEM + 1% penicillin/streptomycin + 1% GlutaMax + 10% FBS. Shortly before transfection, medium was changed to OptiMEM I reduced serum medium supplemented with 5% FBS and no antibiotics. For siRNA transfection the transfection reagent Lipofectamine RNAiMax (Invitrogen, Thermo Fisher Scientific, no. 13778150) was used according to the manufacturer’s protocol. First, 12 pmol of each siRNA (see
Table 1: siRNA target sequences) was diluted in OptiMEM and gently mixed. Then, 6 μL Lipofectamine RNAiMax transfection reagent was diluted in OptiMEM and incubated for 5 min at room temperature. After incubation, diluted siRNA and RNAiMax were combined 1:1 and incubated for 20 min at room temperature and added to cells (final concentration of 8 nM per siRNA). After 6 h medium was changed to OptiMEM I reduced serum medium supplemented with 5% FBS and 48 h later, cells were synchronized as described and harvested according to the parameter investigated.
2.11. Statistical and Circadian Analysis
For statistical analysis Graph Pad Prism Software was used (GraphPad Software Inc. version 5.02, San Diego, CA, USA). Cell culture data are presented as means ± SD, animal data are presented as means ± SEM. One-way ANOVA and Dunnett’s multiple comparison tests were performed to compare all columns to a control group. One-way ANOVA and Tukey’s comparison test was performed to compare all the time points, especially the peak versus trough of a circadian cycle. Student t-tests were used to compare the mean of two groups. The statistical differences are represented by the p-value: * p < 0.05, ** p < 0.01, *** p <0.001 and **** p < 0.0001.
Data from circadian experiments were considered for further analysis with a specific algorithm for rhythmic transcripts to confirm the circadian nature of the cycles. For this, the JTK-Cycle algorithm implemented in R [
23] was used as described [
24]. A
p-value of <0.05 was considered as statistically significant and therefore circadian. When data were considered as circadian, curves were generated in Graph Pad Prism using a standard curve fit function (five parameters).
The complete circadian and statistical analysis, with the corresponding
p-values, can be find in the
Supplementary Table S1.
4. Discussion
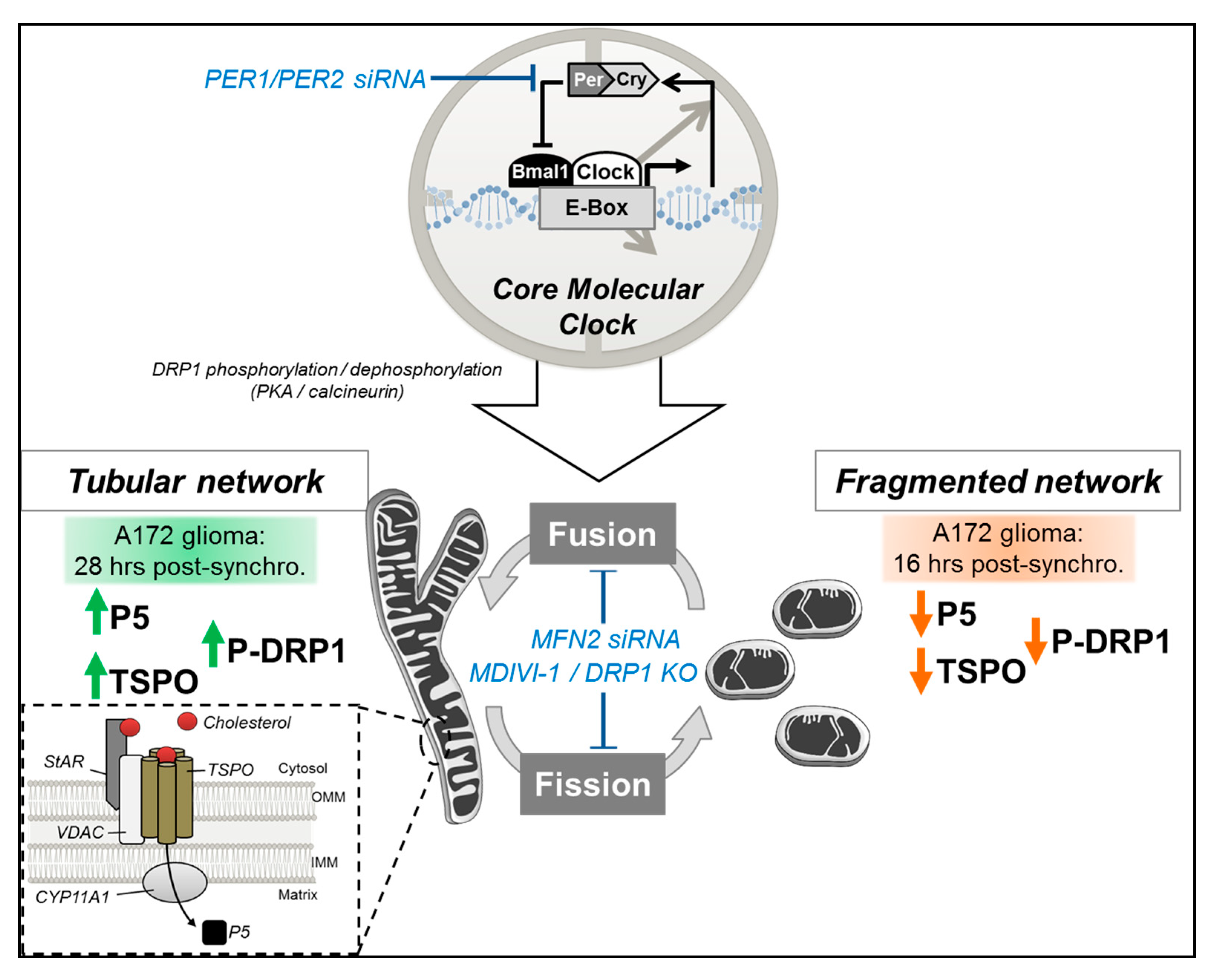
We previously showed that mitochondrial dynamics are clock-controlled via circadian regulation of DRP1 phosphorylation and that abrogation of DRP1 activity abolished the rhythmicity of mitochondrial dynamics and the associated mitochondrial bioenergetic functions, such as respiratory activity and ATP production [
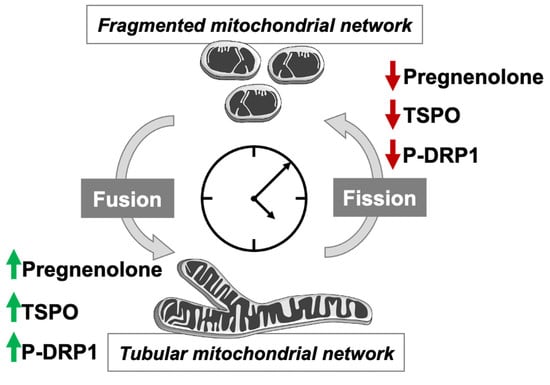
12]. Here, we aimed to investigate whether mitochondrial neurosteroid production follows a circadian rhythm, and to explore whether clock-controlled mitochondrial dynamics regulate mitochondrial neurosteroid synthesis as well. Our key findings are as follows (see also
Figure 6): (i) P5 and TSPO levels exhibit a circadian pattern; (ii) circadian changes in mitochondrial morphology correlate with rhythmic production of P5; and (iii) circadian variations of P5 and TSPO depend on both a functional molecular clock and functional mitochondrial dynamics.
Daily variation in steroid synthesis has already been studied in peripheral tissues. For instance, adrenal synthesis and secretion of glucocorticoids is well-known to follow a circadian rhythm, with a peak around the onset of the active period of the day [
27]. Glucocorticoids, such as cortisol, control a variety of physiological processes such as metabolism, cardiovascular activity, immune response, and brain functions. Baburski and colleagues recently showed daily changes in serum testosterone and dihydrotestosterone levels (the two main male sex steroid hormones), as well as in the expression of some steroidogenic related genes—including
StAR,
CYP11A1, and
CYP17A1 (Cytochrome P450 17A1 or 17α-hydroxylase)—but not
TSPO, in Leydig cells of rat testis [
28]. Of note, in this study, animals were kept in light/dark conditions (12 h light/12 h darkness), suggesting an influence of light input on the parameters measured. In our study, we showed that
StAR and
CYP11A1 did not display any significant circadian variations in glioma cells, whereas TSPO levels followed a circadian rhythm in vitro and in vivo (mouse brain lysates). Peripheral tissues are regulated differently by the circadian clock with metabolic information being a key factor influencing the rhythm in peripheral tissues [
29] and properties of the circadian molecular machinery, which vary among tissues [
30,
31]. Hence, components of the steroidogenesis pathway could be regulated in a tissue-specific fashion by the circadian clock explaining the discrepancy in the observations. In addition, there could be subtle differences in regulation of steroid and neurosteroid production. Interestingly, daily variation in neurosteroid levels was first shown by Robel et al. in 1987 who found a persisting rhythm of brain dehydroepiandrosterone (DHEA) and DHEA sulfates (two neurosteroids modulating neurotransmission) levels after removal of steroidogenic peripheral glands (adrenal glands and gonads) [
32], confirming the existence of autonomous brain mechanisms coordinating neurosteroid production.
The link between mitochondrial dynamics and steroidogenesis was previously highlighted by Duarte and colleagues [
13,
14]. They showed that hormone-triggered mitochondrial fusion correlates with increased steroid production, particularly progesterone, in Leydig cells. This study provided first evidence that mitochondrial fusion is an essential step of steroid production, a process which depends on protein kinase A (PKA) activity. Especially, MFN2 expression was upregulated following hormone stimulation, and its knockdown was sufficient to impair steroidogenesis, suggesting an essential role for mitochondrial fusion during steroidogenesis. More recently, Park and colleagues showed that steroid production in Leydig cells correlates with the protein kinase A (PKA)-dependent DRP1 phosphorylation (Ser 637) and mitochondrial elongation [
33]. In line with these findings, our study shows for the first time a correlation between steroidogenesis and mitochondrial dynamics in brain-derived cells, in particular a cyclic production of P5, which parallels the circadian rhythm of mitochondrial fusion/fission activity.
In light of our new data, the following mechanisms may explain the circadian rhythm in P5 levels that we observed in our study.
We showed that circadian variations of mitochondrial fusion correlate with rhythmic P5 levels, and that disturbing the molecular clock abolishes both rhythms in mitochondrial dynamics [
12] and P5 levels. A link between steroidogenesis and mitochondrial dynamics was previously shown in Leydig cells with mitochondrial shaping proteins, DRP1 and MFN2, playing important roles in the regulation of steroid production [
14,
33]. Namely, PKA activation by dibutyryl cyclic AMP was shown to induce DRP1 phosphorylation at Serine 637, decreasing DRP1 mitochondrial localization and leading to mitochondrial elongation and steroidogenesis [
33]. Strikingly, DRP1 dephosphorylation at Ser 637, which leads to its translocation to mitochondria and mitochondrial fission, is regulated through clock-dependent calcineurin activation [
34] with pharmacological inhibition of calcineurin increasing DRP1 phosphorylation and abrogating circadian DRP1 phosphorylation patterns [
12]. Based on these findings, we can hypothesize that cyclic steroidogenesis (i.e., P5 circadian synthesis) is regulated by mitochondrial dynamics via clock-controlled calcineurin activity, which governs DRP1 phosphorylation and mitochondrial elongation.
In addition, we showed that when mitochondrial dynamics are impaired (MDIVI-1 treatment or MFN2 siRNA), P5 and TSPO expression become arrhythmic. This suggests that mitochondrial dynamics may modulate TSPO expression. We previously demonstrated that DRP1-mediated mitochondrial bioenergetic oscillations may send signals back to the clock (via adenosine monophosphate-activated protein kinase (AMPK) pathways, sirtuins) thereby modulating parameters of the circadian rhythm, such as period length [
12]. Therefore, it is not excluded that this feedback control on the molecular clock influences gene expression, resulting in arrhythmic TSPO expression when mitochondrial dynamics are impaired. Furthermore, when the molecular clock is disturbed (PER1/PER2 knockdown), rhythmic variations of TSPO and P5 are abolished. In line with this observation, TSPO was shown to recruit PKA to mitochondrial to phosphorylate the voltage-dependent anion channel (VDAC1) [
35]. One can hypothesize that this mechanism is also involved in the circadian regulation of P5 synthesis, with TSPO recruiting PKA to mitochondria for DRP1 phosphorylation, triggering mitochondrial elongation and steroidogenesis.
Further studies are now necessary to elucidate these underlying mechanisms and to clarify the connection between mitochondrial dynamics, neurosteroidogenesis, and the circadian clock.
Neurosteroids are also able to modulate neuronal bioenergetics by increasing mitochondrial respiration and ATP production, at least in part via nuclear steroid receptor activation [
36]. Our recent findings revealed that the P5 levels were decreased in cellular models of AD, together with impairments in mitochondrial bioenergetics [
37,
38]. Strikingly, treatment with TSPO ligands increased P5 production and improved mitochondrial energy production in the presence of AD-related pathological proteins, amyloid-β peptide, and abnormal Tau protein. These data showed that neurosteroids play an important role in controlling neuronal function by modulating mitochondrial physiology, and that impaired neurosteroidogenesis may be involved in mitochondrial dysfunction underlying neurodegenerative disorders. A better understanding about how neurosteroid synthesis is regulated in the brain may offer the possibility to develop therapeutic strategies against neurodegenerative processes.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}