Cellular Senescence in Renal and Urinary Tract Disorders

 , and
, and
Abstract
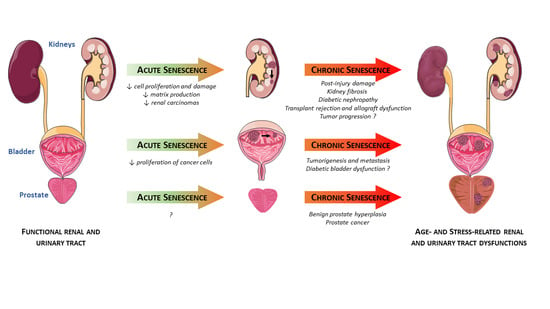
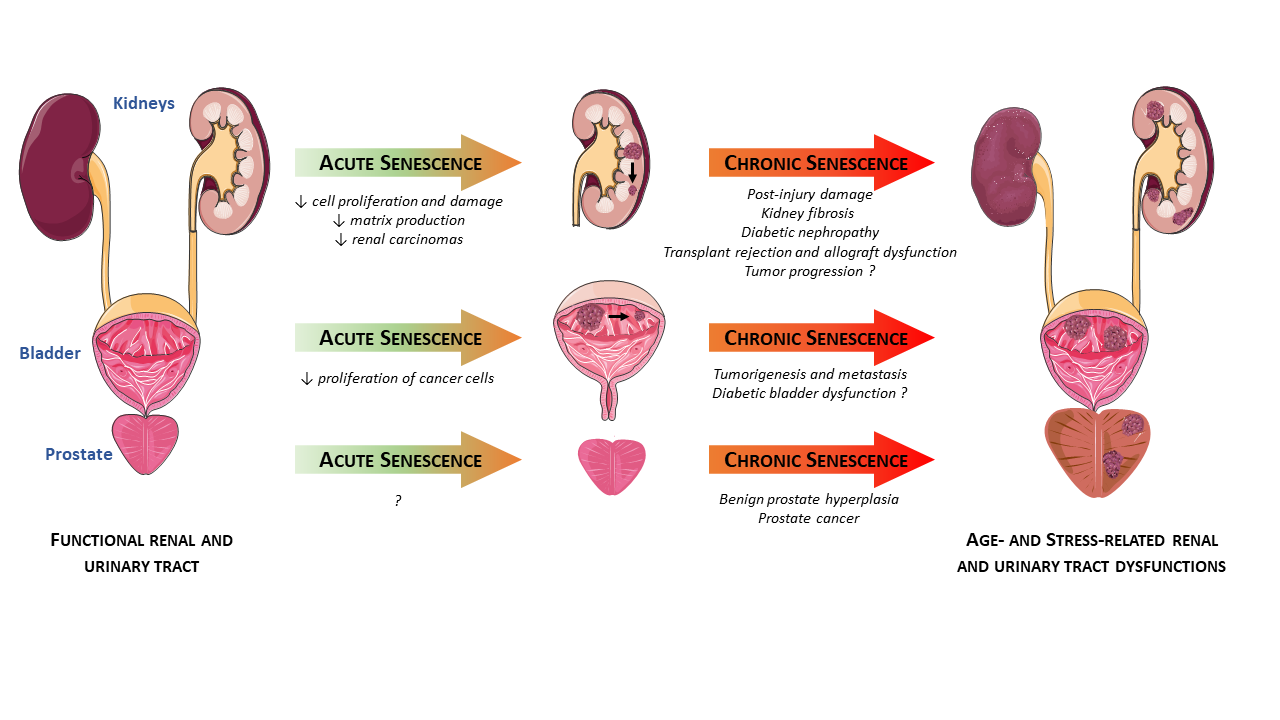
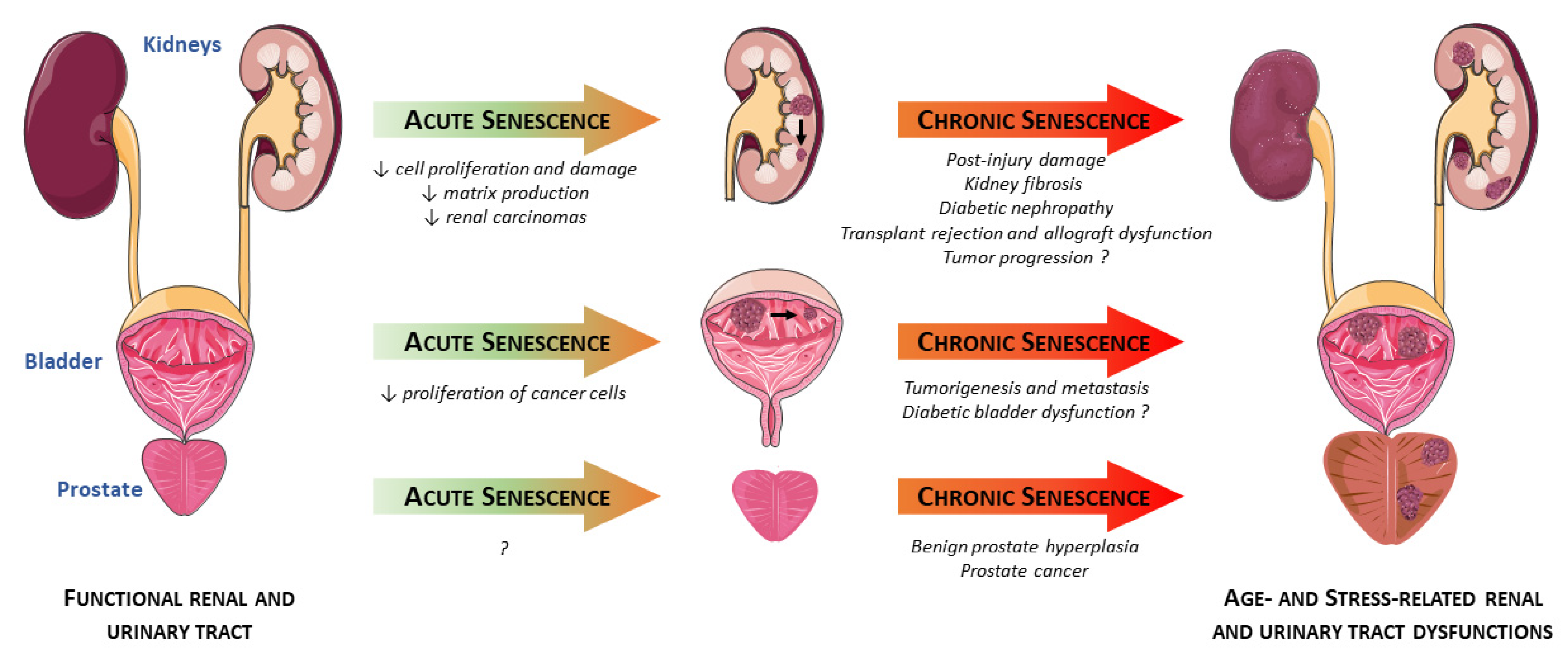
:
{kind=link}
{kind=link}
{kind=link}
1. Overview of Cellular Senescence
2. Senescence in the Kidneys
2.1. Acute Renal Injuries
2.2. Chronic Renal Diseases
2.3. Cancers
3. Senescence in the Prostate
3.1. Benign Prostate Hyperplasia (BPH)
3.2. Prostate Cancer (PCa)
4. Senescence in the Bladder
4.1. Aging Bladder
4.2. Pathological Conditions Associated with Bladder Dysfunction
5. Senotherapeutics in Renal and Urinary Tract Disorders
5.1. Senolytics
5.2. Senostatics/Senomorphics
6. Discussion
Funding
Conflicts of Interest
References
- Lecot, P.; Alimirah, F.; Desprez, P.-Y.; Campisi, J.; Wiley, C. Context-dependent effects of cellular senescence in cancer development. Br. J. Cancer 2016, 114, 1180–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Khosla, S.; Farr, J.N.; Tchkonia, T.; Kirkland, J.L. The role of cellular senescence in ageing and endocrine disease. Nat. Rev. Endocrinol. 2020, 16, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; Fagagna, F. d’Adda di Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [Green Version]
- Kuilman, T.; Peeper, D.S. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer 2009, 9, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef]
- Sadaie, M.; Salama, R.; Carroll, T.; Tomimatsu, K.; Chandra, T.; Young, A.R.J.; Narita, M.; Pérez-Mancera, P.A.; Bennett, D.C.; Chong, H.; et al. Redistribution of the Lamin B1 genomic binding profile affects rearrangement of heterochromatic domains and SAHF formation during senescence. Genes Dev. 2013, 27, 1800–1808. [Google Scholar] [CrossRef] [Green Version]
- Itahana, K.; Itahana, Y.; Dimri, G.P. Colorimetric Detection of Senescence-Associated β Galactosidase. Methods Mol. Biol. 2013, 965, 143–156. [Google Scholar] [CrossRef] [Green Version]
- Georgakopoulou, E.A.; Tsimaratou, K.; Evangelou, K.; Fernandez Marcos, P.J.; Zoumpourlis, V.; Trougakos, I.P.; Kletsas, D.; Bartek, J.; Serrano, M.; Gorgoulis, V.G. Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging 2013, 5, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Dörr, J.R.; Yu, Y.; Milanovic, M.; Beuster, G.; Zasada, C.; Däbritz, J.H.M.; Lisec, J.; Lenze, D.; Gerhardt, A.; Schleicher, K.; et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 2013, 501, 421–425. [Google Scholar] [CrossRef]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef] [Green Version]
- Korolchuk, V.I.; Miwa, S.; Carroll, B.; von Zglinicki, T. Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? EBioMedicine 2017, 21, 7–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakula, D.; Scheibye-Knudsen, M. MitophAging: Mitophagy in Aging and Disease. Front. Cell Dev. Biol. 2020, 8, 239. [Google Scholar] [CrossRef] [Green Version]
- Tchkonia, T.; Kirkland, J.L. Aging, Cell Senescence, and Chronic Disease: Emerging Therapeutic Strategies. JAMA 2018, 320, 1319–1320. [Google Scholar] [CrossRef]
- Palmer, A.K.; Xu, M.; Zhu, Y.; Pirtskhalava, T.; Weivoda, M.M.; Hachfeld, C.M.; Prata, L.G.; van Dijk, T.H.; Verkade, E.; Casaclang-Verzosa, G.; et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 2019, 18, e12950. [Google Scholar] [CrossRef]
- Barnes, P.J.; Baker, J.; Donnelly, L.E. Cellular Senescence as a Mechanism and Target in Chronic Lung Diseases. Am. J. Respir. Crit. Care Med. 2019, 200, 556–564. [Google Scholar] [CrossRef]
- Song, P.; Zhao, Q.; Zou, M.-H. Targeting senescent cells to attenuate cardiovascular disease progression. Ageing Res. Rev. 2020, 60, 101072. [Google Scholar] [CrossRef] [PubMed]
- Musi, N.; Valentine, J.M.; Sickora, K.R.; Baeuerle, E.; Thompson, C.S.; Shen, Q.; Orr, M.E. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 2018, 17, e12840. [Google Scholar] [CrossRef]
- Riessland, M.; Kolisnyk, B.; Kim, T.W.; Cheng, J.; Ni, J.; Pearson, J.A.; Park, E.J.; Dam, K.; Acehan, D.; Ramos-Espiritu, L.S.; et al. Loss of SATB1 induces p21-dependent cellular senescence in post-mitotic dopaminergic neurons. Cell Stem Cell 2019, 25, 514–530.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkenkamp, B.; Susnik, N.; Baisantry, A.; Kuznetsova, I.; Jacobi, C.; Sörensen-Zender, I.; Broecker, V.; Haller, H.; Melk, A.; Schmitt, R. In vivo and in vitro analysis of age-associated changes and somatic cellular senescence in renal epithelial cells. PLoS ONE 2014, 9, e88071. [Google Scholar] [CrossRef] [Green Version]
- Melk, A.; Schmidt, B.M.W.; Takeuchi, O.; Sawitzki, B.; Rayner, D.C.; Halloran, P.F. Expression of p16INK4a and other cell cycle regulator and senescence associated genes in aging human kidney. Kidney Int. 2004, 65, 510–520. [Google Scholar] [CrossRef] [Green Version]
- Klee, N.S.; McCarthy, C.G.; Lewis, S.; McKenzie, J.L.; Vincent, J.E.; Webb, R.C. Urothelial Senescence in the Pathophysiology of Diabetic Bladder Dysfunction—A Novel Hypothesis. Front. Surg. 2018, 5, 5. [Google Scholar] [CrossRef]
- Choi, J.; Shendrik, I.; Peacocke, M.; Peehl, D.; Buttyan, R.; Ikeguchi, E.F.; Katz, A.E.; Benson, M.C. Expression of senescence-associated beta-galactosidase in enlarged prostates from men with benign prostatic hyperplasia. Urology 2000, 56, 160–166. [Google Scholar] [CrossRef]
- Sturmlechner, I.; Durik, M.; Sieben, C.J.; Baker, D.J.; van Deursen, J.M. Cellular senescence in renal ageing and disease. Nat. Rev. Nephrol. 2017, 13, 77–89. [Google Scholar] [CrossRef]
- Karam, Z.; Tuazon, J. Anatomic and physiologic changes of the aging kidney. Clin. Geriatr. Med. 2013, 29, 555–564. [Google Scholar] [CrossRef]
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O’Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.D.R. Global Prevalence of Chronic Kidney Disease–A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef] [PubMed]
- Goligorsky, M.S. Chronic Kidney Disease: A Vicarious Relation to Premature Cell Senescence. Am. J. Pathol. 2020, 190, 1164–1171. [Google Scholar] [CrossRef]
- Zhou, B.; Wan, Y.; Chen, R.; Zhang, C.; Li, X.; Meng, F.; Glaser, S.; Wu, N.; Zhou, T.; Li, S.; et al. The emerging role of cellular senescence in renal diseases. J. Cell. Mol. Med. 2020, 24, 2087–2097. [Google Scholar] [CrossRef]
- Wang, W.-J.; Cai, G.-Y.; Chen, X.-M. Cellular senescence, senescence-associated secretory phenotype, and chronic kidney disease. Oncotarget 2017, 8, 64520–64533. [Google Scholar] [CrossRef]
- Schmitt, R.; Coca, S.; Kanbay, M.; Tinetti, M.E.; Cantley, L.G.; Parikh, C.R. Recovery of kidney function after acute kidney injury in the elderly: A systematic review and meta-analysis. Am. J. Kidney Dis. 2008, 52, 262–271. [Google Scholar] [CrossRef]
- Sörensen-Zender, I.; Rong, S.; Susnik, N.; Zender, S.; Pennekamp, P.; Melk, A.; Haller, H.; Schmitt, R. Renal tubular Notch signaling triggers a prosenescent state after acute kidney injury. Am. J. Physiol. Ren. Physiol. 2014, 306, F907–F915. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Fan, X.; Lawson, W.E.; Paueksakon, P.; Harris, R.C. Telomerase deficiency delays renal recovery in mice after ischemia reperfusion injury by impairing autophagy. Kidney Int. 2015, 88, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.H.; Wolstein, J.M.; Pudasaini, B.; Plotkin, M. INK4a deletion results in improved kidney regeneration and decreased capillary rarefaction after ischemia-reperfusion injury. Am. J. Physiol. Ren. Physiol. 2011, 302, F183–F191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferenbach, D.A.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Megyesi, J.; Andrade, L.; Vieira, J.M.; Safirstein, R.L.; Price, P.M. Positive effect of the induction of p21WAF1/CIP1 on the course of ischemic acute renal failure. Kidney Int. 2001, 60, 2164–2172. [Google Scholar] [CrossRef] [Green Version]
- DiRocco, D.P.; Bisi, J.; Roberts, P.; Strum, J.; Wong, K.-K.; Sharpless, N.; Humphreys, B.D. CDK4/6 inhibition induces epithelial cell cycle arrest and ameliorates acute kidney injury. Am. J. Physiol. Ren. Physiol. 2014, 306, F379–F388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verzola, D.; Gandolfo, M.T.; Gaetani, G.; Ferraris, A.; Mangerini, R.; Ferrario, F.; Villaggio, B.; Gianiorio, F.; Tosetti, F.; Weiss, U.; et al. Accelerated senescence in the kidneys of patients with type 2 diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2008, 295, F1563–F1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitada, K.; Nakano, D.; Ohsaki, H.; Hitomi, H.; Minamino, T.; Yatabe, J.; Felder, R.A.; Mori, H.; Masaki, T.; Kobori, H.; et al. Hyperglycemia causes cellular senescence via a SGLT2- and p21-dependent pathway in proximal tubules in the early stage of diabetic nephropathy. J. Diabetes Complicat. 2014, 28, 604–611. [Google Scholar] [CrossRef] [Green Version]
- Prattichizzo, F.; De Nigris, V.; Mancuso, E.; Spiga, R.; Giuliani, A.; Matacchione, G.; Lazzarini, R.; Marcheselli, F.; Recchioni, R.; Testa, R.; et al. Short-term sustained hyperglycaemia fosters an archetypal senescence-associated secretory phenotype in endothelial cells and macrophages. Redox Biol. 2017, 15, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Perlman, A.S.; Chevalier, J.M.; Wilkinson, P.; Liu, H.; Parker, T.; Levine, D.M.; Sloan, B.J.; Gong, A.; Sherman, R.; Farrell, F.X. Serum Inflammatory and Immune Mediators Are Elevated in Early Stage Diabetic Nephropathy. Ann. Clin. Lab. Sci. 2015, 45, 256–263. [Google Scholar] [PubMed]
- Lujambio, A. To clear, or not to clear (senescent cells)? That is the question. Bioessays 2016, 38 (Suppl. 1), S56–S64. [Google Scholar] [CrossRef] [PubMed]
- Clements, M.E.; Chaber, C.J.; Ledbetter, S.R.; Zuk, A. Increased Cellular Senescence and Vascular Rarefaction Exacerbate the Progression of Kidney Fibrosis in Aged Mice Following Transient Ischemic Injury. PLoS ONE 2013, 8, e70464. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, C.; Zhou, S.; Zhou, Z.; Liu, Y.; Yang, L.; Liu, J.; Zhang, Y.; Li, H.; Liu, Y.; Hou, F.F.; et al. Wnt9a Promotes Renal Fibrosis by Accelerating Cellular Senescence in Tubular Epithelial Cells. JASN 2018, 29, 1238–1256. [Google Scholar] [CrossRef] [Green Version]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [Green Version]
- Wolstein, J.M.; Lee, D.H.; Michaud, J.; Buot, V.; Stefanchik, B.; Plotkin, M.D. INK4a knockout mice exhibit increased fibrosis under normal conditions and in response to unilateral ureteral obstruction. Am. J. Physiol. Ren. Physiol. 2010, 299, F1486–F1495. [Google Scholar] [CrossRef] [Green Version]
- Melk, A.; Schmidt, B.M.W.; Vongwiwatana, A.; Rayner, D.C.; Halloran, P.F. Increased expression of senescence-associated cell cycle inhibitor p16INK4a in deteriorating renal transplants and diseased native kidney. Am. J. Transplant. 2005, 5, 1375–1382. [Google Scholar] [CrossRef]
- Ferlicot, S.; Durrbach, A.; Bâ, N.; Desvaux, D.; Bedossa, P.; Paradis, V. The role of replicative senescence in chronic allograft nephropathy. Hum. Pathol. 2003, 34, 924–928. [Google Scholar] [CrossRef]
- Joosten, S.A.; van Ham, V.; Nolan, C.E.; Borrias, M.C.; Jardine, A.G.; Shiels, P.G.; van Kooten, C.; Paul, L.C. Telomere Shortening and Cellular Senescence in a Model of Chronic Renal Allograft Rejection. Am. J. Pathol. 2003, 162, 1305–1312. [Google Scholar] [CrossRef] [Green Version]
- Braun, H.; Schmidt, B.M.W.; Raiss, M.; Baisantry, A.; Mircea-Constantin, D.; Wang, S.; Gross, M.-L.; Serrano, M.; Schmitt, R.; Melk, A. Cellular Senescence Limits Regenerative Capacity and Allograft Survival. JASN 2012, 23, 1467–1473. [Google Scholar] [CrossRef] [Green Version]
- Domański, L.; Kłoda, K.; Kwiatkowska, E.; Borowiecka, E.; Safranow, K.; Drozd, A.; Ciechanowicz, A.; Ciechanowski, K. Effect of delayed graft function, acute rejection and chronic allograft dysfunction on kidney allograft telomere length in patients after transplantation: A prospective cohort study. BMC Nephrol. 2015, 16, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Willigenburg, H.; de Keizer, P.L.J.; de Bruin, R.W.F. Cellular senescence as a therapeutic target to improve renal transplantation outcome. Pharmacol. Res. 2018, 130, 322–330. [Google Scholar] [CrossRef]
- Melk, A.; Halloran, P.F. Cell Senescence and Its Implications for Nephrology. JASN 2001, 12, 385–393. [Google Scholar]
- McGlynn, L.M.; Stevenson, K.; Lamb, K.; Zino, S.; Brown, M.; Prina, A.; Kingsmore, D.; Shiels, P.G. Cellular senescence in pretransplant renal biopsies predicts postoperative organ function. Aging Cell 2009, 8, 45–51. [Google Scholar] [CrossRef]
- Lee, S.; Lee, J.-S. Cellular senescence: A promising strategy for cancer therapy. BMB Rep. 2019, 52, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Cole, A.M.; Ridgway, R.A.; Derkits, S.E.; Parry, L.; Barker, N.; Clevers, H.; Clarke, A.R.; Sansom, O.J. p21 loss blocks senescence following Apc loss and provokes tumourigenesis in the renal but not the intestinal epithelium. EMBO Mol. Med. 2010, 2, 472–486. [Google Scholar] [CrossRef]
- Shen, Y.; Yu, D.; Qi, P.; Wang, X.; Guo, X.; Zhang, A. Calcitriol induces cell senescence of kidney cancer through JMJD3 mediated histone demethylation. Oncotarget 2017, 8, 100187–100195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, A.P.; Schlisio, S.; Minamishima, Y.A.; Zhang, Q.; Li, L.; Grisanzio, C.; Signoretti, S.; Kaelin, W.G. VHL loss actuates a HIF-independent senescence programme mediated by Rb and p400. Nat. Cell Biol. 2008, 10, 361–369. [Google Scholar] [CrossRef]
- Macher-Goeppinger, S.; Bermejo, J.L.; Schirmacher, P.; Pahernik, S.; Hohenfellner, M.; Roth, W. Senescence-associated protein p400 is a prognostic marker in renal cell carcinoma. Oncol. Rep. 2013, 30, 2245–2253. [Google Scholar] [CrossRef] [Green Version]
- Schosserer, M.; Grillari, J.; Breitenbach, M. The Dual Role of Cellular Senescence in Developing Tumors and Their Response to Cancer Therapy. Front. Oncol. 2017, 7, 278. [Google Scholar] [CrossRef] [Green Version]
- Zeng, S.; Shen, W.H.; Liu, L. Senescence and Cancer. Cancer Transl. Med. 2018, 4, 70–74. [Google Scholar] [CrossRef]
- Isaacs, J.T. Etiology of benign prostatic hyperplasia. Eur. Urol. 1994, 25 (Suppl. 1), 6–9. [Google Scholar] [CrossRef]
- Liu, T.T.; Thomas, S.; Mclean, D.T.; Roldan-Alzate, A.; Hernando, D.; Ricke, E.A.; Ricke, W.A. Prostate enlargement and altered urinary function are part of the aging process. Aging 2019, 11, 2653–2669. [Google Scholar] [CrossRef]
- Jiang, S.; Song, C.S.; Chatterjee, B. Stimulation of prostate cells by the senescence phenotype of epithelial and stromal cells: Implication for benign prostate hyperplasia. FASEB BioAdv. 2019, 1, 353–363. [Google Scholar] [CrossRef]
- Vital, P.; Castro, P.; Tsang, S.; Ittmann, M. The senescence-associated secretory phenotype promotes benign prostatic hyperplasia. Am. J. Pathol. 2014, 184, 721–731. [Google Scholar] [CrossRef] [Green Version]
- Giri, D.; Ittmann, M. Interleukin-1α Is a Paracrine Inducer of FGF7, a Key Epithelial Growth Factor in Benign Prostatic Hyperplasia. Am. J. Pathol. 2000, 157, 249–255. [Google Scholar] [CrossRef]
- Jerde, T.J.; Bushman, W. IL-1 induces IGF-dependent epithelial proliferation in organ development and reactive hyperplasia. Sci. Signal. 2009, 2, ra49. [Google Scholar] [CrossRef] [Green Version]
- Giri, D.; Ittmann, M. Interleukin-8 Is a Paracrine Inducer of Fibroblast Growth Factor 2, a Stromal and Epithelial Growth Factor in Benign Prostatic Hyperplasia. Am. J. Pathol. 2001, 159, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Castro, P.; Xia, C.; Gomez, L.; Lamb, D.J.; Ittmann, M. Interleukin-8 expression is increased in senescent prostatic epithelial cells and promotes the development of benign prostatic hyperplasia. Prostate 2004, 60, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Ewald, J.A.; Desotelle, J.A.; Church, D.R.; Yang, B.; Huang, W.; Laurila, T.A.; Jarrard, D.F. Androgen Deprivation Induces Senescence Characteristics in Prostate Cancer Cells In vitro and In vivo. Prostate 2013, 73, 337–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, D.G.A.; Giribaldi, M.G.; Munoz, A.; Halvorsen, K.; Patel, A.; Jorda, M.; Perez-Stable, C.; Rai, P. Androgen Deprivation-Induced Senescence Promotes Outgrowth of Androgen-Refractory Prostate Cancer Cells. PLoS ONE 2013, 8, e68003. [Google Scholar] [CrossRef]
- Pernicová, Z.; Slabáková, E.; Kharaishvili, G.; Bouchal, J.; Král, M.; Kunická, Z.; Machala, M.; Kozubík, A.; Souček, K. Androgen depletion induces senescence in prostate cancer cells through down-regulation of Skp2. Neoplasia 2011, 13, 526–536. [Google Scholar] [CrossRef] [Green Version]
- Blute, M.L.; Damaschke, N.; Wagner, J.; Yang, B.; Gleave, M.; Fazli, L.; Shi, F.; Abel, E.J.; Downs, T.M.; Huang, W.; et al. Persistence of senescent prostate cancer cells following prolonged neoadjuvant androgen deprivation therapy. PLoS ONE 2017, 12, e0172048. [Google Scholar] [CrossRef] [PubMed]
- Siroky, M.B. The Aging Bladder. Rev. Urol. 2004, 6, S3–S7. [Google Scholar]
- Lluel, P.; Palea, S.; Barras, M.; Grandadam, F.; Heudes, D.; Bruneval, P.; Corman, B.; Martin, D.J. Functional and morphological modifications of the urinary bladder in aging female rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 278, R964–R972. [Google Scholar] [CrossRef] [Green Version]
- Chun, A.L.; Wallace, L.J.; Gerald, M.C.; Levin, R.M.; Wein, A.J. Effect of age on in vivo urinary bladder function in the rat. J. Urol. 1988, 139, 625–627. [Google Scholar] [CrossRef]
- Chamorro, C.I.; Asghar, M.; Ekblad, Å.; Färnert, A.; Götherström, C.; Fossum, M. Urothelial cell senescence is not linked with telomere shortening. J. Tissue Eng. Regen. Med. 2019, 13, 1518–1527. [Google Scholar] [CrossRef]
- Chen, L.; He, P.-L.; Yang, J.; Yang, Y.-F.; Wang, K.; Amend, B.; Stenzl, A.; Zhang, Y.-M.; Wang, Z.-L.; Xing, S.-S.; et al. NLRP3/IL1β inflammasome associated with the aging bladder triggers bladder dysfunction in female rats. Mol. Med. Rep. 2019, 19, 2960–2968. [Google Scholar] [CrossRef]
- Chen, T.; Wang, H.; Zhang, Z.; Li, Q.; Yan, K.; Tao, Q.; Ye, Q.; Xiong, S.; Wang, Y.; Zhai, Z. A Novel Cellular Senescence Gene, SENEX, Is Involved in Peripheral Regulatory T Cells Accumulation in Aged Urinary Bladder Cancer. PLoS ONE 2014, 9, e87774. [Google Scholar] [CrossRef] [Green Version]
- Aljabery, F.; Shabo, I.; Gimm, O.; Jahnson, S.; Olsson, H. The expression profile of p14, p53 and p21 in tumour cells is associated with disease-specific survival and the outcome of postoperative chemotherapy treatment in muscle-invasive bladder cancer. Urol. Oncol. Semin. Orig. Investig. 2018, 36, 530.e7–530.e18. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Cha, H.-J.; Choi, E.O.; Lee, H.; Hwang-Bo, H.; Ji, S.Y.; Kim, M.Y.; Kim, S.Y.; Hong, S.H.; Cheong, J.; et al. Isorhamnetin Induces Cell Cycle Arrest and Apoptosis Via Reactive Oxygen Species-Mediated AMP-Activated Protein Kinase Signaling Pathway Activation in Human Bladder Cancer Cells. Cancers 2019, 11, 1494. [Google Scholar] [CrossRef] [Green Version]
- Felipe, K.B.; Benites, J.; Glorieux, C.; Sid, B.; Valenzuela, M.; Kviecinski, M.R.; Pedrosa, R.C.; Valderrama, J.A.; Levêque, P.; Gallez, B.; et al. Antiproliferative effects of phenylaminonaphthoquinones are increased by ascorbate and associated with the appearance of a senescent phenotype in human bladder cancer cells. Biochem. Biophys. Res. Commun. 2013, 433, 573–578. [Google Scholar] [CrossRef]
- Althubiti, M.; Lezina, L.; Carrera, S.; Jukes-Jones, R.; Giblett, S.M.; Antonov, A.; Barlev, N.; Saldanha, G.S.; Pritchard, C.A.; Cain, K.; et al. Characterization of novel markers of senescence and their prognostic potential in cancer. Cell Death Dis. 2014, 5, e1528. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16 Ink4a -positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [Green Version]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147.e16. [Google Scholar] [CrossRef] [Green Version]
- Kirkland, J.L.; Tchkonia, T. Senolytic drugs: From discovery to translation. J. Intern. Med. 2020, 288, 518–536. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T. Clinical strategies and animal models for developing senolytic agents. Exp. Gerontol. 2015, 68, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Docherty, M.H.; Baird, D.P.; Hughes, J.; Ferenbach, D.A. Cellular Senescence and Senotherapies in the Kidney: Current Evidence and Future Directions. Front. Pharmacol. 2020, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018, 562, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; Deursen, J.M. van Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016, 354, 472–477. [Google Scholar] [CrossRef]
- Dookun, E.; Walaszczyk, A.; Redgrave, R.; Palmowski, P.; Tual-Chalot, S.; Suwana, A.; Chapman, J.; Jirkovsky, E.; Donastorg Sosa, L.; Gill, E.; et al. Clearance of senescent cells during cardiac ischemia–reperfusion injury improves recovery. Aging Cell 2020, 19. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.-M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Kaefer, A.; Yang, J.; Noertersheuser, P.; Mensing, S.; Humerickhouse, R.; Awni, W.; Xiong, H. Mechanism-based pharmacokinetic/pharmacodynamic meta-analysis of navitoclax (ABT-263) induced thrombocytopenia. Cancer Chemother. Pharmacol. 2014, 74, 593–602. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Cavalcante, M.B.; Saccon, T.D.; Nunes, A.D.C.; Kirkland, J.L.; Tchkonia, T.; Schneider, A.; Masternak, M.M. Dasatinib plus quercetin prevents uterine age-related dysfunction and fibrosis in mice. Aging 2020, 12, 2711–2722. [Google Scholar] [CrossRef]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Misra Sen, J.; Gorospe, M.; et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef]
- Kim, S.R.; Jiang, K.; Ogrodnik, M.; Chen, X.; Zhu, X.-Y.; Lohmeier, H.; Ahmed, L.; Tang, H.; Tchkonia, T.; Hickson, L.J.; et al. Increased renal cellular senescence in murine high-fat diet: Effect of the senolytic drug quercetin. Transl. Res. 2019, 213, 112–123. [Google Scholar] [CrossRef]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef] [Green Version]
- Hickson, L.J.; Prata, L.G.L.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.-C.; Kim, A.J.-R. Senotherapeutics: Emerging strategy for healthy aging and age-related disease. BMB Rep. 2019, 52, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knoppert, S.N.; Valentijn, F.A.; Nguyen, T.Q.; Goldschmeding, R.; Falke, L.L. Cellular Senescence and the Kidney: Potential Therapeutic Targets and Tools. Front. Pharmacol. 2019, 10, 770. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, S.; Kuruvilla, V.; Wilbur, K.; Munusamy, S. Nephroprotective Effects of Metformin in Diabetic Nephropathy. J. Cell. Physiol. 2017, 232, 731–742. [Google Scholar] [CrossRef]
- Sultuybek, G.K.; Soydas, T.; Yenmis, G. NF-κB as the mediator of metformin’s effect on ageing and ageing-related diseases. Clin. Exp. Pharmacol. Physiol. 2019, 46, 413–422. [Google Scholar] [CrossRef] [Green Version]
- Falke, L.L.; van Vuuren, S.H.; Kazazi-Hyseni, F.; Ramazani, F.; Nguyen, T.Q.; Veldhuis, G.J.; Maarseveen, E.M.; Zandstra, J.; Zuidema, J.; Duque, L.F.; et al. Local therapeutic efficacy with reduced systemic side effects by rapamycin-loaded subcapsular microspheres. Biomaterials 2015, 42, 151–160. [Google Scholar] [CrossRef]
- Prata, L.G.P.L.; Ovsyannikova, I.G.; Tchkonia, T.; Kirkland, J.L. Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Semin. Immunol. 2018, 40, 101275. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santin, Y.; Lluel, P.; Rischmann, P.; Gamé, X.; Mialet-Perez, J.; Parini, A. Cellular Senescence in Renal and Urinary Tract Disorders. Cells 2020, 9, 2420. https://doi.org/10.3390/cells9112420
Santin Y, Lluel P, Rischmann P, Gamé X, Mialet-Perez J, Parini A. Cellular Senescence in Renal and Urinary Tract Disorders. Cells. 2020; 9(11):2420. https://doi.org/10.3390/cells9112420
Chicago/Turabian StyleSantin, Yohan, Philippe Lluel, Pascal Rischmann, Xavier Gamé, Jeanne Mialet-Perez, and Angelo Parini. 2020. "Cellular Senescence in Renal and Urinary Tract Disorders" Cells 9, no. 11: 2420. https://doi.org/10.3390/cells9112420
APA StyleSantin, Y., Lluel, P., Rischmann, P., Gamé, X., Mialet-Perez, J., & Parini, A. (2020). Cellular Senescence in Renal and Urinary Tract Disorders. Cells, 9(11), 2420. https://doi.org/10.3390/cells9112420