Identification of New Genetic Clusters in Glioblastoma Multiforme: EGFR Status and ADD3 Losses Influence Prognosis

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients, Samples and Clinical Study
2.2. DNA Extraction, Molecular Analysis of IDH1/2, TP53 and MLPA
2.3. Status of EGFR: EGFRvIII, Copy Number Alterations and Interphase Fluorescence In Situ Hybridization
2.4. Analysis of Locus 9p21 and Other Glioma and Cancer-Related Genes
2.5. Statistical Analysis
2.6. TCGA Analysis and Functional Protein Associations
3. Results
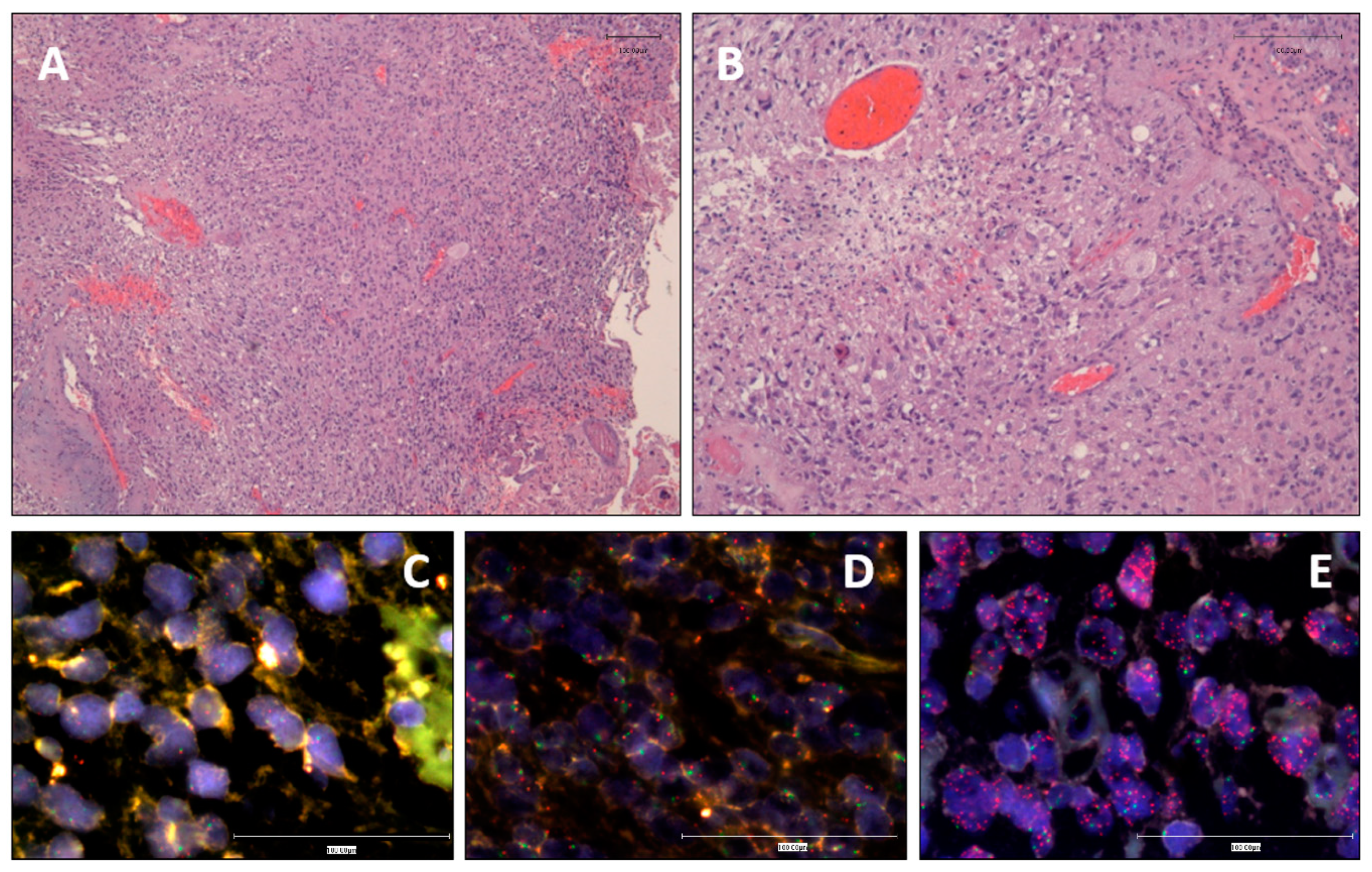
3.1. Clinical and Histopathological Data
3.2. EGFR Status Assessment by iFISH and MLPA
3.3. EGFR Variant III Is More Frequent in Women and Is Associated with Shortened Survival
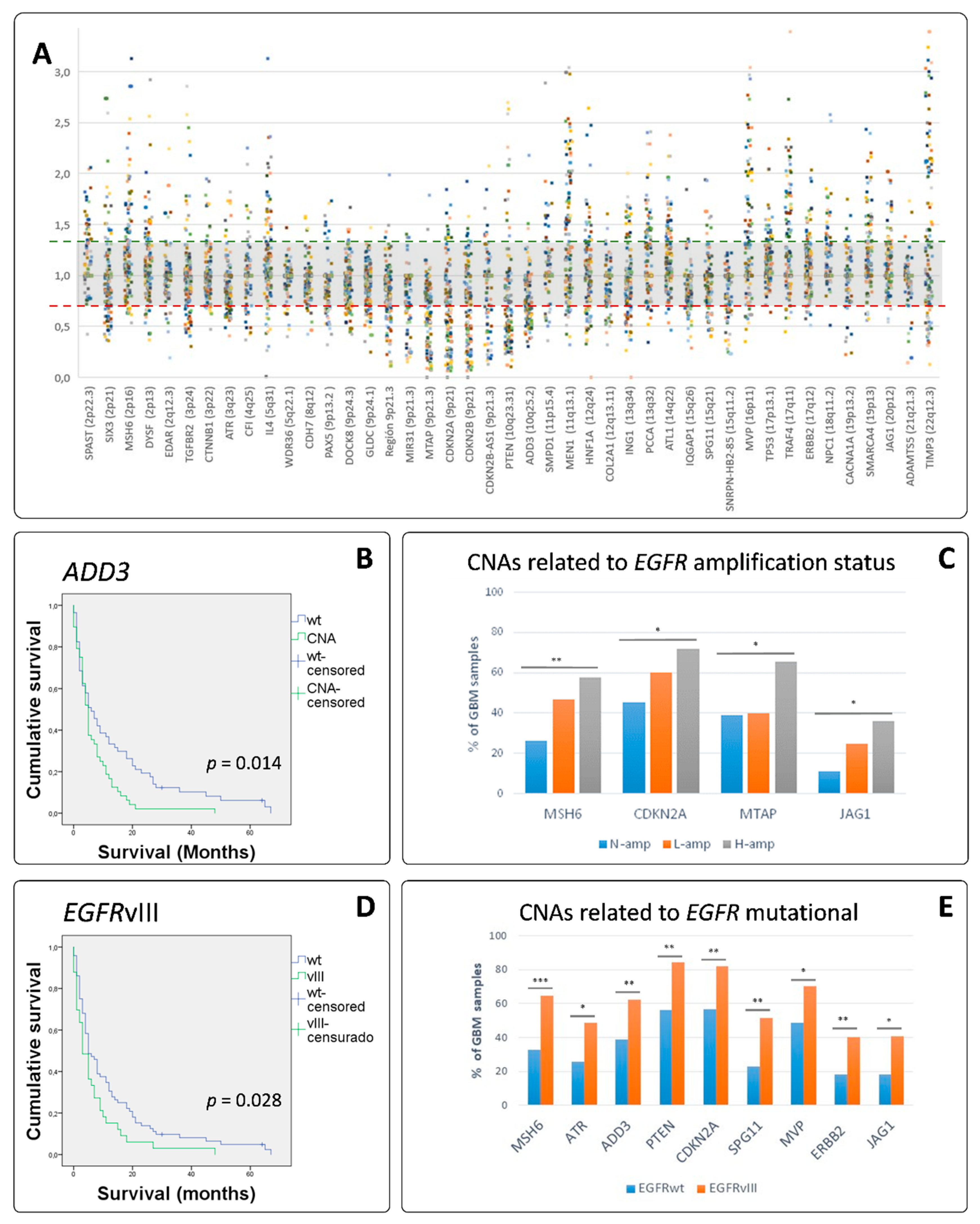
3.4. MLPA Analysis Showed a Great Heterogeneity in GB
3.5. EGFR Amplified GBs Displayed Different SCNAs to Non-EGFR Amplified Cases
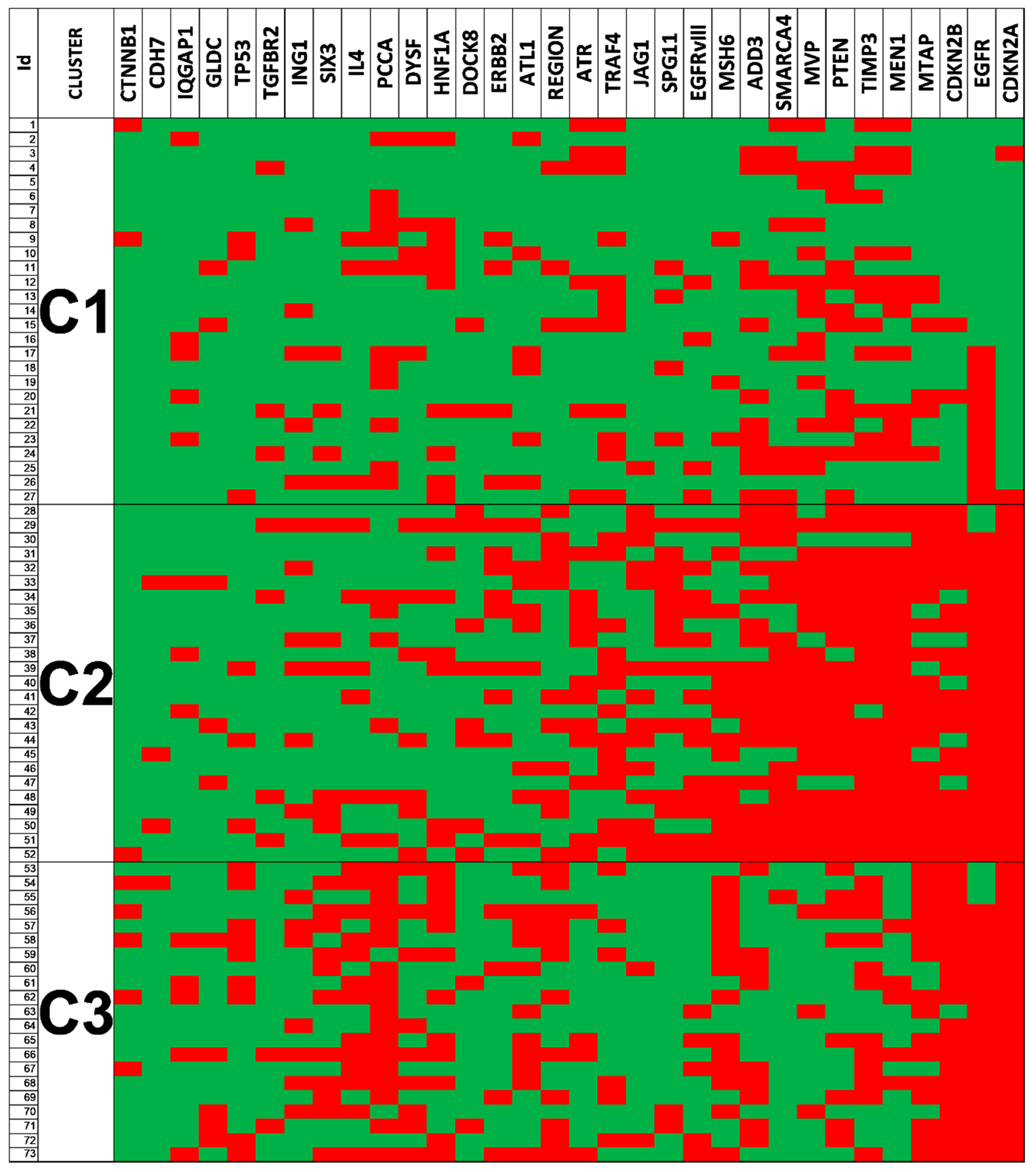
3.6. Clustering Analysis Revealed Different Genetic Glioblastoma Groups
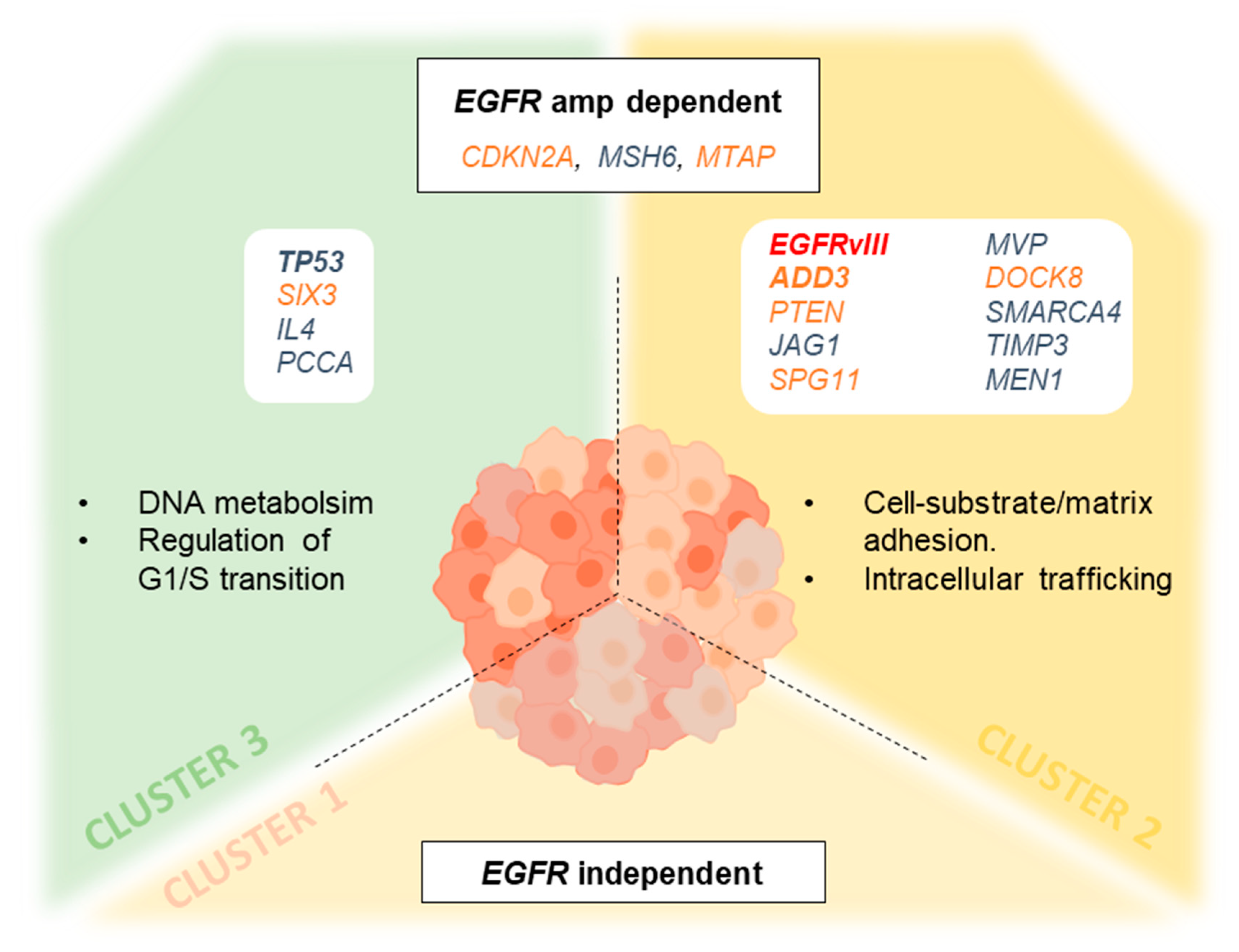
3.7. Genetic Changes According to Clustering Analysis Point to Differentially Altered Pathways
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrom, Q.T.; Gittleman, H.; Liao, P.; Vecchione-Koval, T.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro Oncol. 2017, 19, v1–v88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furnari, F.B.; Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat. Rev. Cancer 2015, 15, 302–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karsy, M.; Gelbman, M.; Shah, P.; Balumbu, O.; Moy, F.; Arslan, E. Established and emerging variants of glioblastoma multiforme: Review of morphological and molecular features. Folia Neuropathol. 2012, 50, 301–321. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Cowperthwaite, M.C.; Burnett, M.G.; Shpak, M. Molecular predictors of long-term survival in glioblastoma multiforme patients. PLoS ONE 2016, 11, e0154313. [Google Scholar] [CrossRef] [Green Version]
- Umehara, T.; Arita, H.; Yoshioka, E.; Shofuda, T.; Kanematsu, D.; Kinoshita, M.; Kodama, Y.; Mano, M.; Kagawa, N.; Fujimoto, Y.; et al. Distribution differences in prognostic copy number alteration profiles in IDH-wild-type glioblastoma cause survival discrepancies across cohorts. Acta Neuropathol. Commun. 2019, 7, 99. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Hochberg, F.H.; Atai, N.A.; Gonda, D.; Hughes, M.S.; Mawejje, B.; Balaj, L.; Carter, R.S. Glioma diagnostics and biomarkers: An ongoing challenge in the field of medicine and science. Expert Rev. Mol. Diagn. 2014, 14, 439–452. [Google Scholar] [CrossRef] [Green Version]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. An integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR and NF1. Cancer Cell 2010, 17, 98. [Google Scholar] [CrossRef] [Green Version]
- Ohgaki, H.; Kleihues, P. Genetic alterations and signaling pathways in the evolution of gliomas. Cancer Sci. 2009, 100, 2235–2241. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. The Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Huse, J.T. Elucidating the oncogenic role of ATRX deficiency in glioma. Neuro Oncol. 2014, 16, iii45. [Google Scholar] [CrossRef] [Green Version]
- Ohgaki, H.; Kleihues, P. Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 2007, 170, 1445–1453. [Google Scholar] [CrossRef] [Green Version]
- Sahm, F.; Reuss, D.E.; Giannini, C. WHO 2016 classification: Changes and advancements in the diagnosis of miscellaneous primary CNS tumours. Neuropathol. Appl. Neurobiol. 2018, 44, 163–171. [Google Scholar] [CrossRef]
- Lassman, A.B.; Roberts-Rapp, L.; Sokolova, I.; Song, M.; Pestova, E.; Kular, R.; Mullen, C.; Zha, Z.; Lu, X.; Gomez, E.; et al. Comparison of biomarker assays for EGFR: Implications for precision medicine in patients with glioblastoma. Clin. Cancer Res. 2019, 25, 3259–3265. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.G.; Falls, H.D.; Mitten, M.J.; Oleksijew, A.; Vaidya, K.S.; Boghaert, E.R.; Gao, W.; Palma, J.P.; Cao, D.; Chia, P.-L.; et al. Targeting Multiple EGFR-expressing Tumors with a Highly Potent Tumor-selective Antibody-Drug Conjugate. Mol. Cancer Ther. 2020, 19, 2117–2125. [Google Scholar] [CrossRef]
- Kim, G.; Ko, Y.T. Small molecule tyrosine kinase inhibitors in glioblastoma. Arch. Pharm. Res. 2020, 43, 385–394. [Google Scholar] [CrossRef]
- Lopez-Gines, C.; Gil-Benso, R.; Ferrer-Luna, R.; Benito, R.; Serna, E.; Gonzalez-Darder, J.; Quilis, V.; Monleon, D.; Celda, B.; Cerdá-Nicolas, M. New pattern of EGFR amplification in glioblastoma and the relationship of gene copy number with gene expression profile. Modern Pathol. 2010, 23, 856–865. [Google Scholar] [CrossRef]
- Stichel, D.; Ebrahimi, A.; Reuss, D.; Schrimpf, D.; Ono, T.; Shirahata, M.; Reifenberger, G.; Weller, M.; Hänggi, D.; Wick, W.; et al. Distribution of EGFR amplification, combined chromosome 7 gain and chromosome 10 loss, and TERT promoter mutation in brain tumors and their potential for the reclassification of IDHwt astrocytoma to glioblastoma. Acta Neuropathol. 2018, 136, 793–803. [Google Scholar] [CrossRef] [Green Version]
- Navarro, L.; Gil-Benso, R.; Megías, J.; Muñoz-Hidalgo, L.; San-Miguel, T.; Callaghan, R.C.; González-Darder, J.M.; López-Ginés, C.; Cerdá-Nicolás, M.J. Alteration of major vault protein in human glioblastoma and its relation with EGFR and PTEN status. Neuroscience 2015, 297, 243–251. [Google Scholar] [CrossRef]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.-W.; Weiss, W.A. Epidermal growth factor receptor (EGFR) and EGFRvIII in glioblastoma (GBM): Signaling pathways and targeted therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef]
- Parker, J.J.; Canoll, P.; Niswander, L.; Kleinschmidt-DeMasters, B.K.; Foshay, K.; Waziri, A. Intratumoral heterogeneity of endogenous tumor cell invasive behavior in human glioblastoma. Sci. Rep. 2018, 8, 18002. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Hidalgo, L.; San-Miguel, T.; Megías, J.; Monleón, D.; Navarro, L.; Roldán, P.; Cerdá-Nicolás, M.; López-Ginés, C. Somatic copy number alterations are associated with EGFR amplification and shortened survival in patients with primary glioblastoma. Neoplasia 2020, 22, 10–21. [Google Scholar] [CrossRef]
- Pedersen, M.W.; Tkach, V.; Pedersen, N.; Berezin, V.; Poulsen, H.S. Expression of a naturally occurring constitutively active variant of the epidermal growth factor receptor in mouse fibroblasts increases motility. Int. J. Cancer 2004, 108, 643–653. [Google Scholar] [CrossRef]
- Feng, H.; Hu, B.; Vuori, K.; Sarkaria, J.N.; Furnari, F.B.; Cavenee, W.K.; Cheng, S.-Y. EGFRvIII stimulates glioma growth and invasion through PKA-dependent serine phosphorylation of Dock180. Oncogene 2014, 33, 2504–2512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, S.; Schmidt, M.H.H. EGFR and EGFRvIII promote angiogenesis and cell invasion in glioblastoma: Combination therapies for an effective treatment. Int. J. Mol. Sci. 2017, 18, 1295. [Google Scholar] [CrossRef] [Green Version]
- Boisselier, B.; Dugay, F.; Belaud-Rotureau, M.-A.; Coutolleau, A.; Garcion, E.; Menei, P.; Guardiola, P.; Rousseau, A. Whole genome duplication is an early event leading to aneuploidy in IDH-wild type glioblastoma. Oncotarget 2018, 9, 36017–36028. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Hamoudi, R.A. Molecular and cytogenetic analysis of glioblastoma multiforme. Cancer Genet. Cytogenet. 2000, 122, 87–92. [Google Scholar] [CrossRef]
- Jeuken, J.; Cornelissen, S.; Boots-Sprenger, S.; Gijsen, S.; Wesseling, P. Multiplex Ligation-Dependent Probe Amplification. J. Mol. Diagn. 2006, 8, 433–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeuken, J.; Sijben, A.; Alenda, C.; Rijntjes, J.; Dekkers, M.; Boots-Sprenger, S.; McLendon, R.; Wesseling, P. Robust Detection of EGFR Copy Number Changes and EGFR Variant III: Technical Aspects and Relevance for Glioma Diagnostics. Brain Pathol. 2009, 19, 661–671. [Google Scholar] [CrossRef] [Green Version]
- González, J.R.; Carrasco, J.L.; Armengol, L.; Villatoro, S.; Jover, L.; Yasui, Y.; Estivill, X. Probe-specific mixed-model approach to detect copy number differences using multiplex ligation-dependent probe amplification (MLPA). BMC Bioinform. 2008, 9, 261. [Google Scholar] [CrossRef] [Green Version]
- Gessi, M.; Hammes, J.; Lauriola, L.; Dörner, E.; Kirfel, J.; Kristiansen, G.; zur Muehlen, A.; Denkhaus, D.; Waha, A.; Pietsch, T. GNA11 and N-RAS mutations: Alternatives for MAPK pathway activating GNAQ mutations in primary melanocytic tumours of the central nervous system. Neuropathol. Appl. Neurobiol. 2013, 39, 417–425. [Google Scholar] [CrossRef]
- Gessi, M.; Gielen, G.H.; Denkhaus, D.; Antonelli, M.; Giangaspero, F.; zur Mühlen, A.; Japp, A.S.; Pietsch, T. Molecular heterogeneity characterizes glioblastoma with lipoblast/adipocyte-like cytology. Virchows Arch. 2015, 467, 105–109. [Google Scholar] [CrossRef]
- Gan, H.K.; Kaye, A.H.; Luwor, R.B. The EGFRvIII variant in glioblastoma multiforme. J. Clin. Neurosci. 2009, 16, 748–754. [Google Scholar] [CrossRef]
- Layfield, L.; Willmore, C.; Tripp, S.; Jones, C.; Jensen, R. Epidermal growth factor receptor gene amplification and protein expression in glioblastoma multiforme. Appl. Immunohistochem. Mol. Morphol. 2006, 14, 91–96. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciriello, G.; Sinha, R.; Hoadley, K.A.; Jacobsen, A.S.; Reva, B.; Perou, C.M.; Sander, C.; Schultz, N. The molecular diversity of Luminal A breast tumors. Breast Cancer Res. Treat. 2013, 141, 409–420. [Google Scholar] [CrossRef] [Green Version]
- Crespo, I.; Vital, A.L.; Nieto, A.B.; Rebelo, O.; Tão, H.; Lopes, M.C.; Oliveira, C.R.; French, P.J.; Orfao, A.; Tabernero, M.D. Detailed characterization of alterations of chromosomes 7, 9, and 10 in glioblastomas as assessed by single-nucleotide polymorphism arrays. J. Mol. Diagn. 2011, 13, 634–647. [Google Scholar] [CrossRef] [PubMed]
- Sahin, A.; Sanchez, C.; Bullain, S.; Waterman, P.; Weissleder, R.; Carter, B.S. Development of third generation anti-EGFRvIII chimeric T cells and EGFRvIII-expressing artificial antigen presenting cells for adoptive cell therapy for glioma. PLoS ONE 2018, 13, e0199414. [Google Scholar] [CrossRef]
- Kiang, K.M.-Y.; Leung, G.K.-K. A review on Adducin from functional to pathological mechanisms: Future direction in cancer. BioMed Res. Int. 2018, 1–14. [Google Scholar] [CrossRef]
- van den Boom, J.; Wolter, M.; Kuick, R.; Misek, D.E.; Youkilis, A.S.; Wechsler, D.S.; Sommer, C.; Reifenberger, G.; Hanash, S.M. Characterization of gene expression profiles associated with glioma progression using oligonucleotide-based microarray analysis and real-time reverse transcription-polymerase chain reaction. Am. J. Pathol. 2003, 163, 1033–1043. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Colella, S.; Kurrer, M.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. Gene expression profiling of low-grade diffuse astrocytomas by cDNA arrays. Cancer Res. 2000, 60, 6868–6874. [Google Scholar]
- Mariani, L.; Beaudry, C.; McDonough, W.S.; Hoelzinger, D.B.; Demuth, T.; Ross, K.R.; Berens, T.; Coons, S.W.; Watts, G.; Trent, J.M.; et al. Glioma cell motility is associated with reduced transcription of proapoptotic and proliferation genes: A cDNA microarray analysis. J. Neurooncol. 2001, 53, 161–176. [Google Scholar] [CrossRef]
- Kiang, K.M.-Y.; Zhang, P.; Li, N.; Zhu, Z.; Jin, L.; Leung, G.K.-K. Loss of cytoskeleton protein ADD3 promotes tumor growth and angiogenesis in glioblastoma multiforme. Cancer Lett. 2020, 474, 118–126. [Google Scholar] [CrossRef]
- Lechuga, S.; Amin, P.H.; Wolen, A.R.; Ivanov, A.I. Adducins inhibit lung cancer cell migration through mechanisms involving regulation of cell-matrix adhesion and cadherin-11 expression. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 395–408. [Google Scholar] [CrossRef]
- Sun, Q.; Pei, C.; Li, Q.; Dong, T.; Dong, Y.; Xing, W.; Zhou, P.; Gong, Y.; Zhen, Z.; Gao, Y.; et al. Up-regulation of MSH6 is associated with temozolomide resistance in human glioblastoma. Biochem. Biophys. Res. Commun. 2018, 496, 1040–1046. [Google Scholar] [CrossRef]
- Skiriute, D.; Vaitkiene, P.; Saferis, V.; Asmoniene, V.; Skauminas, K.; Deltuva, V.P.; Tamasauskas, A. MGMT, GATA6, CD81, DR4, and CASP8 gene promoter methylation in glioblastoma. BMC Cancer 2012, 12, 218. [Google Scholar] [CrossRef] [Green Version]
- Hansen, L.J.; Sun, R.; Yang, R.; Singh, S.X.; Chen, L.H.; Pirozzi, C.J.; Moure, C.J.; Hemphill, C.; Carpenter, A.B.; Healy, P.; et al. MTAP Loss Promotes Stemness in Glioblastoma and Confers Unique Susceptibility to Purine Starvation. Cancer Res. 2019, 79, 3383–3394. [Google Scholar] [CrossRef]
- Lubin, M.; Lubin, A. Selective killing of tumors deficient in methylthioadenosine phosphorylase: A novel strategy. PLoS ONE 2009, 4, e5735. [Google Scholar] [CrossRef] [Green Version]
- Marjon, K.; Cameron, M.J.; Quang, P.; Clasquin, M.F.; Mandley, E.; Kunii, K.; McVay, M.; Choe, S.; Kernytsky, A.; Gross, S.; et al. MTAP Deletions in Cancer Create Vulnerability to Targeting of the MAT2A/PRMT5/RIOK1 Axis. Cell Rep. 2016, 15, 574–587. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.; Lee, H.-O.; An, S.S.; Cai, K.Q.; Kruger, W.D. Specific Targeting of MTAP-Deleted Tumors with a Combination of 2′-Fluoroadenine and 5′-Methylthioadenosine. Cancer Res. 2018, 78, 4386–4395. [Google Scholar] [CrossRef] [Green Version]
- Kesarwani, P.; Prabhu, A.; Kant, S.; Chinnaiyan, P. Metabolic Remodeling Contributes Towards an Immune Suppressive Phenotype in Glioblastoma. Cancer Immunol. Immunother. 2019, 68, 1107–1120. [Google Scholar] [CrossRef]
- Ciaglia, E.; Abate, M.; Laezza, C.; Pisanti, S.; Vitale, M.; Seneca, V.; Torelli, G.; Franceschelli, S.; Catapano, G.; Gazzerro, P.; et al. Antiglioma effects of N6-isopentenyladenosine, an endogenous isoprenoid end product, through the downregulation of epidermal growth factor receptor. Int. J. Cancer 2017, 140, 959–972. [Google Scholar] [CrossRef] [Green Version]
- Ciaglia, E.; Grimaldi, M.; Abate, M.; Scrima, M.; Rodriquez, M.; Laezza, C.; Ranieri, R.; Pisanti, S.; Ciuffreda, P.; Manera, C.; et al. The isoprenoid derivative N6-benzyladenosine CM223 exerts antitumor effects in glioma patient-derived primary cells through the mevalonate pathway. Br. J. Pharmacol. 2017, 174, 2287–2301. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.-X.; Chen, L.; Wang, C.-H.; Lin, Z.-X.; Chen, B.-J.; You, N.; Chen, Y.; Wang, X.-F. The Vascular Notch Ligands Delta-Like Ligand 4 (DLL4) and Jagged1 (JAG1) Have Opposing Correlations with Microvascularization but a Uniform Prognostic Effect in Primary Glioblastoma: A Preliminary Study. World Neurosurg. 2016, 88, 447–458. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Specification | Outcome | Wild-Type IDH1/2 (n = 128) | Mutated IDH1 (n = 9) | p-Value | |
|---|---|---|---|---|---|---|
| Age | Mean (range), in years | 57.7 (24–81) | 59 (24–81) | 40.9 (32–52) | *** <0.001 mw | |
| ≤55 | 40.6% | 36.1% | 100% | *** <0.001 χ2 | ||
| >55 | 59.4% | 63.9% | ||||
| Sex | Male | 54.0% | 56.3% | 22.2% | 0.080 ft | |
| Female | 46.0% | 43.7% | 77.8% | |||
| Tumor location | Parietal | 35.0% | 33.9% | 50.0% | 0.282 kw | |
| Frontal | 20.3% | 19.1% | 37.5% | |||
| Temporal | 36.6% | 38.3% | 12.5% | |||
| Occipital | 4.9% | 5.2% | ||||
| Intraventricular | 0.8% | 0.9% | ||||
| Corpus Callosum | 2.4% | 2.6% | ||||
| Size (cm3) | Mean (range) | 5.2 cm (2–11) | 5.1 cm (2–11) | 6.0 cm (5–7) | 0.210 mw | |
| Initial symptom | Neurological deficit | 30.0% | 32.1% | 0% | 0.146 kw | |
| Epileptic seizure | 21.7% | 21.4% | 25.0% | |||
| Intracranial hypertension | 48.3% | 46.5% | 75.0% | |||
| KPS | ≤85 | 76.9% | 77.0% | 75.0% | 1.000 ft | |
| >85 | 23.1% | 23.0% | 25.0% | |||
| Overall survival | Median (95 CI) | 210 days | 180 days | 3300 days | *** <0.001 lr | |
| TP53 | Mutation | 20.2% | 17.3% | 50.0% | * 0.028 χ2 | |
| EGFR FISH | Alteration | 61.4% | 64.4% | 22.2% | * 0.012 χ2 | |
| N-amp | 38.6% | 35.6% | 77.8% | * 0.011 χ2 | ||
| L-amp | 13.4% | 12.7% | 22.2% | |||
| H-amp | 48.0% | 51.7% | 0.0% | |||
| EGFRvIII | No | 65. 9% | ||||
| Yes | 34.1% | |||||
| SCNA | EGFR | Gain | 65.4% | 70.1% | 0.0% | *** <0.001 χ2 |
| Normal | 34.6% | 29.9% | 100% | *** <0.001 kw | ||
| CDKN2A | Alteration | 63.5% | 65.6% | 33.3% | 0.052 χ2 | |
| Loss | 53.3% | |||||
| Gain | 10.2% | |||||
| CDKN2B | Alteration | 54.0% | 56.3% | 22.2% | 0.080 ft | |
| Loss | 48.9% | |||||
| Gain | 5.1% | |||||
| PTEN | Alteration | 65.9% | 65.9% | 66,7% | 0.961 χ2 | |
| Loss | 55.6% | |||||
| Gain | 10.4% | |||||
| MTAP | Alteration | 52.9% | 53.5% | 44.4% | 0.734 ft | |
| TIMP3 | Alteration | 65.0% | 64.1% | 77.8% | 0.404 χ2 | |
| ERBB2 | Alteration | 26.5% | ||||
| MVP | Alteration | 59.5% | 56.3% | 100% | * 0.020 ft | |
| MEN1 | Alteration | 60.6% | 57.8% | 100% | * 0.012 χ2 | |
| ADD3 | Alteration | 45.3% | 46.9% | 22.2% | 0.183 ft | |
| PCCA | Alteration | 44.1% | 46.6% | 12.5% | 0.075 ft |
| EGFR | |||||
|---|---|---|---|---|---|
| Shallow Deletion | Diploid | Gain | Amplification | ||
| CDKN2A | Deep deletion | 2 | 20 | 131 | 178 |
| Shallow deletion | 3 | 19 | 48 | 38 | |
| Diploid | 1 | 22 | 61 | 35 | |
| Gain | 0 | 2 | 14 | 3 | |
| Amplification | 0 | 1 | 0 | 0 | |
| *** n = 577; p < 0.00001; Chi-square 461.258 | |||||
| JAG1 | Shallow deletion | 1 | 3 | 13 | 1 |
| Diploid | 3 | 52 | 153 | 126 | |
| Gain | 2 | 9 | 88 | 125 | |
| Amplification | 0 | 0 | 0 | 1 | |
| *** n = 576; p < 0.00001; Chi-square 41.9092 | |||||
| MSH6 | Shallow deletion | 0 | 8 | 15 | 14 |
| Diploid | 4 | 55 | 220 | 225 | |
| Gain | 2 | 1 | 19 | 14 | |
| n = 571; p > 0.05; Chi-square 7.2998 | |||||
| MTAP | Deep deletion | 2 | 18 | 114 | 169 |
| Shallow deletion | 3 | 21 | 61 | 46 | |
| Diploid | 1 | 22 | 65 | 35 | |
| Gain | 0 | 2 | 14 | 3 | |
| Amplification | 0 | 1 | 0 | 0 | |
| *** n = 570; p < 0.00001; Chi-square 46.413 | |||||
| Genes Studied | Cluster 1 | Cluster 2 | Cluster 3 | p-Value |
|---|---|---|---|---|
| CTNNB1 | 11.1 | 8.0 | 28.6 | 0.120 (KW) |
| CDH7 | 0.0 | 12.0 | 4.8 | 0.166 (KW) |
| IQGAP1 | 18.5 | 12.0 | 23.8 | 0.581 (KW) |
| GLDC | 7.4 | 12.0 | 23.8 | 0.254 (KW) |
| TP53 | 11.1 | 12.0 | 42.9 | 0.011 * (Chi) |
| TGFBR2 | 11.1 | 16.0 | 9.5 | 0.781 (KW) |
| ING1 | 18.5 | 24.0 | 33.3 | 0.495 (Chi) |
| SIX3 | 14.8 | 24.0 | 52.4 | 0.014 * (Chi) |
| IL4 | 14.8 | 24.0 | 66.7 | <0.001 *** (Chi) |
| PCCA | 44.4 | 24.0 | 85.7 | <0.001 *** (Chi) |
| DYSF | 18.5 | 28.0 | 42.9 | 0.180 (Chi) |
| HNF1A | 37.0 | 28.0 | 57.1 | 0.124 (Chi) |
| DOCK8 | 7.4 | 32.0 | 9.5 | 0.036 * (KW) |
| ERBB2 | 14.8 | 36.0 | 19.0 | 0.169 (Chi) |
| ATL1 | 22.2 | 36.0 | 52.4 | 0.096 (Chi) |
| ATR | 25.9 | 48.0 | 19.0 | 0.080 (Chi) |
| TRAF4 | 44.4 | 52.0 | 28.6 | 0.268 (Chi) |
| JAG1 | 3.7 | 56.0 | 9.5 | <0.001 *** (Chi) |
| SPG11 | 14.8 | 56.0 | 9.5 | <0.001 *** (Chi) |
| EGFRvIII | 14.8 | 56.0 | 23.8 | <0.001 *** (Chi) |
| MSH6 | 11.1 | 60.0 | 66.7 | <0.001 *** (Chi) |
| ADD3 | 40.7 | 72.0 | 42.9 | 0.048 * (Chi) |
| SMARCA4 | 33.3 | 84.0 | 4.8 | <0.001 *** (Chi) |
| MVP | 51.9 | 84.0 | 14.3 | <0.001 *** (Chi) |
| MTAP | 22.2 | 84.0 | 76.2 | <0.001 *** (Chi) |
| PTEN | 44.4 | 88.0 | 38.1 | <0.001 *** (Chi) |
| TIMP3 | 44.4 | 92.0 | 57.1 | 0.001 ** (Chi) |
| EGFR | 40.7 | 92.0 | 85.7 | <0.001 *** (Chi) |
| MEN1 | 44.4 | 96.0 | 23.8 | <0.001 *** (Chi) |
| CDKN2A | 7.4 | 100.0 | 100.0 | <0.001 *** (Chi) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Navarro, L.; San-Miguel, T.; Megías, J.; Santonja, N.; Calabuig, S.; Muñoz-Hidalgo, L.; Roldán, P.; Cerdá-Nicolás, M.; López-Ginés, C. Identification of New Genetic Clusters in Glioblastoma Multiforme: EGFR Status and ADD3 Losses Influence Prognosis. Cells 2020, 9, 2429. https://doi.org/10.3390/cells9112429
Navarro L, San-Miguel T, Megías J, Santonja N, Calabuig S, Muñoz-Hidalgo L, Roldán P, Cerdá-Nicolás M, López-Ginés C. Identification of New Genetic Clusters in Glioblastoma Multiforme: EGFR Status and ADD3 Losses Influence Prognosis. Cells. 2020; 9(11):2429. https://doi.org/10.3390/cells9112429
Chicago/Turabian StyleNavarro, Lara, Teresa San-Miguel, Javier Megías, Nuria Santonja, Silvia Calabuig, Lisandra Muñoz-Hidalgo, Pedro Roldán, Miguel Cerdá-Nicolás, and Concha López-Ginés. 2020. "Identification of New Genetic Clusters in Glioblastoma Multiforme: EGFR Status and ADD3 Losses Influence Prognosis" Cells 9, no. 11: 2429. https://doi.org/10.3390/cells9112429
APA StyleNavarro, L., San-Miguel, T., Megías, J., Santonja, N., Calabuig, S., Muñoz-Hidalgo, L., Roldán, P., Cerdá-Nicolás, M., & López-Ginés, C. (2020). Identification of New Genetic Clusters in Glioblastoma Multiforme: EGFR Status and ADD3 Losses Influence Prognosis. Cells, 9(11), 2429. https://doi.org/10.3390/cells9112429