Therapeutic Strategies to Target Calcium Dysregulation in Alzheimer’s Disease


{kind=link}
{kind=link}
Abstract
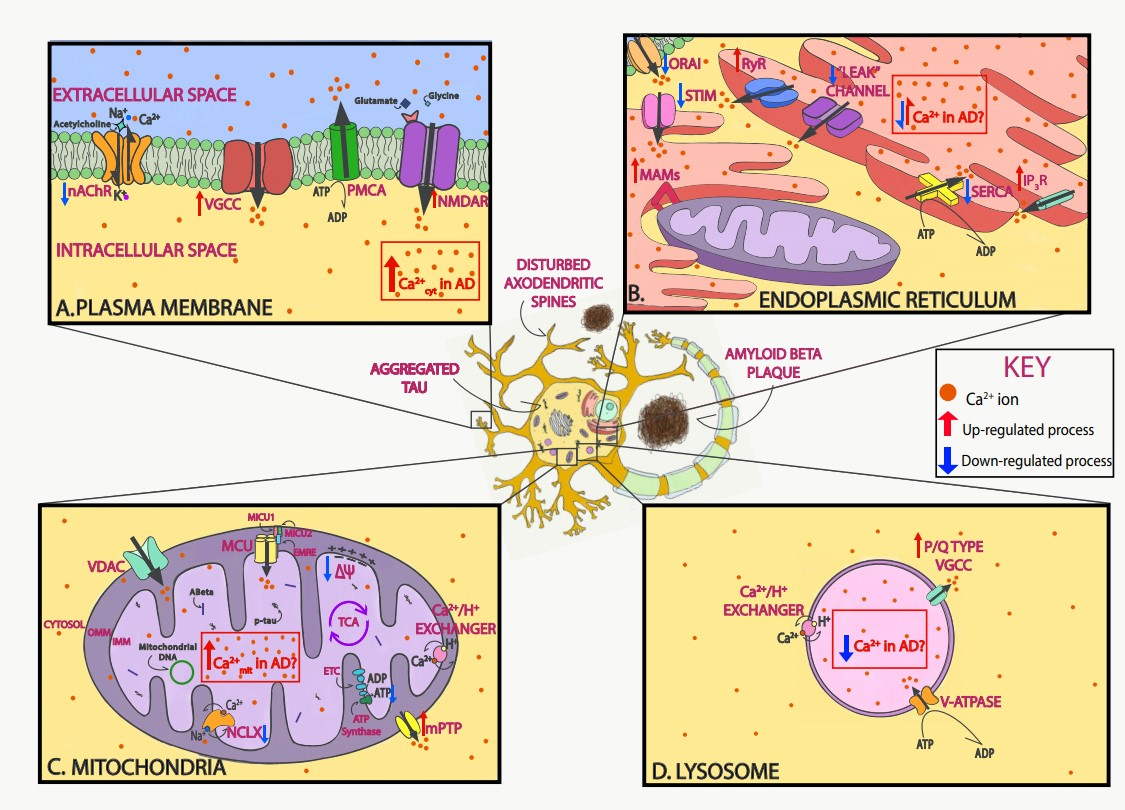
:1. Calcium Dysregulation Is a Hallmark of Alzheimer’s Disease
2. Neuronal Ca2+ as a Therapeutic Target in AD
2.1. Targeting Plasma Membrane Receptors and Cytosolic Ca2+
2.2. Targeting ER Ca2+ and SOCE
2.3. Targeting Mitochondrial Ca2+
2.4. Targeting Lysosomal Ca2+
3. Astrocytic Ca2+ as a Therapeutic Target in AD
4. Microglial Ca2+ as a Therapeutic Target in AD
5. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; E Schmechel, D.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; A Pericak-Vance, M. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- A Hardy, J.; A Higgins, G.; Mayford, M.; Barzilai, A.; Keller, F.; Schacher, S.; Kandel, E. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Davies, P. Selective Loss Of Central Cholinergic Neurons In Alzheimer’s Disease. Lancet 1976, 308, 1403. [Google Scholar] [CrossRef]
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of Tau Misfolding from the Outside to the Inside of a Cell. J. Biol. Chem. 2009, 284, 12845–12852. [Google Scholar] [CrossRef] [Green Version]
- McGeer, P.L.; Rogers, J. Anti-inflammatory agents as a therapeutic approach to Alzheimer’s disease. Neurology 1992, 42, 447. [Google Scholar] [CrossRef] [PubMed]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain—Implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattson, M.P. Calcium and neurodegeneration. Aging Cell 2007, 6, 337–350. [Google Scholar] [CrossRef]
- Khachaturian, Z.S. Hypothesis on the Regulation of Cytosol Calcium Concentration and the Aging Brain. Neurobiol. Aging 1987, 8, 345–346. [Google Scholar] [CrossRef]
- Boada, M.; Antúnez, C.; López-Arrieta, J.; Galán, J.J.; Morón, F.J.; Hernández, I.; Marín, J.; Martínez-Lage, P.; Alegret, M.; Carrasco, J.M.; et al. CALHM1 P86L Polymorphism is Associated with Late-Onset Alzheimer’s Disease in a Recessive Model. J. Alzheimer’s Dis. 2010, 20, 247–251. [Google Scholar] [CrossRef]
- Tolar, M.; Keller, J.N.; Chan, S.; Mattson, M.P.; Marques, M.A.; Crutcher, K.A. Truncated Apolipoprotein E (ApoE) Causes Increased Intracellular Calcium and May Mediate ApoE Neurotoxicity. J. Neurosci. 1999, 19, 7100–7110. [Google Scholar] [CrossRef] [Green Version]
- Zatti, G.; Ghidoni, R.; Barbiero, L.; Binetti, G.; Pozzan, T.; Fasolato, C.; Pizzo, P. The presenilin 2 M239I mutation associated with familial Alzheimer’s disease reduces Ca2+ release from intracellular stores. Neurobiol. Dis. 2004, 15, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Arbel-Ornath, M.; Hudry, E.; Boivin, J.R.; Hashimoto, T.; Takeda, S.; Kuchibhotla, K.V.; Hou, S.; Lattarulo, C.R.; Belcher, A.M.; Trujillo, P.B.; et al. Soluble oligomeric amyloid-beta induces calcium dyshomeostasis that precedes synapse loss in the living mouse brain. Mol. Neurodegener. 2017, 12, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busche, M.A.; Chen, X.; Henning, H.A.; Reichwald, J.; Staufenbiel, M.; Sakmann, B.; Konnerth, A. Critical role of soluble amyloid-beta for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2012, 109, 8740–8745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busche, M.A.; Eichhoff, G.; Adelsberger, H.; Abramowski, D.; Wiederhold, K.-H.; Haass, C.; Staufenbiel, M.; Konnerth, A.; Garaschuk, O. Clusters of Hyperactive Neurons Near Amyloid Plaques in a Mouse Model of Alzheimer’s Disease. Science 2008, 321, 1686–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuchibhotla, K.V.; Goldman, S.T.; Lattarulo, C.R.; Wu, H.Y.; Hyman, B.T.; Bacskai, B.J. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 2008, 59, 214–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busche, M.A.; Wegmann, S.; Dujardin, S.; Commins, C.; Schiantarelli, J.; Klickstein, N.; Kamath, T.V.; Carlson, G.A.; Nelken, I.; Hyman, B.T. Tau impairs neural circuits, dominating amyloid-beta effects, in Alzheimer models in vivo. Nat. Neurosci. 2019, 22, 57–64. [Google Scholar] [CrossRef]
- Delekate, A.; Füchtemeier, M.; Schumacher, T.; Ulbrich, C.; Foddis, M.; Petzold, G.C. Metabotropic P2Y1 receptor signalling mediates astrocytic hyperactivity in vivo in an Alzheimer’s disease mouse model. Nat. Commun. 2014, 5, 5422. [Google Scholar] [CrossRef] [Green Version]
- Kuchibhotla, K.V.; Lattarulo, C.R.; Hyman, B.T.; Bacskai, B.J. Synchronous Hyperactivity and Intercellular Calcium Waves in Astrocytes in Alzheimer Mice. Science 2009, 323, 1211–1215. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Kastanenka, K.V.; Arbel-Ornath, M.; Commins, C.; Kuzuya, A.; Lariviere, A.J.; Krafft, G.A.; Hefti, F.; Jerecic, J.; Bacskai, B.J. An acute functional screen identifies an effective antibody targeting amyloid-beta oligomers based on calcium imaging. Sci. Rep. 2018, 8, 4634. [Google Scholar] [CrossRef]
- Palop, J.J.; Mucke, L. Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2016, 17, 777–792. [Google Scholar] [CrossRef]
- Khachaturian, Z.S. The role of calcium regulation in brain aging: Reexamination of a hypothesis. Aging Clin. Exp. Res. 1989, 1, 17–34. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M. Calcium dyshomeostasis and intracellular signalling in alzheimer’s disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Abeysinghe, A.; Deshapriya, R.; Udawatte, C. Alzheimer’s disease; a review of the pathophysiological basis and therapeutic interventions. Life Sci. 2020, 256, 117996. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Solfrizzi, V.; Imbimbo, B.P.; Logroscino, G. Amyloid-directed monoclonal antibodies for the treatment of Alzheimer’s disease: The point of no return? Expert Opin. Biol. Ther. 2014, 14, 1465–1476. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussiere, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Mattson, M.P. Calcium and neuronal injury in Alzheimer’s disease. Contributions of beta-amyloid precursor protein mismetabolism, free radicals, and metabolic compromise. Ann. N. Y. Acad. Sci. 1994, 747, 50–76. [Google Scholar] [CrossRef]
- Arispe, N.; Rojas, E.; Pollard, H.B. Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: Blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. USA 1993, 90, 567–571. [Google Scholar] [CrossRef] [Green Version]
- De Felice, F.G.; Velasco, P.T.; Lambert, M.P.; Viola, K.; Fernandez, S.J.; Ferreira, S.T.; Klein, W.L. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 2007, 282, 11590–11601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pourbadie, H.G.; Naderi, N.; Mehranfard, N.; Janahmadi, M.; Khodagholi, F.; Motamedi, F. Preventing Effect of L-Type Calcium Channel Blockade on Electrophysiological Alterations in Dentate Gyrus Granule Cells Induced by Entorhinal Amyloid Pathology. PLoS ONE 2015, 10, e0117555. [Google Scholar] [CrossRef] [PubMed]
- Stutzmann, G.E.; Smith, I.; Caccamo, A.; Oddo, S.; LaFerla, F.M.; Parker, I. Enhanced Ryanodine Receptor Recruitment Contributes to Ca2+ Disruptions in Young, Adult, and Aged Alzheimer’s Disease Mice. J. Neurosci. 2006, 26, 5180–5189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawlor, B.A.; Kennelly, S.; O’Dwyer, S.; Cregg, F.; Walsh, C.D.; Cohen, R.; Kenny, R.A.; Howard, R.; Murphy, C.; Adams, J.; et al. NILVAD protocol: A European multicentre double-blind placebo-controlled trial of nilvadipine in mild-to-moderate Alzheimer’s disease. BMJ Open 2014, 4, e006364. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, B.A.; Segurado, R.; Kennelly, S.; Rikkert, M.G.M.O.; Howard, R.; Pasquier, F.; Börjesson-Hanson, A.; Tsolaki, M.; Lucca, U.; Molloy, D.W.; et al. Nilvadipine in mild to moderate Alzheimer disease: A randomised controlled trial. PLoS Med. 2018, 15, e1002660. [Google Scholar] [CrossRef] [PubMed]
- Esteras, N.; Kundel, F.; Amodeo, G.F.; Pavlov, E.V.; Klenerman, D.; Abramov, A.Y. Insoluble tau aggregates induce neuronal death through modification of membrane ion conductance, activation of voltage-gated calcium channels and NADPH oxidase. FEBS J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Dreses-Werringloer, U. A polymorphism in CALHM1 influences Ca2+ homeostasis, Abeta levels, and Alzheimer’s disease risk. Cell 2008, 133, 1149–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minster, R.L.; Demirci, F.Y.; DeKosky, S.T.; Kamboh, M.I. No association betweenCALHM1variation and risk of Alzheimer disease. Hum. Mutat. 2009, 30, E566–E569. [Google Scholar] [CrossRef] [Green Version]
- Tan, E.; Ho, P.; Cheng, S.; Yih, Y.; Li, H.; Fook-Chong, S.; Lee, W.; Zhao, Y. CALHM1 variant is not associated with Alzheimer’s disease among Asians. Neurobiol. Aging 2011, 32, 546.e11–546.e12. [Google Scholar] [CrossRef]
- Vingtdeux, V.; Chang, E.H.; Frattini, S.A.; Zhao, H.; Chandakkar, P.; Adrien, L.; Strohl, J.J.; Gibson, E.L.; Ohmoto, M.; Matsumoto, I.; et al. CALHM1 deficiency impairs cerebral neuron activity and memory flexibility in mice. Sci. Rep. 2016, 6, 24250. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y. The Role of NMDA Receptors in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, I.; Bajouco, L.; Mota, S.; Auberson, Y.; Oliveira, C.; Rego, A.C. Amyloid beta peptide 1–42 disturbs intracellular calcium homeostasis through activation of GluN2B-containing N-methyl-d-aspartate receptors in cortical cultures. Cell Calcium 2012, 51, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Ronicke, R.; Mikhaylova, M.; Ronicke, S.; Meinhardt, J.; Schroder, U.H.; Fandrich, M.; Reiser, G.; Kreutz, M.R.; Reymann, K.G. Early neuronal dysfunction by amyloid beta oligomers depends on activation of NR2B-containing NMDA receptors. Neurobiol. Aging 2011, 32, 2219–2228. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, P.; Feng, J.; Wu, M. Dysfunction of NMDA receptors in Alzheimer’s disease. Neurol. Sci. 2016, 37, 1039–1047. [Google Scholar] [CrossRef] [Green Version]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef] [Green Version]
- Decker, J.M.; Krüger, L.; Sydow, A.; Dennissen, F.J.; Siskova, Z.; Mandelkow, E.; Mandelkow, E. The Tau/A152T mutation, a risk factor for frontotemporal-spectrum disorders, leads to NR 2B receptor-mediated excitotoxicity. EMBO Rep. 2016, 17, 552–569. [Google Scholar] [CrossRef] [Green Version]
- Maeda, S.; Djukic, B.; Taneja, P.; Yu, G.; Lo, I.; Davis, A.; Craft, R.; Guo, W.; Wang, X.; Kim, D.; et al. Expression of A152T human tau causes age-dependent neuronal dysfunction and loss in transgenic mice. EMBO Rep. 2016, 17, 530–551. [Google Scholar] [CrossRef]
- Esclaire, F.; Lesort, M.; Blanchard, C.; Hugon, J. Glutamate toxicity enhances tau gene expression in neuronal cultures. J. Neurosci. Res. 1997, 49, 309–318. [Google Scholar] [CrossRef]
- Sindou, P.; Lesort, M.; Couratier, P.; Yardin, C.; Esclaire, F.; Hugon, J. Glutamate increases tau phosphorylation in primary neuronal cultures from fetal rat cerebral cortex. Brain Res. 1994, 646, 124–128. [Google Scholar] [CrossRef]
- Sun, X.-Y.; Tuo, Q.-Z.; Liuyang, Z.-Y.; Xie, A.-J.; Feng, X.-L.; Yan, X.; Qiu, M.; Li, S.; Wang, X.-L.; Cao, F.-Y.; et al. Extrasynaptic NMDA receptor-induced tau overexpression mediates neuronal death through suppressing survival signaling ERK phosphorylation. Cell Death Dis. 2016, 7, e2449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipton, S.A. Failures and successes of NMDA receptor antagonists: Molecular basis for the use of open-channel blockers like memantine in the treatment of acute and chronic neurologic insults. NeuroRx 2004, 1, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Kishi, T.; Matsunaga, S.; Oya, K.; Nomura, I.; Ikuta, T.; Iwata, N. Memantine for Alzheimer’s Disease: An Updated Systematic Review and Meta-analysis. J. Alzheimer’s Dis. 2017, 60, 401–425. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, C.P.; Clarke, J.R.; Ledo, J.H.; Ribeiro, F.C.; Costa, C.V.; Melo, H.M.; Mota-Sales, A.P.; Saraiva, L.M.; Klein, W.L.; Sebollela, A. et al.; et al. Memantine rescues transient cognitive impairment caused by high-molecular-weight abeta oligomers but not the persistent impairment induced by low-molecular-weight oligomers. J. Neurosci. 2013, 33, 9626–9634. [Google Scholar] [CrossRef] [Green Version]
- Song, M.S.; Rauw, G.; Baker, G.B.; Kar, S. Memantine protects rat cortical cultured neurons against β-amyloid-induced toxicity by attenuating tau phosphorylation. Eur. J. Neurosci. 2008, 28, 1989–2002. [Google Scholar] [CrossRef]
- Wevers, A.; Monteggia, L.; Nowacki, S.; Bloch, W.; Schutz, U.; Lindstrom, J.; Pereira, E.F.R.; Eisenberg, H.; Giacobini, E.; De Vos, R.A.I.; et al. Expression of nicotinic acetylcholine receptor subunits in the cerebral cortex in Alzheimer’s disease: Histotopographical correlation with amyloid plaques and hyperphosphorylated-tau protein. Eur. J. Neurosci. 1999, 11, 2551–2565. [Google Scholar] [CrossRef]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Lahiri, D.K.; Rogers, J.T.; Greig, N.H.; Sambamurti, K. Rationale for the Development of Cholinesterase Inhibitors as Anti- Alzheimer Agents. Curr. Pharm. Des. 2004, 10, 3111–3119. [Google Scholar] [CrossRef]
- Herholz, K. Acetylcholine esterase activity in mild cognitive impairment and Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 25–29. [Google Scholar] [CrossRef]
- Patel, L.; Grossberg, G.T. Combination Therapy for Alzheimerʼs Disease. Drugs Aging 2011, 28, 539–546. [Google Scholar] [CrossRef]
- Gómez-Ramos, A.; Díaz-Hernández, M.; Rubio, A.; Miras-Portugal, M.T.; Avila, J. Extracellular tau promotes intracellular calcium increase through M1 and M3 muscarinic receptors in neuronal cells. Mol. Cell. Neurosci. 2008, 37, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Hartigan, J.A.; Johnson, G.V.W. Transient Increases in Intracellular Calcium Result in Prolonged Site-selective Increases in Tau Phosphorylation through a Glycogen Synthase Kinase 3β-dependent Pathway. J. Biol. Chem. 1999, 274, 21395–21401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacobini, E.; Gold, G. Alzheimer disease therapy—Moving from amyloid-beta to tau. Nat. Rev. Neurol. 2013, 9, 677–686. [Google Scholar] [CrossRef]
- Kang, S.; Son, S.M.; Baik, S.H.; Yang, J.; Mook-Jung, I. Autophagy-Mediated Secretory Pathway is Responsible for Both Normal and Pathological Tau in Neurons. J. Alzheimer’s Dis. 2019, 70, 667–680. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Devos, S.L.; Corjuc, B.T.; Oakley, D.H.; Nobuhara, C.K.; Bannon, R.N.; Chase, A.; Commins, C.; Gonzalez, J.A.; Dooley, P.M.; Frosch, M.P.; et al. Synaptic Tau Seeding Precedes Tau Pathology in Human Alzheimer’s Disease Brain. Front. Neurosci. 2018, 12, 267. [Google Scholar] [CrossRef] [Green Version]
- Friedhoff, P.; Von Bergen, M.; Mandelkow, E.-M.; Davies, P. A nucleated assembly mechanism of Alzheimer paired helical filaments. Proc. Nat. Acad. Sci. USA 1998, 95, 15712–15717. [Google Scholar] [CrossRef] [Green Version]
- Colin, M.; Dujardin, S.; Schraen-Maschke, S.; Meno-Tetang, G.; Duyckaerts, C.; Courade, J.-P.; Buée, L. From the prion-like propagation hypothesis to therapeutic strategies of anti-tau immunotherapy. Acta Neuropathol. 2020, 139, 3–25. [Google Scholar] [CrossRef] [Green Version]
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef]
- Jadhav, S.; Avila, J.; Schöll, M.; Kovacs, G.G.; Kövari, E.; Skrabana, R.; Evans, L.D.; Kontsekova, E.; Malawska, B.; De Silva, R.; et al. A walk through tau therapeutic strategies. Acta Neuropathol. Commun. 2019, 7, 1–31. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 272–293. [Google Scholar] [CrossRef]
- Cirrito, J.R.; Yamada, K.A.; Finn, M.B.; Sloviter, R.S.; Bales, K.R.; May, P.C.; Schoepp, D.D.; Paul, S.M.; Mennerick, S.; Holtzman, D.M. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 2005, 48, 913–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamenetz, F.; Tomita, T.; Hsieh, H.; Seabrook, G.; Borchelt, D.; Iwatsubo, T.; Sisodia, S.; Malinow, R. APP Processing and Synaptic Function. Neuron 2003, 37, 925–937. [Google Scholar] [CrossRef] [Green Version]
- Palop, J.J. Epilepsy and Cognitive Impairments in Alzheimer Disease. Arch. Neurol. 2009, 66, 435–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, P.E.; Zhu, L.; Verret, L.; Vossel, K.A.; Orr, A.G.; Cirrito, J.R.; Devidze, N.; Ho, K.; Yu, G.-Q.; Palop, J.J.; et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc. Nat. Acad. Sci. USA 2012, 109, E2895–E2903. [Google Scholar] [CrossRef] [Green Version]
- Kastanenka, K.V.; Bussiere, T.; Shakerdge, N.; Qian, F.; Weinreb, P.H.; Rhodes, K.; Bacskai, B.J. Immunotherapy with Aducanumab Restores Calcium Homeostasis in Tg2576 Mice. J. Neurosci. 2016, 36, 12549–12558. [Google Scholar] [CrossRef] [Green Version]
- Schneider, L. A resurrection of aducanumab for Alzheimer’s disease. Lancet Neurol. 2020, 19, 111–112. [Google Scholar] [CrossRef] [Green Version]
- Mekahli, D.; Bultynck, G.; Parys, J.B.; De Smedt, H.; Missiaen, L. Endoplasmic-Reticulum Calcium Depletion and Disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a004317. [Google Scholar] [CrossRef]
- Ito, E.; Oka, K.; Etcheberrigaray, R.; Nelson, T.J.; McPHIE, D.L.; Tofel-Grehl, B.; Gibson, G.E.; Alkon, D.L. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc. Nat. Acad. Sci. USA 1994, 91, 534–538. [Google Scholar] [CrossRef] [Green Version]
- Etcheberrigaray, R.; Hirashima, N.; Nee, L.; Prince, J.; Govoni, S.; Racchi, M.; Tanzi, R.E.; Alkon, D.L. Calcium responses in fibroblasts from asymptomatic members of Alzheimer’s disease families. Neurobiol. Dis. 1998, 5, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Stutzmann, G.E.; Caccamo, A.; LaFerla, F.M.; Parker, I. Dysregulated IP3 Signaling in Cortical Neurons of Knock-In Mice Expressing an Alzheimer’s-Linked Mutation in Presenilin1 Results in Exaggerated Ca2+ Signals and Altered Membrane Excitability. J. Neurosci. 2004, 24, 508–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shilling, D.; Müller, M.; Takano, H.; Mak, D.-O.D.; Abel, T.; Coulter, D.A.; Foskett, J.K. Suppression of InsP3 Receptor-Mediated Ca2+ Signaling Alleviates Mutant Presenilin-Linked Familial Alzheimer’s Disease Pathogenesis. J. Neurosci. 2014, 34, 6910–6923. [Google Scholar] [CrossRef] [Green Version]
- De Strooper, B.; Annaert, W.; Cupers, P.; Saftig, P.; Craessaerts, K.; Mumm, J.S.; Schroeter, E.H.; Schrijvers, V.; Wolfe, M.S.; Ray, W.J.; et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature 1999, 398, 518–522. [Google Scholar] [CrossRef]
- Struhl, G.; Greenwald, I. Presenilin is required for activity and nuclear access of Notch in Drosophila. Nat. Cell Biol. 1999, 398, 522–525. [Google Scholar] [CrossRef]
- A Saura, C.; Choi, S.-Y.; Beglopoulos, V.; Malkani, S.; Zhang, D.; Rao, B.S.S.; Chattarji, S.; Kelleher, R.J.; Kandel, E.R.; Duff, K.; et al. Loss of Presenilin Function Causes Impairments of Memory and Synaptic Plasticity Followed by Age-Dependent Neurodegeneration. Neuron 2004, 42, 23–36. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, H.; Xia, D.; Kanekiyo, T.; Kelleher, R.J.; Shen, J. Familial Frontotemporal Dementia-Associated Presenilin-1 c.548G>T Mutation Causes Decreased mRNA Expression and Reduced Presenilin Function in Knock-In Mice. J. Neurosci. 2012, 32, 5085–5096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, W.; Zhang, J.; Kholodenko, D.; Citron, M.; Podlisny, M.B.; Teplow, D.B.; Haass, C.; Seubert, P.; Koo, E.H.; Selkoe, D.J. Enhanced Production and Oligomerization of the 42-residue Amyloid β-Protein by Chinese Hamster Ovary Cells Stably Expressing Mutant Presenilins. J. Biol. Chem. 1997, 272, 7977–7982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duff, K.; Eckman, C.; Zehr, C.; Yu, X.; Prada, C.M.; Perez-tur, J.; Hutton, M.; Buee, L.; Harigaya, Y.; Yager, D.; et al. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin. Nature 1996, 383, 710–713. [Google Scholar] [CrossRef]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef]
- Borchelt, D.R.; Thinakaran, G.; Eckman, C.B.; Lee, M.K.; Davenport, F.; Ratovitsky, T.; Prada, C.M.; Kim, G.; Seekins, S.; Yager, D.; et al. Familial Alzheimer’s disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron 1996, 17, 1005–1013. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Iii, R.J.K. The presenilin hypothesis of Alzheimer’s disease: Evidence for a loss-of-function pathogenic mechanism. Proc. Nat. Acad. Sci. USA 2007, 104, 403–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, H.; Nelson, O.; Bezprozvanny, A.; Wang, Z.; Lee, S.-F.; Hao, Y.-H.; Serneels, L.; De Strooper, B.; Yu, G.; Bezprozvanny, I. Presenilins Form ER Ca2+ Leak Channels, a Function Disrupted by Familial Alzheimer’s Disease-Linked Mutations. Cell 2006, 126, 981–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCombs, J.E.; Gibson, E.A.; Palmer, A.E. Using a genetically targeted sensor to investigate the role of presenilin-1 in ER Ca2+ levels and dynamics. Mol. Biosyst. 2010, 6, 1640–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shilling, D.; Mak, D.-O.D.; Kang, D.E.; Foskett, J.K. Lack of Evidence for Presenilins as Endoplasmic Reticulum Ca2+ Leak Channels. J. Biol. Chem. 2012, 287, 10933–10944. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.C.; Zheng, Q.; Tan, H.; Zhang, B.; Li, X.; Yang, Y.; Yu, J.; Liu, Y.; Chai, H.; Wang, X.; et al. TMCO1 Is an ER Ca2+ Load-Activated Ca2+ Channel. Cell 2016, 165, 1454–1466. [Google Scholar] [CrossRef] [Green Version]
- Bruno, A.M.; Huang, J.Y.; Bennett, D.A.; Marr, R.A.; Hastings, M.L.; Stutzmann, G.E. Altered ryanodine receptor expression in mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging 2012, 33, 1001.e1–1001.e6. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.L.; Mayne, M.; Holden, C.P.; Geiger, J.D.; Mattson, M.P. Presenilin-1 Mutations Increase Levels of Ryanodine Receptors and Calcium Release in PC12 Cells and Cortical Neurons. J. Biol. Chem. 2000, 275, 18195–18200. [Google Scholar] [CrossRef] [Green Version]
- Briggs, C.A.; Chakroborty, S.; Stutzmann, G.E. Emerging pathways driving early synaptic pathology in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2017, 483, 988–997. [Google Scholar] [CrossRef] [Green Version]
- Rybalchenko, V.; Hwang, S.-Y.; Rybalchenko, N.; Koulen, P. The cytosolic N-terminus of presenilin-1 potentiates mouse ryanodine receptor single channel activity. Int. J. Biochem. Cell Biol. 2008, 40, 84–97. [Google Scholar] [CrossRef]
- Wu, B.; Yamaguchi, H.; Lai, F.A.; Shen, J. Presenilins regulate calcium homeostasis and presynaptic function via ryanodine receptors in hippocampal neurons. Proc. Nat. Acad. Sci. USA 2013, 110, 15091–15096. [Google Scholar] [CrossRef] [Green Version]
- Oulès, B.; Del Prete, D.; Greco, B.; Zhang, X.; Lauritzen, I.; Sevalle, J.; Moreno, S.; Paterlini-Bréchot, P.; Trebak, M.; Checler, F.; et al. Ryanodine receptor blockade reduces amyloid-beta load and memory impairments in Tg2576 mouse model of Alzheimer disease. J. Neurosci. 2012, 32, 11820–11834. [Google Scholar] [CrossRef] [PubMed]
- Zahradník, I.; Györke, S.; Zahradníková, A. Calcium Activation of Ryanodine Receptor Channels—Reconciling RyR Gating Models with Tetrameric Channel Structure. J. Gen. Physiol. 2005, 126, 515–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreiro, E.; Oliveira, C.R.; Pereira, C. Involvement of endoplasmic reticulum Ca2+ release through ryanodine and inositol 1,4,5-triphosphate receptors in the neurotoxic effects induced by the amyloid-beta peptide. J. Neurosci. Res. 2004, 76, 872–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supnet, C.; Grant, J.; Kong, H.; Westaway, D.; Mayne, M. Amyloid-β-(1-42) Increases Ryanodine Receptor-3 Expression and Function in Neurons of TgCRND8 Mice. J. Biol. Chem. 2006, 281, 38440–38447. [Google Scholar] [CrossRef] [Green Version]
- Bussiere, R.; Lacampagne, A.; Reiken, S.; Liu, X.; Scheuerman, V.; Zalk, R.; Martin, C.; Checler, F.; Marks, A.R.; Chami, M. Amyloid beta production is regulated by beta2-adrenergic signaling-mediated post-translational modifications of the ryanodine receptor. J. Biol. Chem. 2017, 292, 10153–10168. [Google Scholar] [CrossRef] [Green Version]
- Lacampagne, A.; Liu, X.; Reiken, S.; Bussiere, R.; Meli, A.C.; Lauritzen, I.; Teich, A.F.; Zalk, R.; Saint, N.; Arancio, O.; et al. Post-translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer’s disease-like pathologies and cognitive deficits. Acta Neuropathol. 2017, 134, 749–767. [Google Scholar] [CrossRef]
- Brunello, L.; Zampese, E.; Florean, C.; Pozzan, T.; Pizzo, P.; Fasolato, C. Presenilin-2 dampens intracellular Ca2+ stores by increasing Ca2+ leakage and reducing Ca2+ uptake. J. Cell. Mol. Med. 2009, 13, 3358–3369. [Google Scholar] [CrossRef] [Green Version]
- Green, K.N.; Demuro, A.; Akbari, Y.; Hitt, B.D.; Smith, I.F.; Parker, I.; LaFerla, F.M. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J. Cell Biol. 2008, 181, 1107–1116. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Liang, G.; Inan, S.; Wu, Z.; Joseph, D.J.; Meng, Q.; Peng, Y.; Eckenhoff, M.F.; Wei, H. Dantrolene ameliorates cognitive decline and neuropathology in Alzheimer triple transgenic mice. Neurosci. Lett. 2012, 516, 274–279. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Yang, B.; Liu, C.; Liang, G.; Liu, W.; Pickup, S.; Meng, Q.; Tian, Y.; Li, S.; Eckenhoff, M.F.; et al. Long-term Dantrolene Treatment Reduced Intraneuronal Amyloid in Aged Alzheimer Triple Transgenic Mice. Alzheimer Dis. Assoc. Disord. 2015, 29, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Bollimuntha, S.; Pani, B.; Singh, B.B. Neurological and Motor Disorders: Neuronal Store-Operated Ca2+ Signaling: An Overview and Its Function. Atherosclerosis 2017, 993, 535–556. [Google Scholar] [CrossRef] [Green Version]
- Kraft, R. STIM and ORAI proteins in the nervous system. Channels 2015, 9, 245–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Putney, J.W. The Physiological Function of Store-operated Calcium Entry. Neurochem. Res. 2011, 36, 1157–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leissring, M.A.; Akbari, Y.; Fanger, C.M.; Cahalan, M.D.; Mattson, M.P.; LaFerla, F.M. Capacitative Calcium Entry Deficits and Elevated Luminal Calcium Content in Mutant Presenilin-1 Knockin Mice. J. Cell Biol. 2000, 149, 793–798. [Google Scholar] [CrossRef] [Green Version]
- Smith, I.; Boyle, J.; Vaughan, P.; Pearson, H.; Cowburn, R.; Peers, C. Ca2+ Stores and capacitative Ca2+ entry in human neuroblastoma (SH-SY5Y) cells expressing a familial Alzheimer’s disease presenilin-1 mutation. Brain Res. 2002, 949, 105–111. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Hernando-Perez, E.; Nunez, L.; Villalobos, C. Amyloid beta Oligomers Increase ER-Mitochondria Ca2+ Cross Talk in Young Hippocampal Neurons and Exacerbate Aging-Induced Intracellular Ca2+ Remodeling. Front. Cell. Neurosci. 2019, 13, 22. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhang, H.; Liu, J.; Popugaeva, E.; Xu, N.-J.; Feske, S.; White, C.L.; Bezprozvanny, I. Reduced Synaptic STIM2 Expression and Impaired Store-Operated Calcium Entry Cause Destabilization of Mature Spines in Mutant Presenilin Mice. Neuron 2014, 82, 79–93. [Google Scholar] [CrossRef] [Green Version]
- Yoo, A.S.; Cheng, I.; Chung, S.; Grenfell, T.Z.; Lee, H.; Pack-Chung, E.; Handler, M.; Shen, J.; Xia, W.; Tesco, G.; et al. Presenilin-Mediated Modulation of Capacitative Calcium Entry. Neuron 2000, 27, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wu, L.; Pchitskaya, E.; Zakharova, O.D.; Saito, T.; Saido, T.C.; Bezprozvanny, I.B. Neuronal Store-Operated Calcium Entry and Mushroom Spine Loss in Amyloid Precursor Protein Knock-In Mouse Model of Alzheimer’s Disease. J. Neurosci. 2015, 35, 13275–13286. [Google Scholar] [CrossRef] [Green Version]
- Tsukamoto, A.; Kaneko, Y. Thapsigargin, a Ca2+-ATPase inhibitor, depletes the intracellular Ca2+ pool and induces apoptosis in human hepatoma cells. Cell Biol. Int. 1993, 17, 969–970. [Google Scholar] [CrossRef]
- Soboloff, J.; Berger, S.A. Sustained ER Ca2+ Depletion Suppresses Protein Synthesis and Induces Activation-enhanced Cell Death in Mast Cells. J. Biol. Chem. 2002, 277, 13812–13820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popugaeva, E.; Pchitskaya, E.; Speshilova, A.B.; Alexandrov, S.; Zhang, H.; Vlasova, O.; Bezprozvanny, I.B. STIM2 protects hippocampal mushroom spines from amyloid synaptotoxicity. Mol. Neurodegener. 2015, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, E.; Zhou, Q.; Li, S.; Wang, X.; Liu, Y.; Wang, L.; Sun, D.; Ye, J.; Gao, Y.; et al. TRPC1 Null Exacerbates Memory Deficit and Apoptosis Induced by Amyloid-beta. J. Alzheimer’s Dis. JAD 2018, 63, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Werth, J.L.; A Thayer, S. Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. J. Neurosci. 1994, 14, 348–356. [Google Scholar] [CrossRef] [Green Version]
- Billups, B.; Forsythe, I.D. Presynaptic Mitochondrial Calcium Sequestration Influences Transmission at Mammalian Central Synapses. J. Neurosci. 2002, 22, 5840–5847. [Google Scholar] [CrossRef] [Green Version]
- Wescott, A.P.; Kao, J.P.Y.; Lederer, W.J.; Boyman, L. Voltage-energized calcium-sensitive ATP production by mitochondria. Nat. Metab. 2019, 1, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; A Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nat. Cell Biol. 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nat. Cell Biol. 2011, 476, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Raffaello, A.; De Stefani, D.; Sabbadin, D.; Teardo, E.; Merli, G.; Picard, A.; Checchetto, V.; Moro, S.; Szabò, I.; Rizzuto, R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013, 32, 2362–2376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.P.; Luongo, T.S.; Tomar, D.; Jadiya, P.; Gao, E.; Zhang, X.; Lucchese, A.M.; Kolmetzky, D.W.; Shah, N.S.; Elrod, J.W. MCUB Regulates the Molecular Composition of the Mitochondrial Calcium Uniporter Channel to Limit Mitochondrial Calcium Overload During Stress. Circulation 2019, 140, 1720–1733. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Markhard, A.L.; Kitami, T.; Kovács-Bogdán, E.; Kamer, K.J.; Udeshi, N.D.; Carr, S.A.; Chaudhuri, D.; Clapham, D.E.; Li, A.A.; et al. EMRE Is an Essential Component of the Mitochondrial Calcium Uniporter Complex. Science 2013, 342, 1379–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perocchi, F.; Gohil, V.M.; Girgis, H.S.; Bao, X.R.; McCombs, J.E.; Palmer, A.E.; Mootha, V.K. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nat. Cell Biol. 2010, 467, 291–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallilankaraman, K.; Doonan, P.J.; Cárdenas, C.; Chandramoorthy, H.C.; Müller, M.; Miller, R.; Hoffman, N.E.; Gandhirajan, R.K.; Molgó, J.; Birnbaum, M.J.; et al. MICU1 Is an Essential Gatekeeper for MCU-Mediated Mitochondrial Ca2+ Uptake that Regulates Cell Survival. Cell 2012, 151, 630–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patron, M.; Checchetto, V.; Raffaello, A.; Teardo, E.; Reane, D.V.; Mantoan, M.; Granatiero, V.; Szabò, I.; De Stefani, D.; Rizzuto, R. MICU1 and MICU2 Finely Tune the Mitochondrial Ca2+ Uniporter by Exerting Opposite Effects on MCU Activity. Mol. Cell 2014, 53, 726–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csordás, G.; Golenár, T.; Seifert, E.L.; Kamer, K.J.; Sancak, Y.; Perocchi, F.; Moffat, C.; Weaver, D.; Perez, S.D.L.F.; Bogorad, R.; et al. MICU1 Controls Both the Threshold and Cooperative Activation of the Mitochondrial Ca2+ Uniporter. Cell Metab. 2013, 17, 976–987. [Google Scholar] [CrossRef] [Green Version]
- Plovanich, M.; Bogorad, R.L.; Sancak, Y.; Kamer, K.J.; Strittmatter, L.; Li, A.A.; Girgis, H.S.; Kuchimanchi, S.; De Groot, J.; Speciner, L.; et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS ONE 2013, 8, e55785. [Google Scholar] [CrossRef] [Green Version]
- Patron, M.; Granatiero, V.; Espino, J.; Rizzuto, R.; De Stefani, D. MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ. 2018, 26, 179–195. [Google Scholar] [CrossRef] [Green Version]
- Mallilankaraman, K.; Cárdenas, C.; Doonan, P.J.; Chandramoorthy, H.C.; Irrinki, K.M.; Golenár, T.; Csordás, G.; Madireddi, P.; Yang, J.; Müller, M.; et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat. Cell Biol. 2012, 14, 1336–1343. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, N.E.; Chandramoorthy, H.C.; Shanmughapriya, S.; Zhang, X.Q.; Vallem, S.; Doonan, P.J.; Malliankaraman, K.; Guo, S.; Rajan, S.; Elrod, J.W.; et al. SLC25A23 augments mitochondrial Ca2+ uptake, interacts with MCU, and induces oxidative stress–mediated cell death. Mol. Biol. Cell 2014, 25, 936–947. [Google Scholar] [CrossRef]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Nat. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, D.; Zhao, L.; Clapham, D.E. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 2009, 326, 144–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumgartner, H.K.; Gerasimenko, J.V.; Thorne, C.; Ferdek, P.; Pozzan, T.; Tepikin, A.V.; Petersen, O.H.; Sutton, R.; Watson, A.J.; Gerasimenko, O.V.; et al. Calcium elevation in mitochondria is the main Ca2+ requirement for mitochondrial permeability transition pore (mPTP) opening. J. Biol. Chem. 2009, 284, 20796–20803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hengartner, M.O. The biochemistry of apoptosis. Nat. Cell Biol. 2000, 407, 770–776. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Khan, S.M. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 2004, 63, 8–20. [Google Scholar] [CrossRef]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial Abnormalities in Alzheimer’s Disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [CrossRef] [Green Version]
- Rice, A.C.; Keeney, P.M.; Algarzae, N.K.; Ladd, A.C.; Thomas, R.R.; Bennett, J.P., Jr. Mitochondrial DNA Copy Numbers in Pyramidal Neurons are Decreased and Mitochondrial Biogenesis Transcriptome Signaling is Disrupted in Alzheimer’s Disease Hippocampi. J. Alzheimer’s Dis. 2014, 40, 319–330. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Wang, J.; Markesbery, W.R.; Lovell, M.A. Increased oxidative damage in nuclear and mitochondrial DNA in mild cognitive impairment. J. Neurochem. 2006, 96, 825–832. [Google Scholar] [CrossRef]
- Area-Gomez, E.; De Groof, A.; Bonilla, E.; Montesinos, J.; Tanji, K.; Boldogh, I.; Pon, L.; Schon, E.A. A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death Dis. 2018, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Calkins, M.J.; Reddy, P.H. Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer’s disease neurons. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2011, 1812, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cieri, D.; Vicario, M.; Vallese, F.; D’Orsi, B.; Berto, P.; Grinzato, A.; Catoni, C.; De Stefani, D.; Rizzuto, R.; Brini, M.; et al. Tau localises within mitochondrial sub-compartments and its caspase cleavage affects ER-mitochondria interactions and cellular Ca2+ handling. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1864, 3247–3256. [Google Scholar] [CrossRef] [PubMed]
- Amadoro, G.; Corsetti, V.; Stringaro, A.; Colone, M.; D’Aguanno, S.; Meli, G.; Ciotti, M.; Sancesario, G.; Cattaneo, A.; Bussani, R.; et al. A NH2 Tau Fragment Targets Neuronal Mitochondria at AD Synapses: Possible Implications for Neurodegeneration. J. Alzheimer’s Dis. 2010, 21, 445–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo-Rodríguez, M.; García-Durillo, M.; Villalobos, C.; Núñez, L. Aging Enables Ca2+ Overload and Apoptosis Induced by Amyloid-beta Oligomers in Rat Hippocampal Neurons: Neuroprotection by Non-Steroidal Anti-Inflammatory Drugs and R-Flurbiprofen in Aging Neurons. J. Alzheimer’s Dis. JAD 2016, 54, 207–221. [Google Scholar] [CrossRef]
- Sanz-Blasco, S.; Valero, R.A.; Rodriguez-Crespo, I.; Villalobos, C.; Nunez, L. Mitochondrial Ca2+ overload underlies Abeta oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PLoS ONE 2008, 3, e2718. [Google Scholar] [CrossRef] [Green Version]
- Calvo-Rodriguez, M.; Hou, S.S.; Snyder, A.C.; Kharitonova, E.K.; Russ, A.N.; Das, S.; Fan, Z.; Muzikansky, A.; Garcia-Alloza, M.; Serrano-Pozo, A.; et al. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Yan, Y.; Gong, K.; Ma, T.; Zhang, L.; Zhao, N.; Zhang, X.; Tang, P.; Gong, Y. Protective effect of edaravone against Alzheimer’s disease-relevant insults in neuroblastoma N2a cells. Neurosci. Lett. 2012, 531, 160–165. [Google Scholar] [CrossRef]
- Moreira, P.I.; Santos, M.S.; Moreno, A.; Oliveira, C. Amyloid β-Peptide Promotes Permeability Transition Pore in Brain Mitochondria. Biosci. Rep. 2001, 21, 789–800. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; A Sosunov, A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef]
- Jadiya, P.; Kolmetzky, D.W.; Tomar, D.; Di Meco, A.; Lombardi, A.A.; Lambert, J.P.; Luongo, T.S.; Ludtmann, M.H.; Praticò, D.; Elrod, J.W. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kern, S.; Skoog, I.; Östling, S.; Kern, J.; Börjesson-Hanson, A. Does low-dose acetylsalicylic acid prevent cognitive decline in women with high cardiovascular risk? A 5-year follow-up of a non-demented population-based cohort of Swedish elderly women. BMJ Open 2012, 2, e001288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Wang, Y.; Wang, D.; Zhang, J.; Zhang, F. NSAID Exposure and Risk of Alzheimer’s Disease: An Updated Meta-Analysis From Cohort Studies. Front. Aging Neurosci. 2018, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Breitner, J.; Meyer, P.-F. Author response: INTREPAD: A randomized trial of naproxen to slow progress of presymptomatic Alzheimer disease. Neurology 2020, 94, 594. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Blasco, S.; Calvo-Rodriguez, M.; Caballero, E.; Garcia-Durillo, M.; Nunez, L.; Villalobos, C. Is it All Said for NSAIDs in Alzheimer’s Disease? Role of Mitochondrial Calcium Uptake. Curr. Alzheimer Res. 2018, 15, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Gutknecht, J. Salicylates and proton transport through lipid bilayer membranes: A model for salicylate-induced uncoupling and swelling in mitochondria. J. Membr. Biol. 1990, 115, 253–260. [Google Scholar] [CrossRef]
- Núñez, L.; Valero, R.A.; Senovilla, L.; Sanz-Blasco, S.; García-Sancho, J.; Villalobos, C. Cell proliferation depends on mitochondrial Ca2+ uptake: Inhibition by salicylate. J. Physiol. 2006, 571, 57–73. [Google Scholar] [CrossRef]
- Calvo-Rodríguez, M.; Sanz-Blasco, S.; Caballero, E.; Villalobos, C.; Núñez, L. Susceptibility to excitotoxicity in aged hippocampal cultures and neuroprotection by non-steroidal anti-inflammatory drugs: Role of mitochondrial calcium. J. Neurochem. 2015, 132, 403–417. [Google Scholar] [CrossRef]
- Angelova, P.R.; Vinogradova, D.; Neganova, M.E.; Serkova, T.P.; Sokolov, V.V.; Bachurin, S.O.; Shevtsova, E.F.; Abramov, A.Y. Pharmacological Sequestration of Mitochondrial Calcium Uptake Protects Neurons Against Glutamate Excitotoxicity. Mol. Neurobiol. 2019, 56, 2244–2255. [Google Scholar] [CrossRef] [Green Version]
- Dubey, M.; Chaudhury, P.; Kabiru, H.; Shea, T.B. Tau inhibits anterograde axonal transport and perturbs stability in growing axonal neurites in part by displacing kinesin cargo: Neurofilaments attenuate tau-mediated neurite instability. Cell Motil. Cytoskelet. 2008, 65, 89–99. [Google Scholar] [CrossRef]
- DuBoff, B.; Götz, J.; Feany, M.B. Tau Promotes Neurodegeneration via DRP1 Mislocalization In Vivo. Neuron 2012, 75, 618–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintanilla, R.A.; Matthews-Roberson, T.A.; Dolan, P.J.; Johnson, G.V. Caspase-cleaved Tau Expression Induces Mitochondrial Dysfunction in Immortalized Cortical Neurons. J. Biol. Chem. 2009, 284, 18754–18766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britti, E.; Ros, J.; Esteras, N.; Abramov, A.Y. Tau inhibits mitochondrial calcium efflux and makes neurons vulnerable to calcium-induced cell death. Cell Calcium 2020, 86, 102150. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Reddy, P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef] [Green Version]
- García-Pérez, C.; Hajnóczky, G.; Csordás, G. Physical Coupling Supports the Local Ca2+ Transfer between Sarcoplasmic Reticulum Subdomains and the Mitochondria in Heart Muscle. J. Biol. Chem. 2008, 283, 32771–32780. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Su, T.-P. Sigma-1 Receptor Chaperones at the ER- Mitochondrion Interface Regulate Ca2+ Signaling and Cell Survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [Green Version]
- Csordas, G.; Varnai, P.; Golenar, T.; Roy, S.; Purkins, G.; Schneider, T.G.; Balla, T.; Hajnoczky, G. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cell 2010, 39, 121–132. [Google Scholar] [CrossRef]
- Area-Gomez, E.; Schon, E.A. On the Pathogenesis of Alzheimer’s Disease: The MAM Hypothesis. FASEB J. 2017, 31, 864–867. [Google Scholar] [CrossRef] [Green Version]
- Hedskog, L.; Pinho, C.M.; Filadi, R.; Rönnbäck, A.; Hertwig, L.; Wiehager, B.; Larssen, P.; Gellhaar, S.; Sandebring, A.; Westerlund, M.; et al. Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Nat. Acad. Sci. USA 2013, 110, 7916–7921. [Google Scholar] [CrossRef] [Green Version]
- Perreault, S.; Bousquet, O.; Lauzon, M.; Paiement, J.; Leclerc, N. Increased Association Between Rough Endoplasmic Reticulum Membranes and Mitochondria in Transgenic Mice That Express P301L Tau. J. Neuropathol. Exp. Neurol. 2009, 68, 503–514. [Google Scholar] [CrossRef] [Green Version]
- Area-Gomez, E.; Castillo, M.D.C.L.; Tambini, M.D.; Guardia-Laguarta, C.; De Groof, A.J.C.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012, 31, 4106–4123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreiro, E.; Oliveira, C.R.; Pereira, C.F. The release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathway. Neurobiol. Dis. 2008, 30, 331–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zampese, E.; Fasolato, C.; Kipanyula, M.J.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Nat. Acad. Sci. USA 2011, 108, 2777–2782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Smith, M.A.; Perry, G.H.; Aliev, G. Mitochondrial failures in Alzheimer’s disease. Am. J. Alzheimer’s Dis. Other Dement. 2004, 19, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Cenini, G.; Voos, W. Mitochondria as Potential Targets in Alzheimer Disease Therapy: An Update. Front. Pharmacol. 2019, 10, 902. [Google Scholar] [CrossRef] [PubMed]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.J.; Murphy, M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, J.M.P.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br. J. Pharmacol. 2019, 176, 3489–3507. [Google Scholar] [CrossRef]
- Szeto, H.H. Cell-permeable, mitochondrial-targeted, peptide antioxidants. AAPS J. 2006, 8, E277–E283. [Google Scholar] [CrossRef]
- Birk, A.V.; Chao, W.M.; Bracken, C.; Warren, J.D.; Szeto, H.H. Targeting mitochondrial cardiolipin and the cytochromec/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br. J. Pharmacol. 2014, 171, 2017–2028. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H.; Manczak, M.; Kandimalla, R. Mitochondria-targeted small molecule SS31: A potential candidate for the treatment of Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 1597. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H.; Manczak, M.; Yin, X.; Reddy, A.P. Synergistic Protective Effects of Mitochondrial Division Inhibitor 1 and Mitochondria-Targeted Small Peptide SS31 in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1549–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkins, H.M.; Morris, J.K. New Therapeutics to Modulate Mitochondrial Function in Neurodegenerative Disorders. Curr. Pharm. Des. 2017, 23, 731–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd-Evans, E.; Platt, F.M. Lysosomal Ca2+ homeostasis: Role in pathogenesis of lysosomal storage diseases. Cell Calcium 2011, 50, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Gala, U.; Zhang, Y.; Shang, W.; Jaiswal, S.N.; Di Ronza, A.; Jaiswal, M.; Yamamoto, S.; Sandoval, H.; DuRaine, L.; et al. A voltage-gated calcium channel regulates lysosomal fusion with endosomes and autophagosomes and is required for neuronal homeostasis. PLoS Biol. 2015, 13, e1002103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.; Docampo, R. Acidic calcium stores open for business: Expanding the potential for intracellular Ca2+ signaling. Trends Cell Biol. 2010, 20, 277–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrity, A.G.; Wang, W.; Collier, C.M.; A Levey, S.; Gao, Q.; Xu, H. The endoplasmic reticulum, not the pH gradient, drives calcium refilling of lysosomes. eLife 2016, 5, e15887. [Google Scholar] [CrossRef] [PubMed]
- Mindell, J.A. Lysosomal Acidification Mechanisms. Annu. Rev. Physiol. 2012, 74, 69–86. [Google Scholar] [CrossRef] [Green Version]
- Bach, G.; Chen, C.-S.; E Pagano, R. Elevated lysosomal pH in Mucolipidosis type IV cells. Clin. Chim. Acta 1999, 280, 173–179. [Google Scholar] [CrossRef]
- Soyombo, A.A.; Tjon-Kon-Sang, S.; Rbaibi, Y.; Bashllari, E.; Bisceglia, J.; Muallem, S.; Kiselyov, K. TRP-ML1 Regulates Lysosomal pH and Acidic Lysosomal Lipid Hydrolytic Activity. J. Biol. Chem. 2006, 281, 7294–7301. [Google Scholar] [CrossRef] [Green Version]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-H.; McBrayer, M.K.; Wolfe, D.M.; Haslett, L.J.; Kumar, A.; Sato, Y.; Lie, P.P.Y.; Mohan, P.S.; Coffey, E.E.; Kompella, U.B.; et al. Presenilin 1 Maintains Lysosomal Ca2+ Homeostasis via TRPML1 by Regulating vATPase-Mediated Lysosome Acidification. Cell Rep. 2015, 12, 1430–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neely, K.M.; Green, K.N.; LaFerla, F.M. Presenilin is necessary for efficient proteolysis through the autophagy-lysosome system in a gamma-secretase-independent manner. J. Neurosci. 2011, 31, 2781–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greotti, E.; Capitanio, P.; Wong, A.; Pozzan, T.; Pizzo, P.; Pendin, D. Familial Alzheimer’s disease-linked presenilin mutants and intracellular Ca2+ handling: A single-organelle, FRET-based analysis. Cell Calcium 2019, 79, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; MacLeod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive Involvement of Autophagy in Alzheimer Disease: An Immuno-Electron Microscopy Study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [CrossRef] [Green Version]
- Wolfe, D.M.; Lee, J.-H.; Kumar, A.; Lee, S.; Orenstein, S.J.; Nixon, R.A. Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Eur. J. Neurosci. 2013, 37, 1949–1961. [Google Scholar] [CrossRef] [Green Version]
- Medeiros, R.; LaFerla, F.M. Astrocytes: Conductors of the Alzheimer disease neuroinflammatory symphony. Exp. Neurol. 2013, 239, 133–138. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Parpura, V. Recent advances in (patho)physiology of astroglia. Acta Pharmacol. Sin. 2010, 31, 1044–1054. [Google Scholar] [CrossRef] [Green Version]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Guerra-Gomes, S.; Sousa, N.; Pinto, L.; Oliveira, J.F. Functional Roles of Astrocyte Calcium Elevations: From Synapses to Behavior. Front. Cell. Neurosci. 2018, 11, 427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef] [PubMed]
- Perez-Nievas, B.G.; Serrano-Pozo, A. Deciphering the Astrocyte Reaction in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, D.; Iyer, A.; Ronco, V.; A Grolla, A.; Canonico, P.L.; Aronica, E.; Genazzani, A.A. Amyloid beta deregulates astroglial mGluR5-mediated calcium signaling via calcineurin and Nf-kB. Glia 2013, 61, 1134–1145. [Google Scholar] [CrossRef]
- Grolla, A.A.; Sim, J.A.; Lim, D.; Rodriguez, J.J.; Genazzani, A.A.; Verkhratsky, A. Amyloid-beta and Alzheimer’s disease type pathology differentially affects the calcium signalling toolkit in astrocytes from different brain regions. Cell Death Dis. 2013, 4, e623. [Google Scholar] [CrossRef]
- Alberdi, E.; Wyssenbach, A.; Alberdi, M.; Sanchez-Gomez, M.V.; Cavaliere, F.; Rodriguez, J.J.; Verkhratsky, A.; Matute, C. Ca2+-dependent endoplasmic reticulum stress correlates with astrogliosis in oligomeric amyloid beta-treated astrocytes and in a model of Alzheimer’s disease. Aging Cell 2013, 12, 292–302. [Google Scholar] [CrossRef] [Green Version]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Changes in Intracellular Calcium and Glutathione in Astrocytes as the Primary Mechanism of Amyloid Neurotoxicity. J. Neurosci. 2003, 23, 5088–5095. [Google Scholar] [CrossRef] [Green Version]
- Toivari, E.; Manninen, T.; Nahata, A.K.; Jalonen, T.O.; Linne, M.-L. Effects of Transmitters and Amyloid-Beta Peptide on Calcium Signals in Rat Cortical Astrocytes: Fura-2AM Measurements and Stochastic Model Simulations. PLoS ONE 2011, 6, e17914. [Google Scholar] [CrossRef] [Green Version]
- Larramona-Arcas, R.; González-Arias, C.; Perea, G.; Gutiérrez, A.; Vitorica, J.; García-Barrera, T.; Gómez-Ariza, J.L.; Pascua-Maestro, R.; Ganfornina, M.D.; Kara, E.; et al. Sex-dependent calcium hyperactivity due to lysosomal-related dysfunction in astrocytes from APOE4 versus APOE3 gene targeted replacement mice. Mol. Neurodegener. 2020, 15, 1–23. [Google Scholar] [CrossRef]
- Hirase, H.; Qian, L.; Barthó, P.; Buzsáki, G. Calcium Dynamics of Cortical Astrocytic Networks In Vivo. PLoS Biol. 2004, 2, e96. [Google Scholar] [CrossRef] [Green Version]
- Torres, A.; Wang, F.; Xu, Q.; Fujita, T.; Dobrowolski, R.; Willecke, K.; Takano, T.; Nedergaard, M. Extracellular Ca2+ Acts as a Mediator of Communication from Neurons to Glia. Sci. Signal. 2012, 5, ra8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Bernhardi, R.; Tichauer, J.E.; Eugenín, J. Aging-dependent changes of microglial cells and their relevance for neurodegenerative disorders. J. Neurochem. 2010, 112, 1099–1114. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Kann, O.; Ohlemeyer, C.; Hanisch, U.-K.; Kettenmann, H. Elevation of Basal Intracellular Calcium as a Central Element in the Activation of Brain Macrophages (Microglia): Suppression of Receptor-Evoked Calcium Signaling and Control of Release Function. J. Neurosci. 2003, 23, 4410–4419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franciosi, S.; Choi, H.B.; Kim, S.U.; McLarnon, J.G. Interferon-? acutely induces calcium influx in human microglia. J. Neurosci. Res. 2002, 69, 607–613. [Google Scholar] [CrossRef]
- Goghari, V.; Franciosi, S.; Kim, S.U.; Lee, Y.B.; McLarnon, J.G. Acute application of interleukin-1beta induces Ca2+ responses in human microglia. Neurosci. Lett. 2000, 281, 83–86. [Google Scholar] [CrossRef]
- McLarnon, J.G.; Franciosi, S.; Wang, X.; Bae, J.; Choi, H.; Kim, S. Acute actions of tumor necrosis factor-α on intracellular Ca2+ and K+ currents in human microglia. Neuroscience 2001, 104, 1175–1184. [Google Scholar] [CrossRef]
- Turovskaya, M.V.; Turovsky, E.A.; Zinchenko, V.P.; Levin, S.G.; Godukhin, O.V. Interleukin-10 modulates [Ca2+]i response induced by repeated NMDA receptor activation with brief hypoxia through inhibition of InsP3-sensitive internal stores in hippocampal neurons. Neurosci. Lett. 2012, 516, 151–155. [Google Scholar] [CrossRef]
- Itagaki, S.; McGeer, P.; Akiyama, H.; Zhu, S.; Selkoe, D. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J. Neuroimmunol. 1989, 24, 173–182. [Google Scholar] [CrossRef]
- Meyer-Luehmann, M.; Spires-Jones, T.L.; Prada, C.; Garcia-Alloza, M.; de Calignon, A.; Rozkalne, A.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Bacskai, B.J.; Hyman, B.T. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature 2008, 451, 720–724. [Google Scholar] [CrossRef] [Green Version]
- Perez-Nievas, B.G.; Stein, T.D.; Tai, H.-C.; Dols-Icardo, O.; Scotton, T.C.; Barroeta-Espar, I.; Fernandez-Carballo, L.; De Munain, E.L.; Perez, J.; Marquie, M.; et al. Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain 2013, 136, 2510–2526. [Google Scholar] [CrossRef] [PubMed]
- Bezzi, P.; Domercq, M.; Brambilla, L.; Galli, R.; Schols, D.; De Clercq, E.; Vescovi, A.; Bagetta, G.; Kollias, G.; Meldolesi, J.; et al. CXCR4-activated astrocyte glutamate release via TNFalpha: Amplification by microglia triggers neurotoxicity. Nat. Neurosci. 2001, 4, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Agulhon, C.; Sun, M.-Y.; Murphy, T.; Myers, T.; Lauderdale, K.; Fiacco, T.A. Calcium Signaling and Gliotransmission in Normal vs. Reactive Astrocytes. Front. Pharmacol. 2012, 3, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brawek, B.; Garaschuk, O. Network-wide dysregulation of calcium homeostasis in Alzheimer’s disease. Cell Tissue Res. 2014, 357, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Silei, V.; Fabrizi, C.; Venturini, G.; Salmona, M.; Bugiani, O.; Tagliavini, F.; Lauro, G.M. Activation of microglial cells by PrP and beta-amyloid fragments raises intracellular calcium through L-type voltage sensitive calcium channels. Brain Res. 1999, 818, 168–170. [Google Scholar] [CrossRef]
- McLarnon, J.G.; Choi, H.B.; Lue, L.-F.; Walker, D.G.; Kim, S.U. Perturbations in calcium-mediated signal transduction in microglia from Alzheimer’s disease patients. J. Neurosci. Res. 2005, 81, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Eichhoff, G.; Brawek, B.; Garaschuk, O. Microglial calcium signal acts as a rapid sensor of single neuron damage in vivo. Biochim. Biophys. Acta (BBA) Bioenerg. 2011, 1813, 1014–1024. [Google Scholar] [CrossRef] [Green Version]
- Heo, D.K.; Lim, H.M.; Nam, J.H.; Lee, M.G.; Kim, J.Y. Regulation of phagocytosis and cytokine secretion by store-operated calcium entry in primary isolated murine microglia. Cell. Signal. 2015, 27, 177–186. [Google Scholar] [CrossRef]
- Michaelis, M.; Nieswandt, B.; Stegner, D.; Eilers, J.; Kraft, R. STIM1, STIM2, and Orai1 regulate store-operated calcium entry and purinergic activation of microglia. Glia 2015, 63, 652–663. [Google Scholar] [CrossRef]
- Klegeris, A.; Choi, H.B.; McLarnon, J.G.; McGeer, P.L. Functional ryanodine receptors are expressed by human microglia and THP-1 cells: Their possible involvement in modulation of neurotoxicity. J. Neurosci. Res. 2007, 85, 2207–2215. [Google Scholar] [CrossRef]
- Lim, H.M.; Woon, H.; Han, J.W.; Baba, Y.; Kurosaki, T.; Lee, M.G.; Kim, J.Y. UDP-Induced Phagocytosis and ATP-Stimulated Chemotactic Migration Are Impaired in STIM1(-/-) Microglia In Vitro and In Vivo. Mediat. Inflamm. 2017, 2017, 8158514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnstock, G. Purinergic Signalling: Therapeutic Developments. Front. Pharmacol. 2017, 8, 661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, H.; Chan, C.M.; Loesch, A.; Unwin, R.; Burnstock, G. Induction of proliferation and apoptotic cell death via P2Y and P2X receptors, respectively, in rat glomerular mesangial cells. Kidney Int. 2000, 57, 949–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLarnon, J.G.; Ryu, J.K.; Walker, D.G.; Choi, H.B. Upregulated expression of purinergic P2X(7) receptor in Alzheimer disease and amyloid-beta peptide-treated microglia and in peptide-injected rat hippocampus. Exp. Neurol. 2006, 65, 1090–1097. [Google Scholar]
- Parvathenani, L.K.; Tertyshnikova, S.; Greco, C.R.; Roberts, S.B.; Robertson, B.; Posmantur, R. P2 × 7 mediates superoxide production in primary microglia and is up-regulated in a transgenic mouse model of Alzheimer’s disease. J. Biol. Chem. 2003, 278, 13309–13317. [Google Scholar] [CrossRef] [Green Version]
- Sanz, J.M.; Chiozzi, P.; Ferrari, D.; Colaianna, M.; Idzko, M.; Falzoni, S.; Fellin, R.; Trabace, L.; Di Virgilio, F. Activation of microglia by amyloid {beta} requires P2 × 7 receptor expression. J. Immunol. Res. 2009, 182, 4378–4385. [Google Scholar]
- Illes, P.; Rubini, P.; Huang, L.; Tang, Y. The P2 × 7 receptor: A new therapeutic target in Alzheimer’s disease. Expert Opin. 2019, 23, 165–176. [Google Scholar]
- Jana, M.K.; Cappai, R.; Ciccotosto, G.D. Oligomeric Amyloid-beta Toxicity Can Be Inhibited by Blocking Its Cellular Binding in Cortical Neuronal Cultures with Addition of the Triphenylmethane Dye Brilliant Blue, G. ACS Chem. Biol. 2016, 7, 1141–1147. [Google Scholar]
- Diaz-Hernandez, J.I.; Gomez-Villafuertes, R.; Leon-Otegui, M.; Hontecillas-Prieto, L.; Del Puerto, A.; Trejo, J.L.; Lucas, J.J.; Garrido, J.J.; Gualix, J.; Miras-Portugal, M.T.; et al. In vivo P2 × 7 inhibition reduces amyloid plaques in Alzheimer’s disease through GSK3beta and secretases. Neurobiol. Aging 2012, 33, 1816–1828. [Google Scholar] [CrossRef] [Green Version]
- Martin, E.; Amar, M.; Dalle, C.; Youssef, I.; Boucher, C.; Le Duigou, C.; Bruckner, M.; Prigent, A.; Sazdovitch, V.; Halle, A.; et al. New role of P2 × 7 receptor in an Alzheimer’s disease mouse model. Mol. Psychiatry 2019, 24, 108–125. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.-S.; Subramanian, N.; Kim, A.I.; Aksentijevich, I.; Goldbach-Mansky, R.; Sacks, D.B.; Germain, R.N.; Kastner, D.L.; Chae, J.J. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nat. Cell Biol. 2012, 492, 123–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Nat. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nat. Cell Biol. 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher-Moser, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 Associated with the Risk of Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Tan, L.; Zhu, X.-C.; Zhang, Q.-Q.; Cao, L.; Tan, M.-S.; Gu, L.-Z.; Wang, H.-F.; Ding, Z.-Z.; Zhang, Y.-D.; et al. Upregulation of TREM2 Ameliorates Neuropathology and Rescues Spatial Cognitive Impairment in a Transgenic Mouse Model of Alzheimer’s Disease. Neuropsychopharmacology 2014, 39, 2949–2962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchon, A.; Hernández-Munain, C.; Cella, M.; Colonna, M. A Dap12-Mediated Pathway Regulates Expression of Cc Chemokine Receptor 7 and Maturation of Human Dendritic Cells. J. Exp. Med. 2001, 194, 1111–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyanohara, A.; Kamizato, K.; Juhas, S.; Juhasova, J.; Navarro, M.; Marsala, S.; Lukacova, N.; Hruska-Plochan, M.; Curtis, E.; Gabel, B.; et al. Potent spinal parenchymal AAV9-mediated gene delivery by subpial injection in adult rats and pigs. Mol. Ther. Methods Clin. Dev. 2016, 3, 16046. [Google Scholar] [CrossRef] [Green Version]
- Challis, R.C.; Kumar, S.R.; Chan, K.Y.; Challis, C.; Beadle, K.; Jang, M.J.; Kim, H.M.; Rajendran, P.S.; Tompkins, J.D.; Shivkumar, K.; et al. Systemic AAV vectors for widespread and targeted gene delivery in rodents. Nat. Protoc. 2019, 14, 379–414. [Google Scholar] [CrossRef] [PubMed]
- Alves, S.; Fol, R.; Cartier, N. Gene Therapy Strategies for Alzheimer’s Disease: An Overview. Hum. Gene Ther. 2016, 27, 100–107. [Google Scholar] [CrossRef]
- Zhao, L.; Gottesdiener, A.J.; Parmar, M.; Li, M.; Kaminsky, S.M.; Chiuchiolo, M.J.; Sondhi, D.; Sullivan, P.M.; Holtzman, D.M.; Crystal, R.G.; et al. Intracerebral adeno-associated virus gene delivery of apolipoprotein E2 markedly reduces brain amyloid pathology in Alzheimer’s disease mouse models. Neurobiol. Aging 2016, 44, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.B.; Kaplitt, M.G.; De, B.P.; Chen, A.; Flagiello, T.; Salami, C.; Pey, E.; Zhao, L.; Ricart Arbona, R.J.; Monette, S.; et al. AAVrh.10-Mediated APOE2 Central Nervous System Gene Therapy for APOE4-Associated Alzheimer’s Disease. Hum. Gen. Ther. Clin. Dev. 2018, 29, 24–47. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calvo-Rodriguez, M.; Kharitonova, E.K.; Bacskai, B.J. Therapeutic Strategies to Target Calcium Dysregulation in Alzheimer’s Disease. Cells 2020, 9, 2513. https://doi.org/10.3390/cells9112513
Calvo-Rodriguez M, Kharitonova EK, Bacskai BJ. Therapeutic Strategies to Target Calcium Dysregulation in Alzheimer’s Disease. Cells. 2020; 9(11):2513. https://doi.org/10.3390/cells9112513
Chicago/Turabian StyleCalvo-Rodriguez, Maria, Elizabeth K. Kharitonova, and Brian J. Bacskai. 2020. "Therapeutic Strategies to Target Calcium Dysregulation in Alzheimer’s Disease" Cells 9, no. 11: 2513. https://doi.org/10.3390/cells9112513
APA StyleCalvo-Rodriguez, M., Kharitonova, E. K., & Bacskai, B. J. (2020). Therapeutic Strategies to Target Calcium Dysregulation in Alzheimer’s Disease. Cells, 9(11), 2513. https://doi.org/10.3390/cells9112513