New Pieces in the Puzzle of uPAR Role in Cell Migration Mechanisms

 and
and 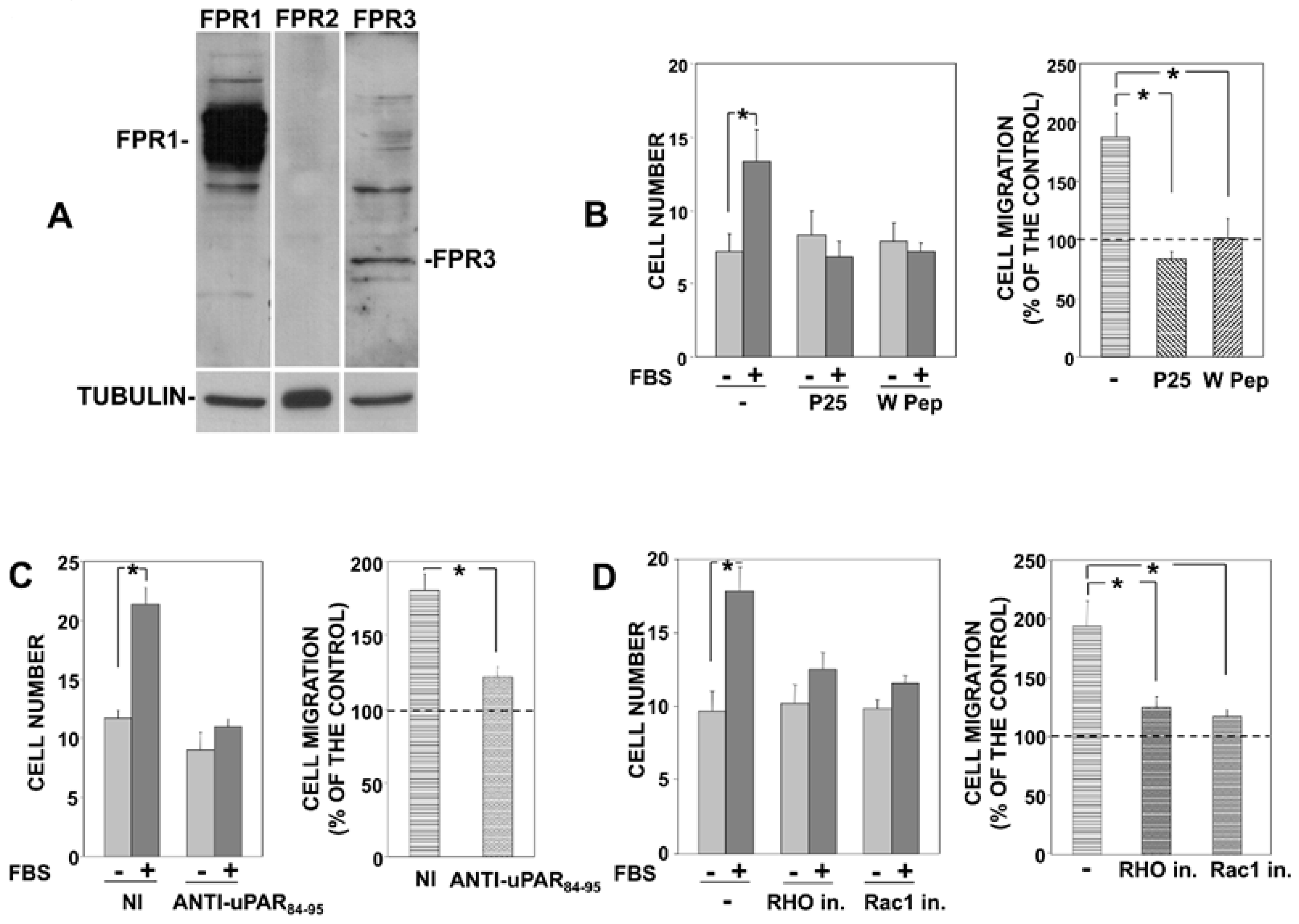{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Cell Migration Assay
2.4. TM-uPAR Construct
2.5. Transfections
2.6. PI-PLC and MCD Treatment
2.7. Lipid Rafts Analysis
2.8. Western Blot Analysis
2.9. Statistical Analysis
3. Results
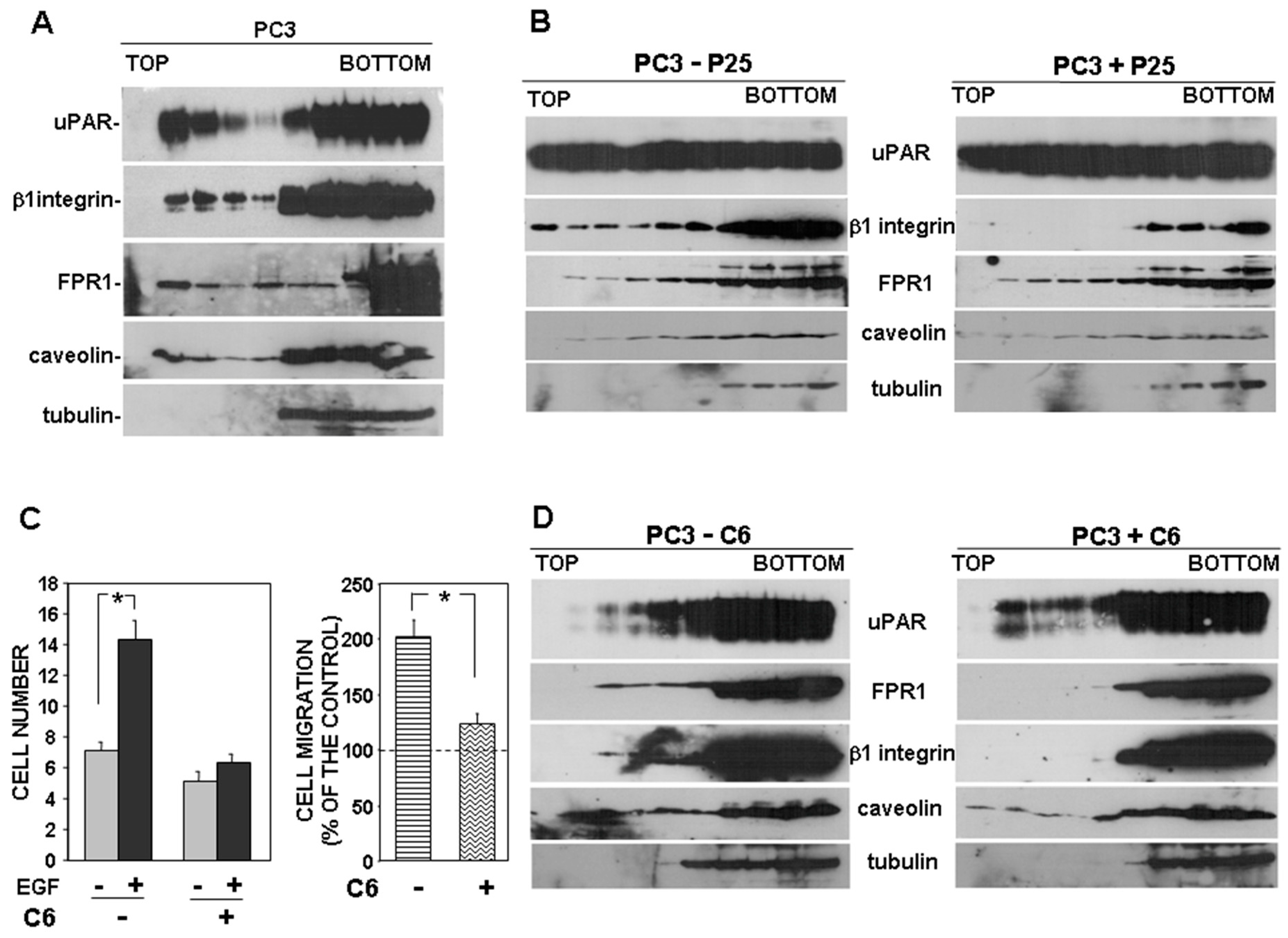
3.1. uPAR Interactions with FPRs and β1 Integrins Are Involved in Migration of Prostate Carcinoma PC3 Cells
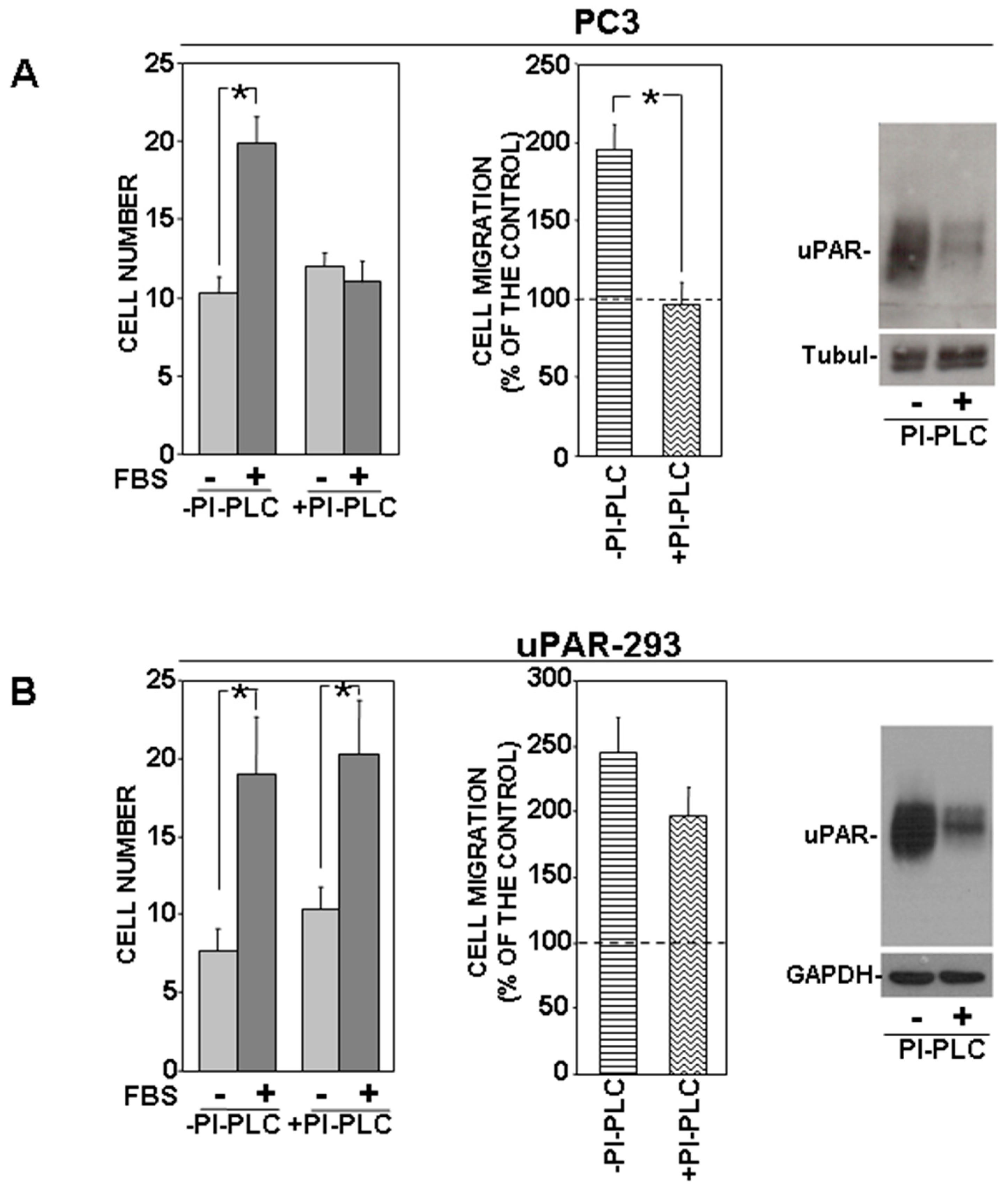
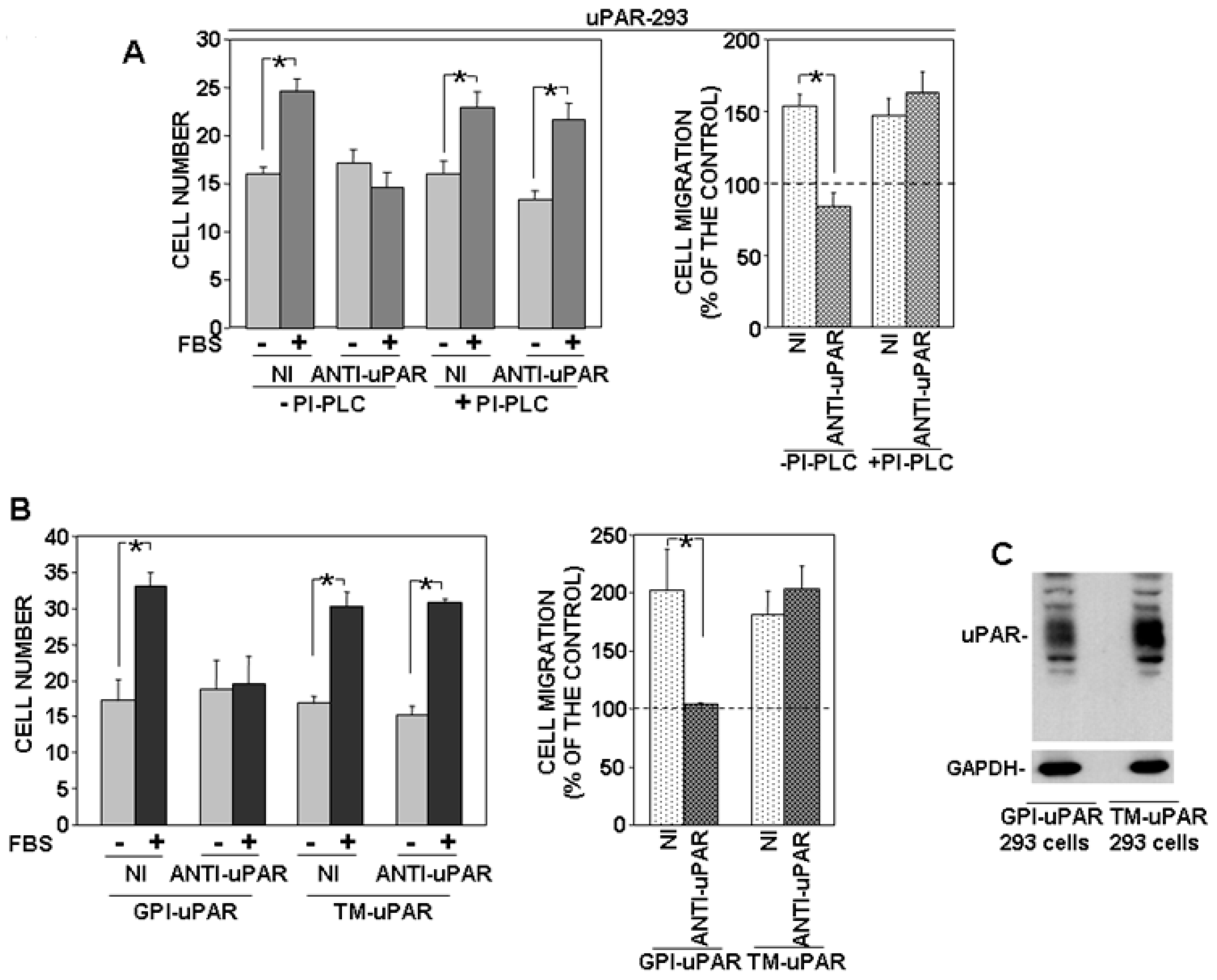
3.2. Removal of uPAR GPI-Anchor Impairs the uPAR-Controlled Mechanism of Cell Migration
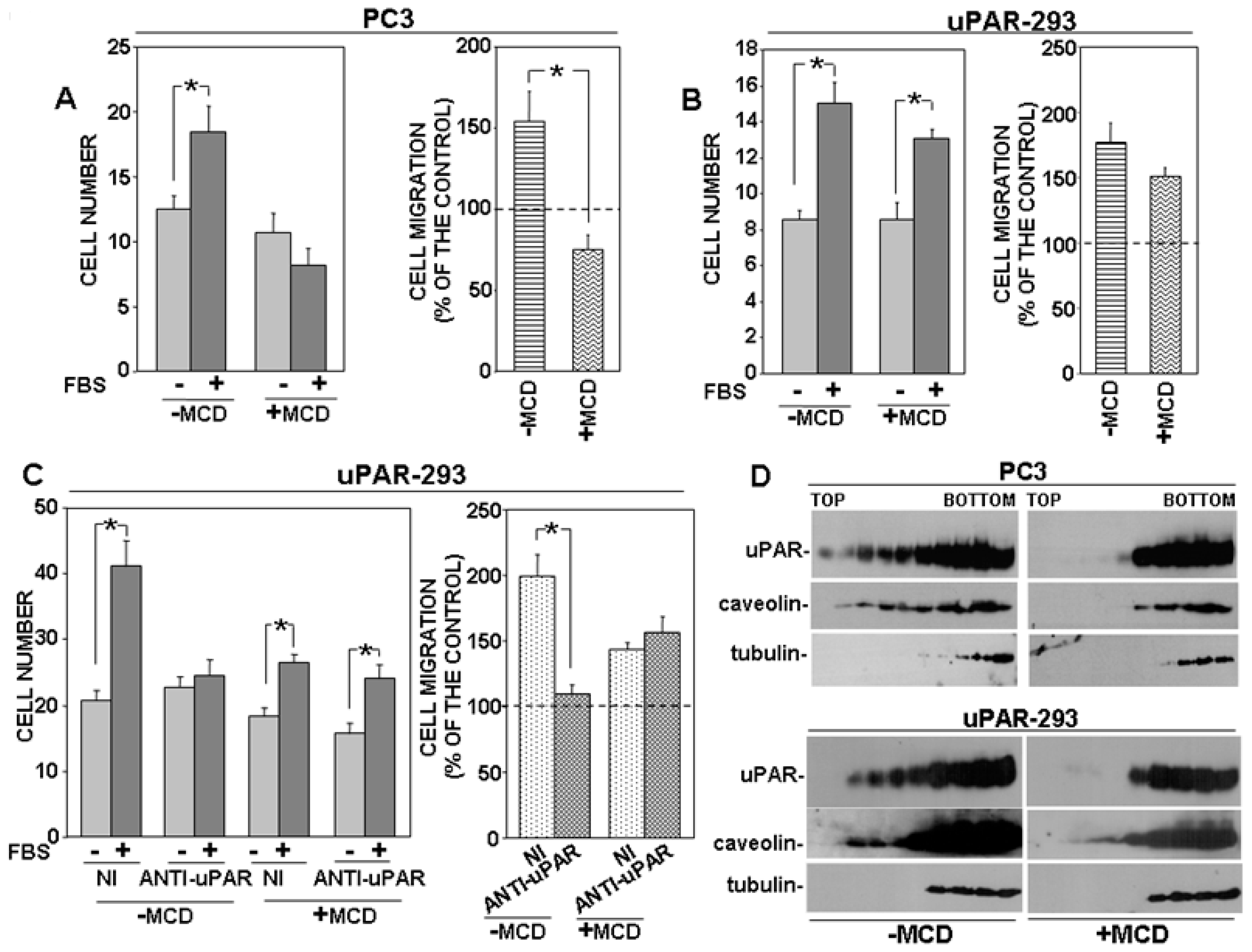
3.3. Disruption of Lipid Rafts Impairs uPAR-Controlled Cell Migration
3.4. uPAR Drives Its Signaling Partners to Lipid Rafts
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Montuori, N.; Ragno, P. Multiple activities of a multifaceted receptor: Roles of cleaved and soluble uPAR. Front. Biosci. 2009, 14, 2494–2503. [Google Scholar] [CrossRef] [Green Version]
- Kjaergaard, M.; Hansen, L.V.; Jacobsen, B.; Gardsvoll, H.; Ploug, M. Structure and ligand interactions of the urokinase receptor (uPAR). Front. Biosci. 2008, 13, 5441–5461. [Google Scholar] [CrossRef]
- Tang, C.H.; Wei, Y. The urokinase receptor and integrins in cancer progression. Cell. Mol. Life Sci. 2008, 65, 1916–1932. [Google Scholar] [CrossRef]
- Montuori, N.; Pesapane, A.; Rossi, F.W.; Giudice, V.; De Paulis, A.; Selleri, C.; Ragno, P. Urokinase type plasminogen activator receptor (uPAR) as a new therapeutic target in cancer. Transl. Med. Unisa 2016, 15, 15–21. [Google Scholar]
- Smith, H.W.; Marshall, C.J. Regulation of cell signalling by uPAR. Nat. Rev. Mol. Cell Biol. 2010, 11, 23–36. [Google Scholar] [CrossRef]
- Baart, V.M.; Houvast, R.D.; de Geus-Oei, L.F.; Quax, P.H.A.; Kuppen, P.J.K.; Vahrmeijer, A.L.; Sier, C.F.M. Molecular imaging of the urokinase plasminogen activator receptor: Opportunities beyond cancer. EJNMMI Res. 2020, 10, 87. [Google Scholar] [CrossRef]
- Hyangsoon, N.; Sungguan, H.; Shuang, H. Role of urokinase receptor in tumor progression and development. Theranostics 2013, 3, 487–495. [Google Scholar]
- Montuori, N.; Bifulco, K.; Carriero, M.V.; La Penna, C.; Visconte, V.; Alfano, D.; Pesapane, A.; Rossi, F.W.; Salzano, S.; Rossi, G.; et al. The cross-talk between the urokinase receptor and fMLP receptors regulates the activity of the CXCR4 chemokine receptor. Cell. Mol. Life Sci. 2011, 68, 2453–2467. [Google Scholar] [CrossRef]
- Gorrasi, A.; Li Santi, A.; Amodio, G.; Alfano, D.; Remondelli, P.; Montuori, N.; Ragno, P. The urokinase receptor takes control of cell migration by recruiting integrins and FPR1 on the cell surface. PLoS ONE 2014, 9, e86352. [Google Scholar] [CrossRef] [Green Version]
- Pulukuri, S.M.; Gondi, C.S.; Lakka, S.S.; Jutla, A.; Estes, N.; Gujrati, M.; Rao, J.S. RNA interference-directed knockdown of urokinase plasminogen activator and urokinase plasminogen activator receptor inhibits prostate cancer cell invasion, survival, and tumorigenicity in vivo. J. Biol. Chem. 2005, 280, 36529–36540. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, O.; Andolfo, A.; Santovito, M.L.; Iuzzolino, L.; Blasi, F.; Sidenius, N. Dimerization controls the lipid raft partitioning of uPAR/CD87 and regulates its biological functions. EMBO J. 2003, 22, 5994–6003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Head, B.P.; Patel, H.H.; Insel, P.A. Interaction of membrane/lipid rafts with the cytoskeleton: Impact on signaling and function: Membrane/lipid rafts, mediators of cytoskeletal arrangement and cell signaling. Biochim. Biophys. Acta 2014, 1838, 532–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Lukashev, M.; Simon, D.I.; Bodary, S.C.; Rosenberg, S.; Doyle, M.V.; Chapman, H.A. Regulation of integrin function by the urokinase receptor. Science 1996, 273, 1551–1555. [Google Scholar] [CrossRef] [PubMed]
- Montuori, N.; Carriero, M.V.; Salzano, S.; Rossi, G.; Ragno, P. The cleavage of the urokinase receptor regulates its multiple functions. J. Biol. Chem. 2002, 277, 46932–46939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, A.Y. Differential Expression of Cell Surface Molecules in Prostate Cancer Cells. Cancer Res. 2000, 60, 3429–3434. [Google Scholar]
- He, H.Q.; Ye, R.D. The Formyl Peptide Receptors: Diversity of Ligands and Mechanism for Recognition. Molecules 2017, 22, 455. [Google Scholar] [CrossRef]
- Levental, I.; Grzybek, M.; Simons, K. Greasing their way: Lipid modifications determine protein association with membrane rafts. Biochemistry 2010, 49, 6305–6316. [Google Scholar] [CrossRef]
- Rea, V.E.; Lavecchia, A.; Di Giovanni, C.; Rossi, F.W.; Gorrasi, A.; Pesapane, A.; de Paulis, A.; Ragno, P.; Montuori, N. Discovery of new small molecules targeting the vitronectin-binding site of the urokinase receptor that block cancer cell invasion. Mol. Cancer Ther. 2013, 12, 1402–1416. [Google Scholar] [CrossRef] [Green Version]
- Sitrin, R.G.; Johnson, D.R.; Pan, P.M.; Harsh, D.M.; Huang, J.; Petty, H.R.; Blackwood, R.A. Lipid raft compartmentalization of urokinase receptor signaling in human neutrophils. Am. J. Respir. Cell Mol. Biol. 2004, 30, 233–241. [Google Scholar] [CrossRef]
- Mazzieri, R.; Pietrogrande, G.; Gerasi, L.; Gandelli, A.; Colombo, P.; Moi, D.; Brombin, C.; Ambrosi, A.; Danese, S.; Mignatti, P.; et al. Urokinase Receptor Promotes Skin Tumor Formation by Preventing Epithelial Cell Activation of Notch1. Cancer Res. 2015, 75, 4895–4909. [Google Scholar] [CrossRef] [Green Version]
- Margheri, F.; Chillà, A.; Laurenzana, A.; Serratì, S.; Mozzanti, B.; Saccaridi, R.; Santosuosso, M.; Danza, G.; Sturli, N.; Rosati, F.; et al. Endothelial progenitor cell-dependent angiogenesis requires localization of the full-length form of uPAR in caveolae. Blood 2011, 118, 3743–3755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahores, M.; Prinetti, A.; Chiabrando, G.; Blasi, F.; Sonnino, S. uPA binding increases UPAR localization to lipid rafts and modifies the receptor microdomain composition. Biochim. Biophys. Acta 2008, 1778, 250–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grove, L.M.; Southern, B.D.; Jin, T.H.; White, K.E.; Paruchuri, S.; Harel, E.; Wei, Y.; Rahaman, S.O.; Gladson, C.L.; Ding, Q.; et al. Urokinase-type plasminogen activator receptor (uPAR) ligation induces a raft-localized integrin signaling switch that mediates the hypermotile phenotype of fibrotic fibroblasts. J. Biol. Chem. 2014, 289, 12791–12804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, D.D.; Fernandez-Borja, M. Leukocyte adhesion and polarization: Role of glycosylphosphatidylinositol-anchored proteins. Bioarchitecture 2015, 5, 61–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leitinger, B.; Hogg, N. The involvement of lipid rafts in the regulation of integrin function. J. Cell Sci. 2002, 115, 963–972. [Google Scholar]
- Hogg, N.; Henderson, R.; Leitinger, B.; McDowall, A.; Porter, J.; Stanley, P. Mechanisms contributing to the activity of integrins on leukocytes. Immunol. Rev. 2002, 186, 164–171. [Google Scholar] [CrossRef]
- Resnati, M.; Pallavicini, I.; Wang, J.M.; Oppenheim, J.; Serhan, C.N.; Romano, M.; Blasi, F. The fibrinolytic receptor for urokinase activates the G protein-coupled chemotactic receptor FPRL1/LXA4R. Proc. Natl. Acad. Sci. USA 2002, 9, 1359–1364. [Google Scholar] [CrossRef] [Green Version]
- Sitrin, R.G.; Emery, S.L.; Sassanella, T.M.; Blackwood, R.A.; Petty, H.R. Selective localization of recognition complexes for leukotriene B4 and formyl-Met-Leu-Phe within lipid raft microdomains of human polymorphonuclear neutrophils. J. Immunol. 2006, 177, 8177–8184. [Google Scholar] [CrossRef] [Green Version]
- Xue, M.; Vines, C.M.; Buranda, T.; Cimino, D.F.; Bennett, T.A.; Prossnitz, E.R. N-formyl peptide receptors cluster in an active raft-associated state prior to phosphorylation. J. Biol. Chem. 2004, 279, 45175–45184. [Google Scholar] [CrossRef] [Green Version]
- de Paulis, A.; Montuori, N.; Prevete, N.; Fiorentino, I.; Rossi, F.W.; Visconte, V.; Rossi, G.; Marone, G.; Ragno, P. Urokinase induces basophil chemotaxis through a urokinase receptor epitope that is an endogenous ligand for formyl peptide receptor-like 1 and -like 2. J. Immunol. 2004, 173, 5739–5748. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, H.M.; Gripentrog, J.M.; Jesaitis, A.J. Chemotaxis of chinese hamster ovary cells expressing the human neutrophil formyl peptide receptor: Role of signal transduction molecules and alpha5beta1 integrin. J. Cell Sci. 1998, 111, 1921–1928. [Google Scholar] [PubMed]
- Sadhu, C.; Masinovsky, B.; Staunton, D.E. Differential regulation of chemoattractant-stimulated beta 2, beta 3, and beta 7 integrin activity. J. Immunol. 1998, 160, 5622–5628. [Google Scholar] [PubMed]
- Campbell, J.J.; Qin, S.; Bacon, K.B.; Mackay, C.R.; Butcher, E.C. Biology of chemokine and classical chemoattractant receptors: Differential requirements for adhesion-triggering versus chemotactic responses in lymphoid cells. J. Cell Biol. 1996, 134, 255–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorrasi, A.; Petrone, A.M.; Li Santi, A.; Alfieri, M.; Montuori, N.; Ragno, P. New Pieces in the Puzzle of uPAR Role in Cell Migration Mechanisms. Cells 2020, 9, 2531. https://doi.org/10.3390/cells9122531
Gorrasi A, Petrone AM, Li Santi A, Alfieri M, Montuori N, Ragno P. New Pieces in the Puzzle of uPAR Role in Cell Migration Mechanisms. Cells. 2020; 9(12):2531. https://doi.org/10.3390/cells9122531
Chicago/Turabian StyleGorrasi, Anna, Anna Maria Petrone, Anna Li Santi, Mariaevelina Alfieri, Nunzia Montuori, and Pia Ragno. 2020. "New Pieces in the Puzzle of uPAR Role in Cell Migration Mechanisms" Cells 9, no. 12: 2531. https://doi.org/10.3390/cells9122531
APA StyleGorrasi, A., Petrone, A. M., Li Santi, A., Alfieri, M., Montuori, N., & Ragno, P. (2020). New Pieces in the Puzzle of uPAR Role in Cell Migration Mechanisms. Cells, 9(12), 2531. https://doi.org/10.3390/cells9122531