Involvement of Bradykinin Receptor 2 in Nerve Growth Factor Neuroprotective Activity

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Mixed Cortical Cultures (CNs), Treatment and Viability Assay
2.3. Enriched Microglial Cultures
2.4. Microarray Analysis
2.5. Immunocytochemistry
2.6. ELISA
2.7. Electrophysiology
2.8. Transgenic Mice
2.9. Intranasal Treatment with NGF and Tissue Processing
2.10. Western Blotting
2.11. Data Analysis
3. Results
3.1. Expression of Bradykinin (BK) and BK Receptors in CNs Following NGF Treatment and Deprivation
3.2. Steady-State Levels of B2R Protein
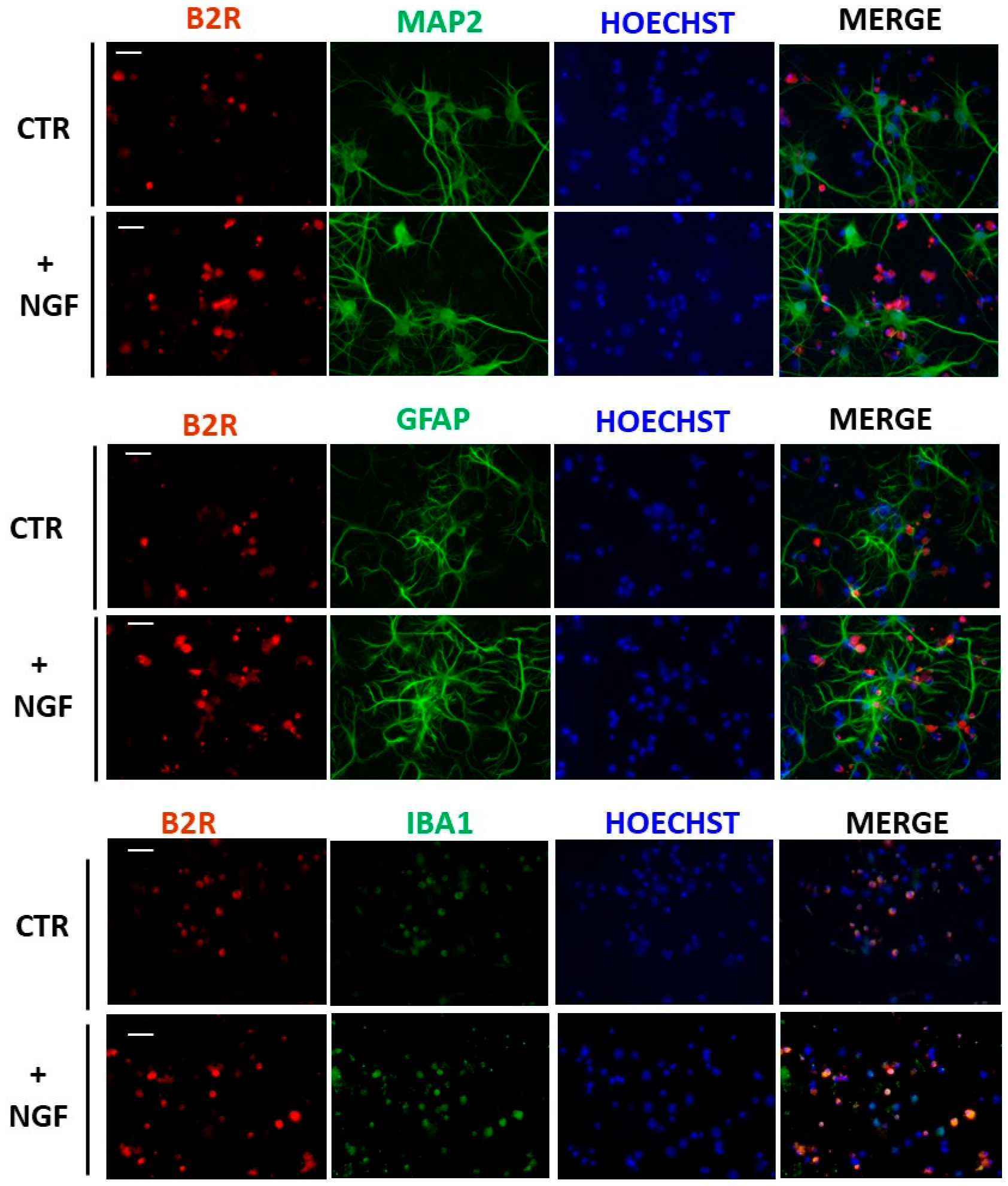
3.3. Expression of B2R in Cultured Microglial Cells
3.4. CNs Viability
3.5. Electrophysiology
3.6. NGF Treated AD Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Levi-Montalcini, R. The nerve growth factor 35 years later. Science 1987, 237, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Delcroix, J.D.; Valletta, J.; Wu, C.; Howe, C.L.; Lai, C.F.; Cooper, J.D.; Belichenko, P.V.; Salehi, A.; Mobley, W.C. Trafficking the NGF signal: Implications for normal and degenerating neurons. Prog. Brain Res. 2004, 146, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Mufson, E.J.; Counts, S.E.; Perez, S.E.; Ginsberg, S.D. Cholinergic system during the progression of Alzheimer’s disease: Therapeutic implications. Expert Rev. Neurother. 2008, 8, 1703–1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuszynski, M.H.; Thal, L.; Pay, M.; Salmon, D.P.; Bakay, R.; Patel, P.; Blesch, A.; Vahlsing, H.L.; Ho, G.; Tong, G.; et al. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat. Med. 2005, 11, 551–555. [Google Scholar] [CrossRef]
- Chao, M.V.; Rajagopal, R.; Lee, F.S. Neurotrophin signalling in health and disease. Clin. Sci. 2006, 110, 167–173. [Google Scholar] [CrossRef]
- Giacobini, E.; Becker, R.E. One Hundred Years After the Discovery of Alzheimer’s Disease. A Turning Point for Therapy? J. Alzheimer Dis. 2007, 12, 37–52. [Google Scholar] [CrossRef]
- Cattaneo, A.; Capsoni, S.; Paoletti, F. Towards non invasive nerve growth factor therapies for Alzheimer’s disease. J. Alzheimer Dis. 2008, 15, 255–283. [Google Scholar] [CrossRef] [Green Version]
- Matrone, C.; Ciotti, M.T.; Mercanti, D.; Marolda, R.; Calissano, P. NGF and BDNF signaling control amyloidogenic route and Abeta production in hippocampal neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 13139–13144. [Google Scholar] [CrossRef] [Green Version]
- Poduslo, J.F.; Curran, G.L.; Berg, C.T. Macromolecular permeability across the blood-nerve and blood-brain barriers. Proc. Natl. Acad. Sci. USA 1994, 91, 5705–5709. [Google Scholar] [CrossRef] [Green Version]
- Petty, B.G.; Cornblath, D.R.; Adornato, B.T.; Chaudhry, V.; Flexner, C.; Wachsman, M.; Sinicropi, D.; Burton, L.E.; Peroutka, S.J. The Effect of Systemically Administered Recombinant Human Nerve Growth Factor in Healthy Human Subjects. Ann. Neurol. 1994, 36, 244–246. [Google Scholar] [CrossRef]
- Pezet, S.; McMahon, S.B. Neurotrophins: Mediators and modulators of pain. Ann. Rev. Neurosci. 2006, 29, 507–538. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.R.; Woolf, C.J. Neuronal plasticity and signal transduction in nociceptive neurons: Implications for the initiation and maintenance of pathological pain. Neurobiol. Dis. 2001, 8, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewin, G.R.; Ritter, A.M.; Mendell, L.M. Nerve growth factor-induced hyperalgesia in the neonatal and adult rat. J. Neurosci. 1993, 13, 2136–2148. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.W.; Dray, A.; McCarson, K.E.; Krause, J.E.; Urban, L. Nerve growth factor induces mechanical allodynia associated with novel A fibre-evoked spinal reflex activity and enhanced neurokinin-1 receptor activation in the rat. Pain 1995, 62, 219–231. [Google Scholar] [CrossRef]
- Severini, C.; Petrocchi Passeri, P.; Ciotti, M.T.; Florenzano, F.; Petrella, C.; Malerba, F.; Bruni, B.; D’Onofrio, M.; Arisi, I.; Brandi, R.; et al. Nerve growth factor derivative NGF61/100 promotes outgrowth of primary sensory neurons with reduced signs of nociceptive sensitization. Neuropharmacology 2017, 117, 134–148. [Google Scholar] [CrossRef]
- Dray, A.; Perkins, M. Bradykinin and inflammatory pain. Trends Neurosci. 1993, 16, 99–104. [Google Scholar] [CrossRef]
- Brown, D.A.; Passmore, G.M. Some New Insights into the Molecular Mechanisms of Pain Perception. J. Clin. Investig. 2010, 120, 1380–1383. [Google Scholar] [CrossRef]
- Mizumura, K.; Sugiura, T.; Katanosaka, K.; Banik, R.K.; Kozaki, Y. Excitation and Sensitization of Nociceptors by Bradykinin: What Do We Know? Exp. Brain Res. 2009, 196, 53–65. [Google Scholar] [CrossRef]
- Regoli, D.; Barabé, J. Pharmacology of bradykinin and related kinins. Pharmacol. Rev. 1980, 32, 1–46. [Google Scholar]
- Regoli, D.; Nsa Allogho, S.; Rizzi, A.; Gobeil, F.J. Bradykinin receptors and their antagonists. Eur. J. Pharmacol. 1998, 348, 1–10. [Google Scholar] [CrossRef]
- Kasai, M.; Kumazawa, T.; Mizumura, K. Nerve Growth Factor Increases Sensitivity to Bradykinin, Mediated Through B2 Receptors, in Capsaicin-Sensitive Small Neurons Cultured from Rat Dorsal Root Ganglia. Neurosci. Res. 1998, 32, 231–239. [Google Scholar] [CrossRef]
- Kasai, M.; Mizumura, K. Endogenous Nerve Growth Factor Increases the Sensitivity to Bradykinin in Small Dorsal Root Ganglion Neurons of Adjuvant Inflamed Rats. Neurosci. Lett. 1999, 272, 41–44. [Google Scholar] [CrossRef]
- Naffah-Mazzacoratti, M.G.; Gouveia, T.L.; Simões, P.S.; Perosa, S.R. What have we learned about the kallikrein-kinin and renin-angiotensin systems in neurological disorders? World J. Biol. Chem. 2014, 5, 130–140. [Google Scholar] [CrossRef]
- Viel, T.A.; Buck, H.S. Kallikrein-kinin system mediated inflammation in Alzheimer’s disease in vivo. Curr. Alzheimer Res. 2011, 8, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Petrella, C.; Di Certo, M.G.; Barbato, C.; Gabanella, F.; Ralli, M.; Greco, A.; Possenti, R.; Severini, C. Neuropeptides in Alzheimer′s Disease: An Update. Curr. Alzheimer Res. 2019, 16, 544–558. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.M.; Lin, T.A.; Sun, G.Y.; Gibson, G.E. Increased Inositol 1,4,5-trisphosphate Accumulation Correlates with an Up-Regulation of Bradykinin Receptors in Alzheimer’s Disease. J. Neurochem. 1995, 64, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Jong, Y.J.I.; Dalemar, L.R.; Seehra, K.; Baenziger, N.L. Bradykinin Receptor Modulation in Cellular Models of Aging and Alzheimer’s Disease. Int. Immunopharmacol. 2002, 2, 1833–1840. [Google Scholar] [CrossRef]
- Viel, T.A.; Caetano, A.L.; Nasello, A.G.; Lancelott, C.L.; Nunes, V.A.; Araujo, M.S.; Buck, H.S. Increases of Kinin B1 and B2 Receptors Binding Sites After Brain Infusion of Amyloid-Beta 1-40 Peptide in Rats. Neurobiol. Aging 2008, 29, 1805–1814. [Google Scholar] [CrossRef]
- Prediger, R.D.S.; Medeiros, R.; Pandolfo, P.; Duarte, F.; Passos, G.; Pesquero, J.; Campos, M.M.; Calixto, J.; Takahashi, R. Genetic Deletion or Antagonism of Kinin B(1) and B(2) Receptors Improves Cognitive Deficits in a Mouse Model of Alzheimer’s Disease. Neuroscience 2008, 151, 631–643. [Google Scholar] [CrossRef]
- Lacoste, B.; Tong, X.-K.; Lahjouji, K.; Couture, R.; Hamel, E. Cognitive and Cerebrovascular Improvements Following Kinin B1 Receptor Blockade in Alzheimer’s Disease Mice. J. Neuroinflamm. 2013, 10, 57. [Google Scholar] [CrossRef] [Green Version]
- Asraf, K.; Torika, N.; Roasso, E.; Fleisher-Berkovich, S. Differential Effect of Intranasally Administrated Kinin B1 and B2 Receptor Antagonists in Alzheimer’s Disease Mice. Biol. Chem. 2016, 397, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Amaral, F.A.; Lemos, M.T.; Dong, K.E.; Bittencourt, M.F.; Caetano, A.L.; Pesquero, J.B.; Viel, T.A.; Buck, H.S. Participation of kinin receptors on memory impairment after chronic infusion of human amyloid-beta 1-40 peptide in mice. Neuropeptides 2010, 44, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Caetano, A.L.; Dong-Creste, K.E.; Amaral, F.A.; Monteiro-Silva, K.C.; Pesquero, J.B.; Araujo, M.S.; Montor, W.R.; Viel, T.A.; Buck, H.S. Kinin B2 receptor can play a neuroprotective role in Alzheimer′s disease. Neuropeptides 2015, 53, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Severini, C.; Lattanzi, R.; Maftei, D.; Marconi, V.; Ciotti, M.T.; Petrocchi Passeri, P.; Florenzano, F.; Del Duca, E.; Caioli, S.; Zona, C.; et al. Bv8/prokineticin 2 is involved in Aβ-induced neurotoxicity. Sci. Rep. 2015, 5, 15301. [Google Scholar] [CrossRef] [Green Version]
- Pieri, M.; Amadoro, G.; Carunchio, I.; Ciotti, M.T.; Quaresima, S.; Florenzano, F.; Calissano, P.; Possenti, R.; Zona, C.; Severini, C. SP protects cerebellar granule cells against beta-amyloid-induced apoptosis by down-regulation and reduced activity of Kv4 potassium channels. Neuropharmacology 2010, 58, 268–276. [Google Scholar] [CrossRef]
- Marolda, R.; Ciotti, M.T.; Matrone, C.; Possenti, R.; Calissano, P.; Cavallaro, S.; Severini, C. Substance P activates ADAM9 mRNA expression and induces α-secretase-mediated amyloid precursor protein cleavage. Neuropharmacology 2012, 62, 1954–1963. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Capsoni, S.; Malerba, F.; Carucci, N.M.; Rizzi, C.; Criscuolo, C.; Origlia, N.; Calvello, M.; Viegi, A.; Meli, G.; Cattaneo, A. The chemokine CXCL12 mediates the anti-amyloidogenic action of painless human nerve growth factor. Brain 2017, 140, 201–217. [Google Scholar] [CrossRef] [Green Version]
- Martins, A.H.; Alves, J.M.; Perez, D.; Carrasco, M.; Torres-Rivera, W.; Eterović, V.A.; Ferchmin, P.A.; Ulrich, H. Kinin-B2 receptor mediated neuroprotection after NMDA excitotoxicity is reversed in the presence of kinin-B1 receptor agonists. PLoS ONE 2012, 7, e30755. [Google Scholar] [CrossRef] [Green Version]
- Eterović, V.A.; Del Valle-Rodriguez, A.; Pérez, D.; Carrasco, M.; Khanfar, M.A.; El Sayed, K.A.; Ferchmin, P.A. Protective activity of (1S,2E,4R,6R,7E,11E)-2,7,11-cembratriene-4,6-diol analogues against diisopropylfluorophosphate neurotoxicity: Preliminary structure-activity relationship and pharmacophore modeling. Bioorg. Med. Chem. 2013, 21, 4678–4686. [Google Scholar] [CrossRef] [Green Version]
- Torres-Rivera, W.; Pérez, D.; Park, K.-Y.; Carrasco, M.; Platt, M.O.; Eterović, V.A.; Ferchmin, P.A.; Ulrich, H.; Martins, A.H. Kinin-B2 receptor exerted neuroprotection after diisopropylfluorophosphate-induced neuronal damage. Neuroscience 2013, 247, 273–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, B.; Cheng, B.; Pan, Y.; Wang, C.; Chen, J.; Bai, B. Neuroprotection of bradykinin/bradykinin B2 receptor system in cerebral ischemia. Biomed. Pharmacother 2017, 94, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Levi-Montalcini, R.; Hamburger, V. A nerve growth-stimulating factor isolated from sarcoma AS 37 and 180. Proc. Natl. Acad. Sci. USA 1954, 40, 1014–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.P.; Schmidt, R.E.; DiStefano, P.S.; Lowry, O.H.; Carter, J.G.; Johnson, E.M., Jr. Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J. Cell Biol. 1998, 106, 829–844. [Google Scholar] [CrossRef] [PubMed]
- Hefty, F. Nerve growth factor promotes survival of septal cholinergic neurons after fimbrial transections. J. Neurosci. 1986, 6, 2155–2162. [Google Scholar] [CrossRef] [Green Version]
- Seiler, M.; Schwab, M.E. Specific retrograde transport of nerve growth factor (NGF) from neocortex to nucleus basalis in the rat. Brain Res. 1984, 300, 33–39. [Google Scholar] [CrossRef]
- Li, Y.; Holtzman, D.M.; Kromer, L.F.; Kaplan, D.R.; Chua-Couzens, J.; Clary, D.O.; Knüsel, B.; Mobley, W.C. Regulation of TrkA and ChAT expression in developing rat basal forebrain: Evidence that both exogenous and endogenous NGF regulate differentiation of cholinergic neurons. J. Neurosci. 1995, 15, 2888–2905. [Google Scholar] [CrossRef] [Green Version]
- Debeir, T.; Saragovi, H.U.; Cuello, A.C. A nerve growth factor mimetic TrkA antagonist causes withdrawal of cortical cholinergic boutons in the adult rat. Proc. Natl. Acad. Sci. USA 1999, 96, 4067–4072. [Google Scholar] [CrossRef] [Green Version]
- Scott, S.A.; Mufson, E.J.; Weingartner, J.A.; Skau, K.A.; Crutcher, K.A. Nerve growth factor in Alzheimer’s disease: Increased levels throughout the brain coupled with declines in nucleus basalis. J. Neurosci. 1995, 15, 6213–6221. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.J.; Zachrisson, O.; Tonge, D.A.; McNaughton, P.A. Upregulation of bradykinin B2 receptor expression by neurotrophic factors and nerve injury in mouse sensory neurons. Mol. Cell Neurosci. 2002, 19, 186–200. [Google Scholar] [CrossRef]
- Chen, E.Y.; Emerich, D.F.; Bartus, R.T.; Kordower, J.H. B2 bradykinin receptor immunoreactivity in rat brain. J. Comp. Neurol. 2000, 427, 1–18. [Google Scholar] [CrossRef]
- Cholewinski, A.J.; Stevens, G.; McDermott, A.M.; Wilkin, G.P. Identification of B2 bradykinin binding sites on cultured cortical astrocytes. J. Neurochem. 1991, 57, 1456–1458. [Google Scholar] [CrossRef]
- Hsieh, H.L.; Yen, M.H.; Jou, M.J.; Yang, C.M. Intracellular signalings underlying bradykinin-induced matrix metalloproteinase-9 expression in rat brain astrocyte-1. Cell Signal. 2004, 16, 1163–1176. [Google Scholar] [CrossRef] [PubMed]
- Torika, N.; Filipovich-Rimon, T.; Asraf, K.; Roasso, E.; Danon, A.; Fleisher-Berkovich, S. Differential regulation of astrocyte prostaglandin response by kinins: Possible role for mitogen activated protein kinases. Eur. J. Pharmacol. 2014, 741, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Noda, M.; Kariura, Y.; Amano, T.; Manago, Y.; Nishikawa, K.; Aoki, S.; Wada, K. Expression and function of bradykinin receptors in microglia. Life Sci. 2003, 72, 1573–1581. [Google Scholar] [CrossRef]
- Noda, M.; Kariura, Y.; Amano, T.; Manago, Y.; Nishikawa, K.; Aoki, S.; Wada, K. Kinin receptors in cultured rat microglia. Neurochem. Int. 2004, 45, 437–442. [Google Scholar] [CrossRef]
- Tang, M.; Cui, M.; Dong, Q.; Ren, H.M.; Xiao, B.G.; Luo, B.Y.; Shao, Y.; Liu, L.; Zhou, H.G. The bradykinin B2 receptor mediates hypoxia/reoxygenation induced neuronal cell apoptosis through the ERK1/2 pathway. Neurosci. Lett. 2009, 450, 40–44. [Google Scholar] [CrossRef]
- Rizzi, C.; Tiberi, A.; Giustizieri, M.; Marrone, M.C.; Gobbo, F.; Carucci, N.M.; Meli, G.; Arisi, I.; D’Onofrio, M.; Marinelli, S.; et al. NGF steers microglia toward a neuroprotective phenotype. Glia 2018, 66, 1395–1416. [Google Scholar] [CrossRef] [Green Version]
- Bhoola, K.D.; Figueroa, C.D.; Worthy, K. Bioregulation of kinins: Kallikreins, kininogens and kininases. Pharmacol. Rev. 1992, 44, 1–80. [Google Scholar]
- Frey, W.H.; Liu, J.; Chen, X.; Thorne, R.G.; Fawcett, J.R.; Ala, T.A.; Rahman, Y.-E. Delivery of 125I-NGF to the brain via the olfactory route. Drug Deliv. 1997, 4, 87–92. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrella, C.; Ciotti, M.T.; Nisticò, R.; Piccinin, S.; Calissano, P.; Capsoni, S.; Mercanti, D.; Cavallaro, S.; Possenti, R.; Severini, C. Involvement of Bradykinin Receptor 2 in Nerve Growth Factor Neuroprotective Activity. Cells 2020, 9, 2651. https://doi.org/10.3390/cells9122651
Petrella C, Ciotti MT, Nisticò R, Piccinin S, Calissano P, Capsoni S, Mercanti D, Cavallaro S, Possenti R, Severini C. Involvement of Bradykinin Receptor 2 in Nerve Growth Factor Neuroprotective Activity. Cells. 2020; 9(12):2651. https://doi.org/10.3390/cells9122651
Chicago/Turabian StylePetrella, Carla, Maria Teresa Ciotti, Robert Nisticò, Sonia Piccinin, Pietro Calissano, Simona Capsoni, Delio Mercanti, Sebastiano Cavallaro, Roberta Possenti, and Cinzia Severini. 2020. "Involvement of Bradykinin Receptor 2 in Nerve Growth Factor Neuroprotective Activity" Cells 9, no. 12: 2651. https://doi.org/10.3390/cells9122651
APA StylePetrella, C., Ciotti, M. T., Nisticò, R., Piccinin, S., Calissano, P., Capsoni, S., Mercanti, D., Cavallaro, S., Possenti, R., & Severini, C. (2020). Involvement of Bradykinin Receptor 2 in Nerve Growth Factor Neuroprotective Activity. Cells, 9(12), 2651. https://doi.org/10.3390/cells9122651