The Close Relationship between the Golgi Trafficking Machinery and Protein Glycosylation




{kind=link}
{kind=link}
{kind=link}
Abstract
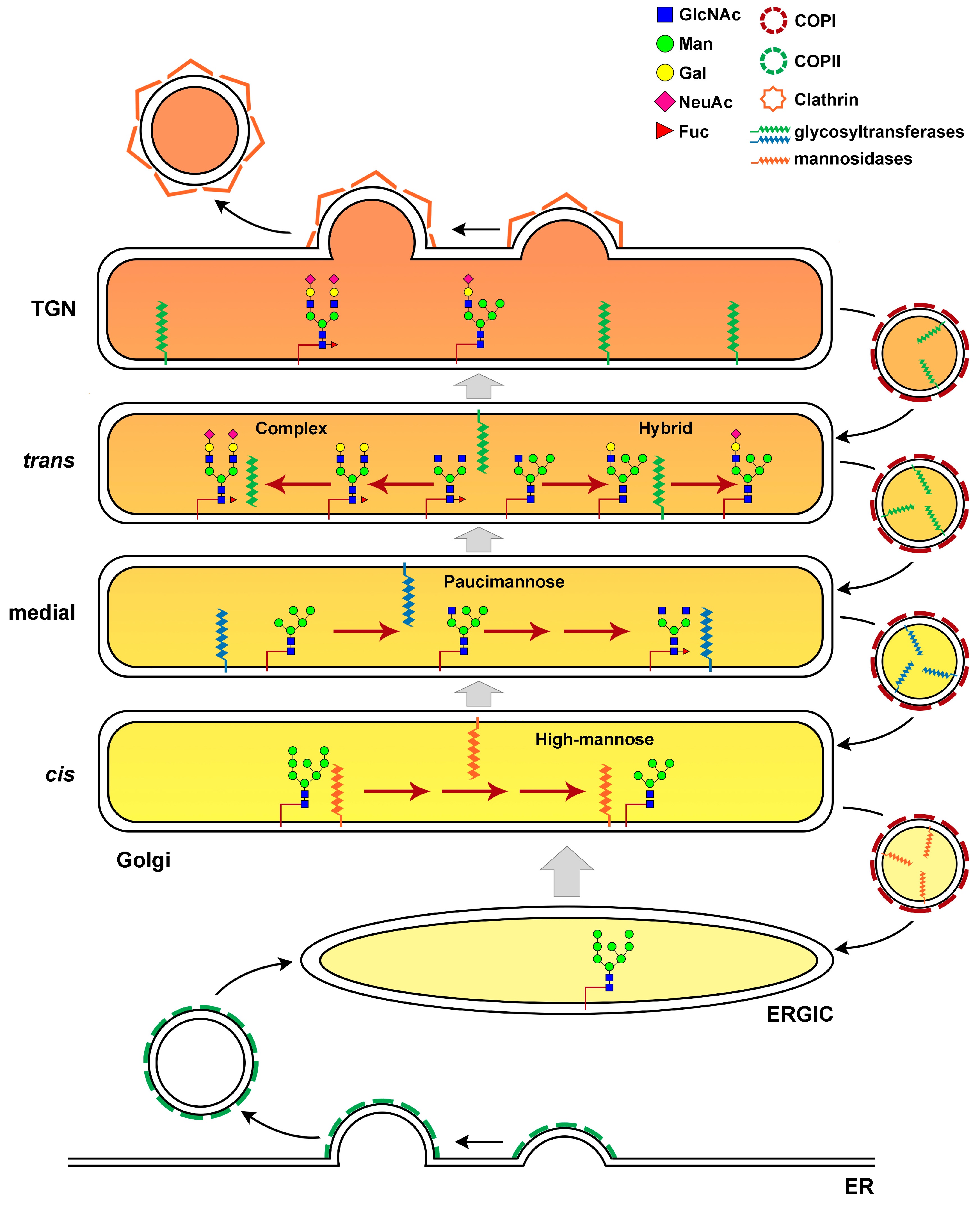
:1. Protein Glycosylation and the Golgi
2. Enzyme Sorting at the Golgi Cisternae
3. Molecular Actors in Vesicle Targeting and Fusion at the Golgi
4. GOLPH3 Interacts with COPI to Recruit Glycosyltransferases to Golgi Compartments
5. GORAB, a Scaffolding Protein for COPI, Is Involved in Gerodermia Osteodysplastica
6. The Vesicle Tethering COG Complex and Other Tethering Factors Are Required for Normal Glycosylation
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Dennis, J.W.; Lau, K.S.; Demetriou, M.; Nabi, I.R. Adaptive regulation at the cell surface by N-glycosylation. Traffic 2009, 10, 1569–1578. [Google Scholar] [CrossRef]
- Haltiwanger, R.S. Regulation of signal transduction by glycosylation. Int. J. Exp. Pathol. 2004, 85, A49–A50. [Google Scholar] [CrossRef]
- Freeze, H.H.; Ng, B.G. Golgi glycosylation and human inherited diseases. Cold Spring Harb. Perspect. Biol. 2011, 3, a005371. [Google Scholar] [CrossRef] [PubMed]
- Khoury, G.A.; Baliban, R.C.; Floudas, C.A. Proteome-wide post- translational modification statistics: Frequency analysis and curation of the swiss-prot database. Sci. Rep. 2011, 1, 90. [Google Scholar] [CrossRef]
- Ohtsubo, K.; Marth, J.D. Glycosylation in cellular mechanisms of health and disease. Cell 2006, 126, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Freeze, H.H. Genetic defects in the human glycome. Nat. Rev. Genet. 2006, 7, 537–551. [Google Scholar] [CrossRef]
- Cummings, R.D. The repertoire of glycan determinants in the human glycome. Mol. Biosyst. 2009, 5, 1087–1104. [Google Scholar] [CrossRef]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [Green Version]
- Burda, P.; Aebi, M. The dolichol pathway of N-linked glycosylation. Biochim. Biophys. Acta 1999, 1426, 239–257. [Google Scholar] [CrossRef]
- Li, H.; Chavan, M.; Schindelin, H.; Lennarz, W.J.; Li, H. Structure of the oligosaccharyl transferase complex at 12 A resolution. Structure 2008, 16, 432–440. [Google Scholar] [CrossRef] [Green Version]
- Zielinska, D.F.; Gnad, F.; Wi’sniewski, J.R.; Mann, M. Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell 2010, 141, 897–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shwartz, F.; Aebi, M. Mechanisms and principles of N-linked protein glycosylation. Curr. Opin. Struct. Biol. 2011, 21, 576–582. [Google Scholar] [CrossRef]
- Helenius, A.; Aebi, M. Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 2004, 73, 1019–1049. [Google Scholar] [CrossRef] [PubMed]
- Lederkremer, G.Z. Glycoprotein folding, quality control and ER-associated degradation. Curr. Opin. Struct. Biol. 2009, 19, 515–523. [Google Scholar] [CrossRef]
- Moremen, K.W.; Tiemeyer, M.; Nairn, A.V. Vertebrate protein glycosylation: Diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 2012, 13, 448–462. [Google Scholar] [CrossRef] [Green Version]
- Rabouille, C.; Hui, N.; Hunte, F.; Kieckbusch, R.; Berger, E.G.; Warren, G.; Nilsson, T. Mapping the distribution of Golgi enzymes involved in the construction of complex oligosaccharides C. J. Cell Sci. 1995, 108, 1617–1627. [Google Scholar] [PubMed]
- Stanley, P. Golgi glycosylation. Cold Spring Harb. Perspect. Biol. 2011, 3, a005199. [Google Scholar] [CrossRef]
- Stanley, P.; Schachter, H.; Taniguchi, N. N-Glycans. In Essentials of Glycobiology; Varki, A., Ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2009; pp. 101–114. [Google Scholar]
- Glick, B.S.; Luini, A. Models for Golgi traffic: A critical assessment. Cold Spring Harb. Perspect. Biol. 2011, 3, a005215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, P.; Ungar, D. Bridging the Gap between Glycosylation and Vesicle Traffic. Front. Cell Dev. Biol. 2016, 4, 15. [Google Scholar] [CrossRef] [Green Version]
- Linders, P.; Peters, E.; Ter Beest, M.; Lefeber, D.J.; Van den Bogaart, G. Sugary Logistics Gone Wrong: Membrane Trafficking and Congenital Disorders of Glycosylation. Int. J. Mol. Sci. 2020, 21, 4654. [Google Scholar] [CrossRef]
- Goreta, S.S.; Dabelic, S.; Dumic, J. Insights into complexity of congenital disorders of glycosylation. Biochem. Med. (Zagreb) 2012, 22, 156–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantazopoulou, A.; Glick, B.S. A Kinetic View of Membrane Traffic Pathways Can Transcend the Classical View of Golgi Compartments. Front. Cell Dev. Biol. 2019, 7, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beznoussenko, G.V.; Parashuraman, S.; Rizzo, R.; Polishchuk, R.; Martella, O.; Di Giandomenico, D.; Fusella, A.; Spaar, A.; Sallese, M.; Capestrano, M.G.; et al. Transport of soluble proteins through the Golgi occurs by diffusion via continuities across cisternae. Elife 2014, 3, e02009. [Google Scholar] [CrossRef] [PubMed]
- Beck, R.; Rawet, M.; Wieland, F.T.; Cassel, D. The COPI system: Molecular mechanisms and function. FEBS Lett. 2009, 583, 2701–2709. [Google Scholar] [CrossRef] [Green Version]
- D’Souza-Schorey, C.; Chavrier, P. ARF proteins: Roles in membrane traffic and beyond. Nat. Rev. Mol. Cell Biol. 2006, 7, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Pucadyil, T.J.; Schmid, S.L. Conserved functions of membrane active GTPases in coated vesicle formation. Science 2009, 325, 1217–1220. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Yang, J.S.; Schmider, A.B.; Soberman, R.J.; Hsu, V.W. Coordinated regulation of bidirectional COPI transport at the Golgi by cdc42. Nature 2015, 521, 529–532. [Google Scholar] [CrossRef] [Green Version]
- Casey, J.R.; Grinstein, S.; Orlowski, J. Sensors and regulators of intracellular pH. Nat. Rev. Mol. Cell Biol. 2010, 11, 50–61. [Google Scholar] [CrossRef]
- Rivinoja, A.; Hassinen, A.; Kokkonen, N.; Kauppila, A.; Kellokumpu, S. Elevated Golgi pH impairs terminal N-glycosylation by inducing mislocalization of Golgi glycosyltransferases. J. Cell Physiol. 2009, 220, 144–154. [Google Scholar] [CrossRef]
- Maeda, Y.; Kinoshita, T. The acidic environment of the Golgi is critical for glycosylation and transport. Methods Enzymol. 2010, 480, 495–510. [Google Scholar] [CrossRef]
- Kornak, U.; Reynders, E.; Dimopoulou, A.; Reeuwijk, J.; Fischer, B.; Rajab, A.; Budde, B.; Nürnberg, P.; Foulquier, F.; Mundlos, A.S. Impaired glycosylation and cutis laxa caused by mutations in the vesicular H+-ATPase subunit ATP6V0A2. Nat. Genet. 2008, 40, 32–34. [Google Scholar] [CrossRef]
- Van Galen, J.; Campelo, F.; Martínez-Alonso, E.; Scarpa, M.; Martínez-Menárguez, J.A.; Malhotra, V. Sphingomyelin homeostasis is required to form functional enzymatic domains at the trans-Golgi network. J. Cell Biol. 2014, 206, 609–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, C.S.; Hung, C.S.; Huoh, Y.S.; Mousley, C.J.; Stefan, C.J.; Bankaitis, V.; Ferguson, K.M.; Burd, C.G. Local control of phosphatidylinositol 4-phosphate signaling in the Golgi apparatus by Vps74 and Sac1 phosphoinositide phosphatase. Mol. Biol. Cell. 2012, 23, 2527–2536. [Google Scholar] [CrossRef] [PubMed]
- Bröcker, C.; Engelbrecht-Vandré, S.; Ungermann, C. Multisubunit tethering complexes and their role in membrane fusion. Curr. Biol. 2010, 20, R943–R952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Hutagalung, A.H.; Novick, P.J. Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 2011, 91, 119–149. [Google Scholar] [CrossRef] [Green Version]
- Barr, F.; Lambright, D.G. Rab GEFs and GAPs. Curr. Opin. Cell Biol. 2010, 22, 461–470. [Google Scholar] [CrossRef]
- Pfeffer, S.R. Rab GTPase regulation of membrane identity. Curr. Opin. Cell Biol. 2013, 25, 414–419. [Google Scholar] [CrossRef] [Green Version]
- Gillingham, A.K. At the ends of their tethers! How coiled-coil proteins capture vesicles at the Golgi. Biochem. Soc. Trans. 2017, 46, 43–50. [Google Scholar] [CrossRef]
- Munro, S. The Golgin coiled-coil proteins of the Golgi apparatus. Cold Spring Harb. Perspect Biol. 2011, 3, a005256. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, S.R. Rab GTPase localization and Rab cascades in Golgi transport. Biochem. Soc. Trans. 2012, 40, 1373–1377. [Google Scholar] [CrossRef] [Green Version]
- Lowe, M. The Physiological Functions of the Golgin Vesicle Tethering Proteins. Front. Cell Dev. Biol. 2019, 7, 94. [Google Scholar] [CrossRef]
- Witkos, T.M.; Lowe, M. The golgin family of coiled-coil tethering proteins. Front. Cell Dev. Biol. 2015, 3, 86. [Google Scholar] [CrossRef] [Green Version]
- Yu, I.-M.; Hughson, F.M. Tethering factors as organizers of intracellular vesicular traffic. Annu. Rev. Cell Dev. Biol. 2010, 26, 137–156. [Google Scholar] [CrossRef]
- Wong, M.; Munro, S. Membrane trafficking. The specificity of vesicle traffic to the Golgi is encoded in the golgin coiled-coil proteins. Science 2014, 346, 1256898. [Google Scholar] [CrossRef] [Green Version]
- Ungermann, C.; Kümmel, D. Structure of membrane tethers and their role in fusion. Traffic 2019, 20, 479–490. [Google Scholar] [CrossRef]
- Kim, J.J.; Lipatova, Z.; Segev, N. TRAPP Complexes in Secretion and Autophagy. Cell Dev. Biol. 2016, 4, 20. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Yu, S.; Menon, S.; Cai, Y.; Lazarova, D.; Fu, C.; Reinisch, K.; Hay, J.C.; Ferro-Novick, S. TRAPPI tethers COPII vesicles by binding the coat subunit Sec23. Nature 2007, 445, 941–944. [Google Scholar] [CrossRef]
- Hong, W.; Lev, S. Tethering the assembly of SNARE complexes. Trends Cell Biol. 2014, 24, 35–43. [Google Scholar] [CrossRef]
- Weber, T.; Zemelman, B.V.; McNew, J.A.; Westermann, B.; Gmachl, M.; Parlati, F.; Söllner, T.H.; Rothman, J.E. SNAREpins: Minimal machinery for membrane fusion. Cell 1998, 92, 759–772. [Google Scholar] [CrossRef] [Green Version]
- Hua, Y.; Scheller, R.H. Three SNARE complexes cooperate to mediate membrane fusion. Proc. Natl. Acad. Sci. USA 2001, 98, 8065–8070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizo, J.; Südhof, T.C. The membrane fusion enigma: SNAREs, Sec1/Munc18 proteins, and their accomplices--guilty as charged? Annu. Rev. Cell Dev. Biol. 2012, 28, 279–308. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Dulubova, I.; Min, S.W.; Chen, X.; Rizo, J.; Südhof, T.C. Sly1 binds to Golgi and ER syntaxins via a conserved N-terminal peptide motif. Dev. Cell 2002, 2, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Carr, C.M.; Grote, E.; Munson, M.; Hughson, F.M.; Novick, P.J. Sec1p binds to SNARE complexes and concentrates at sites of secretion. J. Cell Biol. 1999, 146, 333–344. [Google Scholar] [CrossRef] [Green Version]
- Togneri, J.; Cheng, Y.-S.; Munson, M.; Hughson, F.M.; Carr, C.M. Specific SNARE complex binding mode of the Sec1/Munc-18 protein, Sec1p. Proc. Natl. Acad. Sci. USA 2006, 103, 17730–17735. [Google Scholar] [CrossRef] [Green Version]
- Lobingier, B.T.; Merz, A.J. Sec1/Munc18 protein Vps33 binds to SNARE domains and the quaternary SNARE complex. Biol. Cell 2012, 23, 4611–4622. [Google Scholar] [CrossRef] [PubMed]
- Sechi, S.; Frappaolo, A.; Karimpour-Ghahnavieh, A.; Piergentili, R.; Giansanti, M.G. Oncogenic Roles of GOLPH3 in the Physiopathology of Cancer. Int. J. Mol. Sci. 2020, 21, 933. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.C.; Taylor, R.S.; Lane, D.R.; Ladinsky, M.S.; Weisz, J.A.; Howell, K.E. GMx33: A novel family of trans-Golgi proteins identified by proteomics. Traffic 2000, 1, 963–975. [Google Scholar] [CrossRef]
- Bell, A.W.; Ward, M.A.; Blackstock, W.P.; Freeman, H.N.; Choudhary, J.S.; Lewis, A.P.; Chotai, D.; Fazel, A.; Gushue, J.N.; Paiement, J.; et al. Proteomics characterization of abundant Golgi membrane proteins. J. Biol. Chem. 2001, 276, 5152–5165. [Google Scholar] [CrossRef] [Green Version]
- Dippold, H.C.; Ng, M.M.; Farber-Katz, S.E.; Lee, S.K.; Kerr, M.L.; Peterman, M.C.; Sim, R.; Wiharto, P.A.; Galbraith, K.A.; Madhavarapu, S.; et al. GOLPH3 bridges phosphatidylinositol-4-phosphate and actomyosin to stretch and shape the Golgi to promote budding. Cell 2009, 139, 337–351. [Google Scholar] [CrossRef] [Green Version]
- Snyder, C.M.; Mardones, G.A.; Ladinsky, M.S.; Howell, K.E. GMx33 associates with the trans-Golgi matrix in a dynamic manner and sorts within tubules exiting the Golgi. Mol. Biol. Cell. 2005, 17, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Sechi, S.; Colotti, G.; Belloni, G.; Mattei, V.; Frappaolo, A.; Raffa, G.D.; Fuller, M.T.; Giansanti, M.G. GOLPH3 Is Essential for Contractile Ring Formation and Rab11 Localization to the Cleavage Site during Cytokinesis in Drosophila melanogaster. PLoS Genet. 2014, 10, e1004305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, K.L.; Kabbarah, O.; Liang, M.C.; Ivanova, E.; Anagnostou, V.; Wu, J.; Dhakal, S.; Wu, M.; Chen, S.; Feinberg, T.; et al. GOLPH3 modulates mTOR signaling and rapamycin sensitivity in cancer. Nature 2009, 459, 1085–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckert, E.; Reckmann, I.; Hellwig, A.; Röhling, S.; El-Battari, A.; Wieland, F.T.; Popoff, V. Golgi phosphoprotein 3 triggers signal-mediated incorporation of glycosyltransferases into coatomer-coated (COPI) vesicles. J. Biol. Chem. 2014, 289, 31319–33129. [Google Scholar] [CrossRef] [Green Version]
- Witkos, T.M.; Chan, W.L.; Joensuu, M.; Rhiel, M.; Pallister, E.; Thomas-Oates, J.; Mould, A.P.; Mironov, A.A.; Lowe, M. GORAB scaffolds COPI at the trans-Golgi for efficient enzyme recycling and correct protein glycosylation. Nat. Commun. 2019, 10, 127. [Google Scholar] [CrossRef]
- Schmitz, K.R.; Liu, J.; Li, S.; Setty, T.G.; Wood, C.S.; Burd, C.G.; Ferguson, K.M. Golgi localization of glycosyltransferases requires a Vps74p oligomer. Dev. Cell 2008, 14, 523–534. [Google Scholar] [CrossRef] [Green Version]
- Tu, L.; Tai, W.C.; Chen, L.; Banfield, D.K. Signal-mediated dynamic retention of glycosyltransferases in the Golgi. Science 2008, 321, 404–407. [Google Scholar] [CrossRef]
- Wood, C.S.; Schmitz, K.R.; Bessman, N.J.; Setty, T.G.; Ferguson, K.M.; Burd, C.G. PtdIns4P recognition by Vps74/GOLPH3 links PtdIns 4-kinase signaling to retrograde Golgi trafficking. J. Cell Biol. 2009, 187, 967–975. [Google Scholar] [CrossRef] [Green Version]
- Tu, L.; Chen, L.; Banfield, D.K. A conserved N-terminal arginine-motif in GOLPH3-family proteins mediates binding to coatomer. Traffic 2012, 13, 1496–1507. [Google Scholar] [CrossRef]
- Ali, M.F.; Chachadi, V.B.; Petrosyan, A.; Pi-Wan, C. Golgi phosphoprotein 3 determines cell binding properties under dynamic flow by controlling Golgi localization of core 2 N-acetylglucosaminyltransferase 1. Biol. Chem. 2012, 287, 39564–39577. [Google Scholar] [CrossRef] [Green Version]
- Ochs, H.D.; Wedgwood, R.J.; Heller, S.R.; Beatty, P.G. Complement, membrane glycoproteins, and complement receptors. Their role in regulation of the immune response. Clin. Immunol. Immunopathol. 1986, 40, 94–104. [Google Scholar] [CrossRef]
- Pereira, N.A.; Pu, H.X.; Goh, H.; Song, Z. Golgi phosphoprotein 3 mediates the Golgi localization and function of protein O-linked mannose β-1,2-N-acetlyglucosaminyltransferase 1. Biol. Chem. 2014, 289, 14762–14770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, M.D.; Campbell, K.P. Dystroglycan inside and out. Curr. Opin. Cell Biol. 1999, 11, 602–607. [Google Scholar] [CrossRef]
- Muntoni, F.; Brockington, M.; Godfrey, C.; Ackroyd, M.; Robb, S.; Manzur, A.; Kinali, M.; Mercuri, E.; Kaluarachchi, M.; Feng, L.; et al. Muscular dystrophies due to defective glycosylation of dystroglycan. Acta Myol. 2007, 26, 129–135. [Google Scholar] [PubMed]
- Chang, W.L.; Chang, C.W.; Chang, Y.Y.; Sung, H.H.; Lin, M.D.; Chang, S.C.; Chen, C.H.; Huang, C.W.; Tung, K.S.; Chou, T.B. The Drosophila GOLPH3 homolog regulates the biosynthesis of heparan sulfate proteoglycans by modulating the retrograde trafficking of exostosins. Development 2013, 140, 2798–2807. [Google Scholar] [CrossRef] [Green Version]
- Busse-Wichera, M.; Wicherb, K.B.; Kusche-Gullberg, M. The extostosin family: Proteins with many functions. Matrix Biol. 2014, 35, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Wuyts, W.; Van Hul, W.; De Boulle, K.; Hendrickx, J.; Bakker, E.; Vanhoenacker, F.; Mollica, F.; Lüdecke, H.J.; Sayli, B.S.; Pazzaglia, U.E.; et al. Mutations in the EXT1 and EXT2 genes in hereditary multiple exostoses. Am. J. Hum. Genet. 1998, 62, 346–354. [Google Scholar] [CrossRef] [Green Version]
- Bovée, J.V. EXTra hit for mouse osteochondroma. Proc. Natl. Acad. Sci. USA 2010, 107, 1813–1814. [Google Scholar] [CrossRef] [Green Version]
- Jennes, I.; Pedrini, E.; Zuntini, M.; Mordenti, M.; Balkassmi, S.; Asteggiano, C.G.; Casey, B.; Bakker, B.; Sangiorgi, L.; Wuyts, W. Multiple osteochondromas: Mutation update and description of the multiple osteochondromas mutation database (MOdb). Hum. Mutat. 2009, 30, 1620–1627. [Google Scholar] [CrossRef]
- Isaji, T.; Im, S.; Gu, W.; Wang, Y.; Hangm, Q.; Lu, J.; Fukuda, T.; Hashii, N.; Takakura, D.; Kawasaki, N.; et al. An oncogenic protein Golgi phosphoprotein 3 up-regulates cell migration via sialylation. J. Biol. Chem. 2014, 289, 20694–20705. [Google Scholar] [CrossRef] [Green Version]
- Seales, E.C.; Jurado, G.A.; Brunson, B.A.; Wakefield, J.K.; Frost, A.R.; Bellis, S.L. Hypersialylation of beta1 integrins, observed in colon adenocarcinoma, may contribute to cancer progression by up-regulating cell motility. Cancer Res. 2005, 65, 4645–4652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stowell, S.R.; Ju, T.; Cummings, R.D. Protein Glycosylation in Cancer. Annu. Rev. Pathol. 2015, 10, 473–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, Y.; Li, J.; Fang, J.; Xu, Z.; He, X.; Zhang, F.; Ling, J.; Li, X.; Xu, D.; Li, L.; et al. Cloning and characterization of a novel gene which encodes a protein interacting with the mitosis-associated kinase-like protein NTKL. J. Hum. Genet. 2003, 48, 315–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burman, J.L.; Bourbonniere, L.; Philie, J.; Stroh, T.; Dejgaard, S.Y.; Presley, J.F.; McPherson, P.S. Scyl1, mutated in a recessive form of spinocerebellar neurodegeneration, regulates COPI-mediated retrograde traffic. J. Biol. Chem. 2008, 283, 22774–22786. [Google Scholar] [CrossRef] [Green Version]
- Hamlin, J.N.; Schroeder, L.K.; Fotouhi, M.; Dokainish, H.; Ioannou, M.S.; Girard, M.; Summerfeldt, N.; Melançon, P.; McPherson, P.S. Scyl1 scaffolds class II Arfs to specific subcomplexes of coatomer through the gamma-COP appendage domain. J. Cell. Sci. 2014, 127, 1454–1463. [Google Scholar] [CrossRef] [Green Version]
- Hennies, H.C.; Kornak, U.; Zhang, H.; Egerer, J.; Zhang, X.; Seifert, W.; Kühnisch, J.; Budde, B.; Nätebus, M.; Brancati, F.; et al. Gerodermia osteodysplastica is caused by mutations in SCYL1BP1, a Rab-6 interacting golgin. Nat. Genet. 2008, 40, 1410–1412. [Google Scholar] [CrossRef] [Green Version]
- Hunter, A.G. Is geroderma osteodysplastica underdiagnosed? J. Med. Genet. 1988, 25, 854–857. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, Y.; Lu, X.; Wang, Y.; Duan, Y.; Cheng, C.; Shen, A. SCYL1BP1 modulates neurite outgrowth and regeneration by regulating the Mdm2/p53 pathway. Mol. Biol. Cell 2012, 23, 4506–4514. [Google Scholar] [CrossRef]
- Yan, J.; Zhang, D.; Di, Y.; Shi, H.; Rao, H.; Huo, K. A newly identified Pirh2 substrate SCYL1-BP1 can bind to MDM2 and accelerate MDM2 self-ubiquitination. FEBS Lett. 2010, 584, 3275–3278. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, L.; Chao-Chu, J.; Schneider, S.; Gottardo, M.; Tzolovsky, G.; Dzhindzhev, N.S.; Riparbelli, M.G.; Callaini, G.; Glover, D.M. Gorab is a Golgi protein required for structure and duplication of Drosophila centrioles. Nat. Genet. 2018, 50, 1021–1031. [Google Scholar] [CrossRef]
- Egerer, J.; Emmerich, D.; Fischer-Zirnsak, B.; Meierhofer, D.; Tuysuz, B.; Marschner, K.; Sauer, S.; Barr, F.A.; Mundlos, S.; Kornak, U. GORAB Missense Mutations Disrupt RAB6 and ARF5 Binding and Golgi Targeting. J. Investig. Dermatol. 2015, 135, 2368–2376. [Google Scholar] [CrossRef] [Green Version]
- Gleeson, P.A.; Lock, J.G.; Luke, M.R.; Stow, J.L. Domains of the TGN: Coats, tethers and G proteins. Traffic 2004, 5, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.L.; Steiner, M.; Witkos, T.; Egerer, J.; Busse, B.; Mizumoto, S.; Pestka, J.M.; Zhang, H.; Hausser, I.; Khayal, L.A.; et al. Impaired proteoglycan glycosylation, elevated TGF-beta signaling, and abnormal osteoblast differentiation as the basis for bone fragility in a mouse model for gerodermia osteodysplastica. PLoS Genet. 2018, 14, e1007242. [Google Scholar] [CrossRef] [PubMed]
- Roboti, P.; Sato, K.; Lowe, M. The golgin GMAP-210 is required for efficient membrane trafficking in the early secretory pathway. J. Cell Sci. 2015, 128, 1595–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smits, P.; Bolton, A.D.; Funari, V.; Hong, M.; Boyden, E.D.; Lu, L.; Manning, D.K.; Dwyer, N.D.; Moran, J.L.; Prysak, M.; et al. Lethal skeletal dysplasia in mice and humans lacking the golgin GMAP-210. N. Engl. J. Med. 2010, 362, 206–216. [Google Scholar] [CrossRef] [Green Version]
- Ungar, D.; Oka, T.; Brittle, E.E.; Vasile, E.; Lupashin, V.V.; Chatterton, J.E.; Heuser, E.E.; Krieger, M.; Waters, M.G. Characterization of a mammalian Golgi-localized protein complex, COG, that is required for normal Golgi morphology and function. J. Cell Biol. 2002, 157, 405–415. [Google Scholar] [CrossRef]
- Climer, L.K.; Dobretsov, M.; Lupashin, V. Defects in the COG complex and COG-related trafficking regulators affect neuronal Golgi function. Front. Neurosci. 2015, 9, 405. [Google Scholar] [CrossRef] [Green Version]
- Miller, V.J.; Ungar, D. Re’COG’nition at the Golgi. Traffic 2012, 13, 891–897. [Google Scholar] [CrossRef]
- Smith, R.D.; Lupashin, V.V. Role of the conserved oligomeric Golgi (COG) complex in protein glycosylation. Carbohydr. Res. 2008, 343, 2024–2431. [Google Scholar] [CrossRef] [Green Version]
- Fotso, P.; Koryakina, Y.; Pavliv, O.; Tsiomenko, A.B.; Lupashin, V.V. Cog1p plays a central role in the organization of the yeast conserved oligomeric Golgi complex. J. Biol. Chem. 2005, 280, 27613–27623. [Google Scholar] [CrossRef] [Green Version]
- Ungar, D.; Oka, T.; Vasile, E.; Krieger, M.; Hughson, F.M. Subunit architecture of the conserved oligomeric Golgi complex. J. Biol. Chem. 2005, 280, 32729–32735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackburn, J.B.; D’Souza, Z.; Lupashin, V.V. Maintaining order: COG complex controls Golgi trafficking, processing, and sorting. FEBS Lett. 2019, 593, 2466–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lees, J.A.; Yip, C.K.; Walz, T.; Hughson, F.M. Molecular organization of the COG vesicle tethering complex. Nat. Struct. Mol. Biol. 2010, 17, 1292–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willett, R.; Blackburn, J.B.; Climer, L.; Pokrovskaya, I.; Kudlyk, T.; Wang, W.; Lupashin, V. COG lobe B sub-complex engages v-SNARE GS15 and functions via regulated interaction with lobe A sub-complex. Sci. Rep. 2016, 6, 29139. [Google Scholar] [CrossRef] [Green Version]
- Shestakova, A.; Zolov, S.; Lupashin, V. COG complex-mediated recycling of Golgi glycosyltransferases is essential for normal protein glycosylation. Traffic 2006, 7, 191–204. [Google Scholar] [CrossRef]
- Wuestehube, L.J.; Duden, R.; Eun, A.; Hamamoto, S.; Korn, P.; Ram, R.; Schekman, R. New mutants of Saccharomyces cerevisiae affected in the transport of proteins from the endoplasmic reticulum to the Golgi complex. Genetics 1996, 142, 393–406. [Google Scholar]
- Zolov, S.N.; Lupashin, V.V. Cog3p depletion blocks vesicle-mediated Golgi retrograde trafficking in HeLa cells. J. Cell Biol. 2005, 168, 747–759. [Google Scholar] [CrossRef]
- Cottam, N.P.; Wilson, K.M.; Ng, B.G.; Korner, C.; Freeze, H.H.; Ungar, D. Dissecting functions of the conserved oligomeric Golgi tethering complex using a cell-free assay. Traffic 2014, 15, 12–21. [Google Scholar] [CrossRef]
- Bailey-Blackburn, J.; Pokrovskaya, I.; Fisher, P.; Ungar, D.; Lupashin, V.V. COG complex complexities: Detailed characterization of a complete set of HEK293T cells lacking individual COG subunits. Front. Cell Dev. Biol. 2016, 4, 23. [Google Scholar] [CrossRef] [Green Version]
- Willett, R.; Ungar, D.; Lupashin, V. The Golgi puppet master: COG complex at center stage of membrane trafficking interactions. Histochem. Cell Biol. 2013, 140, 271–283. [Google Scholar] [CrossRef] [Green Version]
- Laufman, O.; Kedan, A.; Hong, W.; Lev, S. Direct interaction between the COG complex and the SM protein, Sly1, is required for Golgi SNARE pairing. EMBO J. 2009, 28, 2006–2017. [Google Scholar] [CrossRef] [Green Version]
- Laufman, O.; Hong, W.; Lev, S. The COG complex interacts directly with Syntaxin 6 and positively regulates endosome-to-TGN retrograde transport. J. Cell Biol. 2011, 194, 459–472. [Google Scholar] [CrossRef] [Green Version]
- Foulquier, F.; Vasile, E.; Schollen, E.; Callewaert, N.; Raemaekers, T.; Quelhas, D.; Jaeken, J.; Mills, P.; Winchester, B.; Krieger, M.; et al. Conserved oligomeric Golgi complex subunit 1 deficiency reveals a previously uncharacterized congenital disorder of glycosylation type II. Proc. Natl. Acad. Sci. USA 2006, 103, 3764–3769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fung, C.W.; Matthijs, G.; Sturiale, L.; Garozzo, D.; Wong, K.Y.; Wong, R. COG5-CDG with a mild neurohepatic presentation. JIMD Rep. 2012, 3, 67–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kranz, C.; Ng, B.G.; Sun, L.; Sharma, V.; Eklund, E.A.; Miura, Y.; Ungar, D.; Lupashin, V.; Winkel, R.D.; Cipollo, J.F.; et al. COG8 deficiency causes new congenital disorder of glycosylation type IIh. Hum. Mol. Genet. 2007, 16, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Kodera, H.; Ando, N.; Yuasa, I.; Wada, Y.; Tsurusaki, Y.; Nakashima, M.; Miyake, N.; Saitoh, S.; Matsumoto, N.; Saitsu, H. Mutations in COG2 encoding a subunit of the conserved oligomeric golgi complex cause a congenital disorder of glycosylation. Clin. Genet. 2015, 87, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Lübbehusen, J.; Thiel, C.; Rind, N.; Ungar, D.; Prinsen, B.H.; De Koning, T.J.; Peter, M.; Hasselt, V.; Körner, C. Fatal outcome due to deficiency of subunit 6 of the conserved oligomeric Golgi complex leading to a new type of congenital disorders of glycosylation. Hum. Mol. Genet. 2010, 19, 3623–3633. [Google Scholar] [CrossRef]
- Morava, E.; Zeevaert, R.; Korsch, E.; Huijben, K.; Wopereis, S.; Matthijs, G.; Keymolen, K.; Lefeber, D.J.; De Meirleir, L.; Wevers, R.A. A common mutation in the COG7 gene with a consistent phenotype including microcephaly, adducted thumbs, growth retardation, VSD and episodes of hyperthermia. Eur. J. Hum. Genet. 2007, 15, 638–645. [Google Scholar] [CrossRef]
- Ng, B.G.; Kranz, C.; Hagebeuk, E.E.; Duran, M.; Abeling, N.G.; Wuyts, B.; Ungar, D.; Lupashin, V.V.; Hartdorff, C.M.; Poll-The, B.T.; et al. Molecular and clinical characterization of a Moroccan Cog7 deficient patient. Mol. Genet. Metab. 2007, 91, 201–214. [Google Scholar] [CrossRef] [Green Version]
- Paesold-Burda, P.; Maag, C.; Troxler, H.; Foulquier, F.; Kleinert, P.; Schnabel, S.; Baumgartner, M.; Hennet, T. Deficiency in COG5 causes a moderate form of congenital disorders of glycosylation. Hum. Mol. Genet. 2009, 18, 4350–4356. [Google Scholar] [CrossRef]
- Reynders, E.; Foulquier, F.; Leão Teles, E.; Quelhas, D.; Morelle, W.; Rabouille, C.; Wim, A.; Gert, M. Golgi function and dysfunction in the first COG4-deficient CDG type II patient. Hum. Mol. Genet. 2009, 18, 3244–3256. [Google Scholar] [CrossRef] [PubMed]
- Spaapen, L.J.; Bakker, J.A.; Van der Meer, S.B.; Sijstermans, H.J.; Steet, R.A.; Wevers, R.A.; Jaeken, J. Clinical and biochemical presentation of siblings with COG-7 deficiency, a lethal multiple O- and N-glycosylation disorder. J. Inherit. Metab. Dis. 2005, 28, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Steet, R.A.; Bohorov, O.; Bakker, J.; Newell, J.; Krieger, M.; Spaapen, L.; Kornfeld, S.; Freeze, H.H. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat. Med. 2004, 10, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Zeevaert, R.; Foulquier, F.; Cheillan, D.; Cloix, I.; Guffon, N.; Sturiale, L.; Garozzo, D.; Matthijsc, M.; Jaeken, J. A new mutation in COG7 extends the spectrum of COG subunit deficiencies. Eur. J. Med. Genet. 2009, 52, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Quental, R.; Azevedo, L.; Matthiesen, R.; Amorim, A. Comparative analyses of the Conserved Oligomeric Golgi (COG) complex in vertebrates. BMC Evol. Biol. 2010, 10, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koumandou, V.L.; Dacks, J.B.; Coulson, M.R.; Field, M.C. Control systems for membrane fusion in the ancestral eukaryote; evolution of tethering complexes and SM proteins. MC Evol. Biol. 2007, 7, 29. [Google Scholar] [CrossRef] [Green Version]
- Kingsley, D.M.; Kozarsky, K.F.; Segal, M.; Krieger, M. Three types of low density lipoprotein receptor-deficient mutant have pleiotropic defects in the synthesis of N-linked, O-linked, and lipid-linked carbohydrate chains. J. Cell Biol. 1986, 102, 1576–1585. [Google Scholar] [CrossRef] [Green Version]
- Struwe, W.B.; Reinhold, V.N. The conserved oligomeric Golgi complex is required for fucosylation of N-glycans in Caenorhabditis elegans. Glycobiology 2012, 22, 863–875. [Google Scholar] [CrossRef] [Green Version]
- Suvorova, E.S.; Duden, R.; Lupashin, V.V. The Sec34/Sec35p complex, a Ypt1p effector required for retrograde intra-Golgi trafficking, interacts with Golgi SNAREs and COPI vesicle coat proteins. J. Cell Biol. 2002, 157, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Podos, S.D.; Reddy, P.; Ashkenas, J.; Krieger, M. LDLC encodes a brefeldin A-sensitive, peripheral Golgi protein required for normal Golgi function. J. Cell. Biol. 1994, 127, 679–691. [Google Scholar] [CrossRef] [Green Version]
- Steet, R.; Kornfeld, S. COG-7-deficient human fibroblasts exhibit altered recycling of Golgi proteins. Mol. Biol. Cell 2006, 17, 2312–2321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pokrovskaya, I.D.; Willett, R.; Smith, R.D.; Morelle, W.; Kudlyk, T.; Lupashin, V.V. Conserved oligomeric Golgi complex specifically regulates the maintenance of Golgi glycosylation machinery. Glycobiology 2011, 21, 1554–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frappaolo, A.; Sechi, S.; Kumagai, T.; Robinson, S.; Fraschini, R.; Karimpour- Ghahnavieh, A.; Belloni, G.; Piergentili, R.; Tiemeyer, K.T.; Tiemeyer, M.; et al. COG7 deficiency in Drosophila generates multifaceted developmental, behavioral and protein glycosylation phenotypes. J. Cell Sci. 2017, 130, 3637–3649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oka, T.; Vasile, E.; Penman, M.; Novina, C.D.; Dykxhoorn, D.M.; Ungar, D.; Hughson, F.M.; Krieger, M. Genetic analysis of the subunit organization and function of the conserved oligomeric golgi (COG) complex: Studies of COG5- and COG7-deficient mammalian cells. J. Biol. Chem. 2005, 280, 32736–32745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peanne, R.; Legrand, D.; Duvet, S.; Mir, A.M.; Matthijs, G.; Rohrer, J.; Foulquier, F. Differential effects of lobe A and lobe B of the Conserved Oligomeric Golgi complex on the stability of {beta}1,4-galactosyltransferase 1 and {alpha}2,6-sialyltransferase 1. Glycobiology 2011, 21, 864–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kingsley, D.M.; Krieger, M. Receptor-mediated endocytosis of low density lipoprotein: Somatic cell mutants define multiple genes required for expression of surface-receptor activity. Proc. Natl. Acad. Sci. USA 1984, 81, 5454–5458. [Google Scholar] [CrossRef] [Green Version]
- Oka, T.; Ungar, D.; Hughson, F.M.; Krieger, M. The COG and COPI complexes interact to control the abundance of GEARs, a subset of Golgi integral membrane proteins. Mol. Biol. Cell 2004, 15, 2423–2435. [Google Scholar] [CrossRef]
- Whyte, J.R.; Munro, S. A yeast homolog of the mammalian mannose 6-phosphate receptors contributes to the sorting of vacuolar hydrolases. Curr. Biol. 2001, 11, 1074–1078. [Google Scholar] [CrossRef] [Green Version]
- Ram, R.J.; Li, B.; Kaiser, C.A. Identification of Sec36p, Sec37p, and Sec38p: Components of yeast complex that contains Sec34p and Sec35p. Mol. Biol. Cell 2002, 13, 1484–1500. [Google Scholar] [CrossRef] [Green Version]
- Conde, R.; Guadalupe, P.; Cueva, R.; Larriba, G. Screening for new yeast mutants affected in mannosylphosphorylation of cell wall mannoproteins. Yeast 2003, 20, 1189–1211. [Google Scholar] [CrossRef] [Green Version]
- Corbacho, I.; Olivero, I.; Hernández, L.M. Identification of low-dye-binding (ldb) mutants of Saccharomyces cerevisiae. S. Yeast Res. 2004, 4, 437–444. [Google Scholar] [CrossRef] [Green Version]
- Kubota, Y.; Sano, M.; Goda, S.; Suzuki, N.; Nishiwaki, K. The conserved oligomeric Golgi complex acts in organ morphogenesis via glycosylation of an ADAM protease in C. elegans. Development 2006, 133, 263–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farkas, R.M.; Giansanti, M.G.; Gatti, M.; Fuller, M.T. The Drosophila Cog5 homologue is required for cytokinesis, cell elongation, and assembly of specialized Golgi architecture during spermatogenesis. Mol. Biol. Cell 2003, 14, 190–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fári, K.; Takács, S.; Ungár, D.; Sinka, R. The role of acroblast formation during Drosophila spermatogenesis. Biol. Open 2016, 5, 1102–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belloni, G.; Sechi, S.; Riparbelli, M.G.; Fuller, M.T.; Callaini, G.; Giansanti, M.G. Mutations in Cog7 affect Golgi structure, meiotic cytokinesis and sperm development during Drosophila spermatogenesis. J. Cell Sci. 2012, 125, 5441–5452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comstra, H.S.; McArthy, J.; Rudin-Rush, S.; Hartwig, C.; Gokhale, A.; Zlatic, S.A.; Blackburn, J.B.; Werner, E.; Petris, M.; D’Souza, P.; et al. The interactome of the copper transporter ATP7A belongs to a network of neurodevelopmental and neurodegeneration factors. Elife 2017, 6, e24722. [Google Scholar] [CrossRef]
- Sechi, S.; Frappaolo, A.; Fraschini, R.; Capalbo, L.; Gottardo, M.; Belloni, G.; Glover, D.M.; Wainman, A.; Giansanti, M.G. Rab1 interacts with GOLPH3 and controls Golgi structure and contractile ring constriction during cytokinesis in Drosophila melanogaster. Open Biol. 2017, 7, 160257. [Google Scholar] [CrossRef]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef]
- Ju, T.; Wang, Y.; Aryal, R.P.; Lehoux, S.D.; Ding, X.; Kudelka, M.R.; Cutler, C.; Zeng, J.; Wang, J.; Sun, X.; et al. Tn and sialyl-Tn antigens, aberrant O-glycomics as human disease markers. Proteomics Clin. Appl. 2013, 7, 618–631. [Google Scholar] [CrossRef] [Green Version]
- Freeze, H.H.; Chong, J.X.; Bamshad, M.J.; Ng, B.G. Solving Glycosylation Disorders: Fundamental Approaches Reveal Complicated Pathways. Am. J. Hum. Genet. 2014, 94, 161–175. [Google Scholar] [CrossRef] [Green Version]
- Frappaolo, A.; Sechi, S.; Kumagai, T.; Karimpour-Ghahnavieh, A.; Tiemeyer, M.; Giansanti, M.G. Modeling Congenital Disorders of N-Linked Glycoprotein Glycosylation in Drosophila melanogaster. Front. Genet. 2018, 9, 436. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frappaolo, A.; Karimpour-Ghahnavieh, A.; Sechi, S.; Giansanti, M.G. The Close Relationship between the Golgi Trafficking Machinery and Protein Glycosylation. Cells 2020, 9, 2652. https://doi.org/10.3390/cells9122652
Frappaolo A, Karimpour-Ghahnavieh A, Sechi S, Giansanti MG. The Close Relationship between the Golgi Trafficking Machinery and Protein Glycosylation. Cells. 2020; 9(12):2652. https://doi.org/10.3390/cells9122652
Chicago/Turabian StyleFrappaolo, Anna, Angela Karimpour-Ghahnavieh, Stefano Sechi, and Maria Grazia Giansanti. 2020. "The Close Relationship between the Golgi Trafficking Machinery and Protein Glycosylation" Cells 9, no. 12: 2652. https://doi.org/10.3390/cells9122652
APA StyleFrappaolo, A., Karimpour-Ghahnavieh, A., Sechi, S., & Giansanti, M. G. (2020). The Close Relationship between the Golgi Trafficking Machinery and Protein Glycosylation. Cells, 9(12), 2652. https://doi.org/10.3390/cells9122652