Type 3 Inositol 1,4,5-Trisphosphate Receptor is a Crucial Regulator of Calcium Dynamics Mediated by Endoplasmic Reticulum in HEK Cells


 and
and 
Abstract
:1. Introduction
2. Results and Discussion
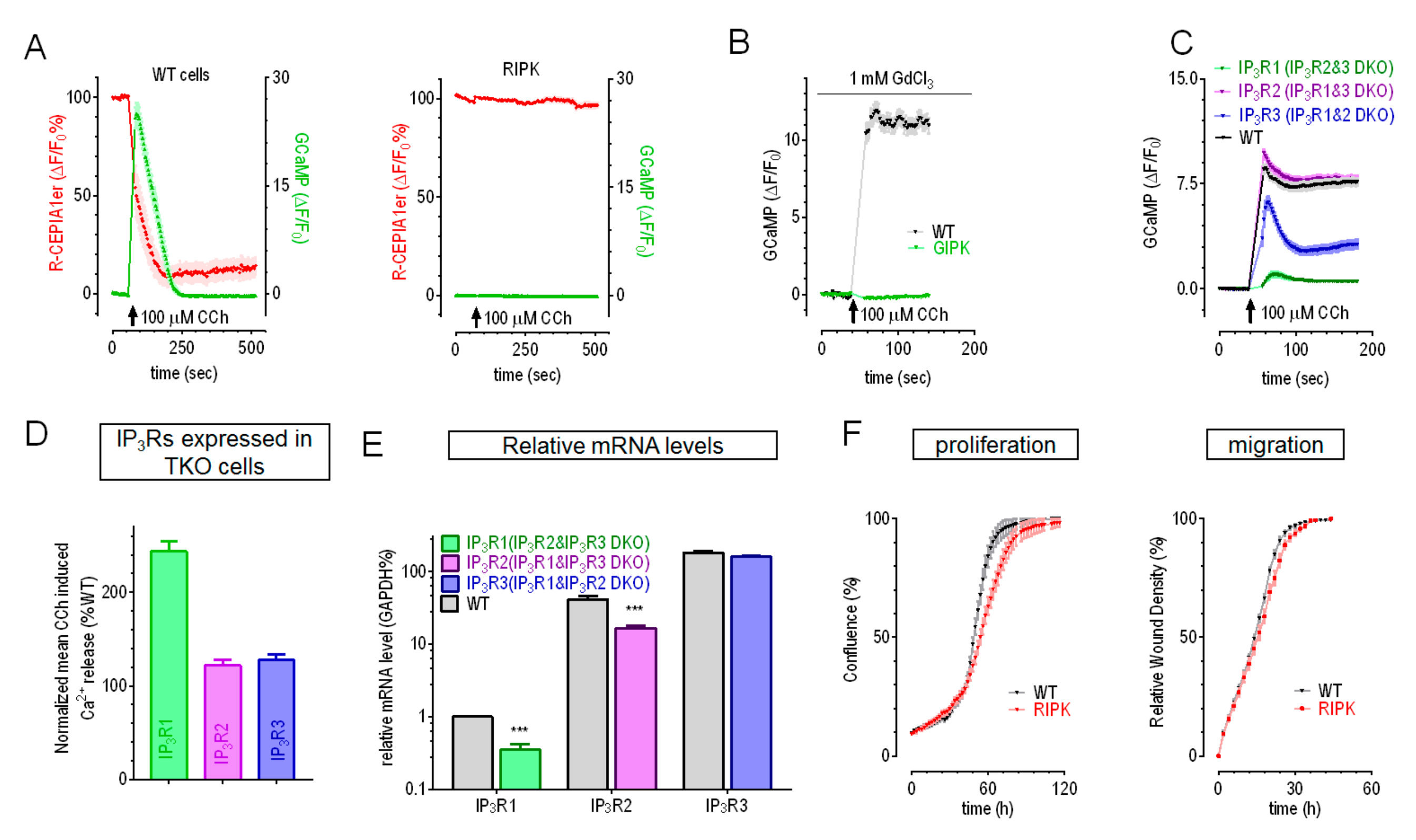
2.1. With IP3R2 Being the Dominant Isoform, IP3Rs Regulate Growth and Migration of HEK Cells
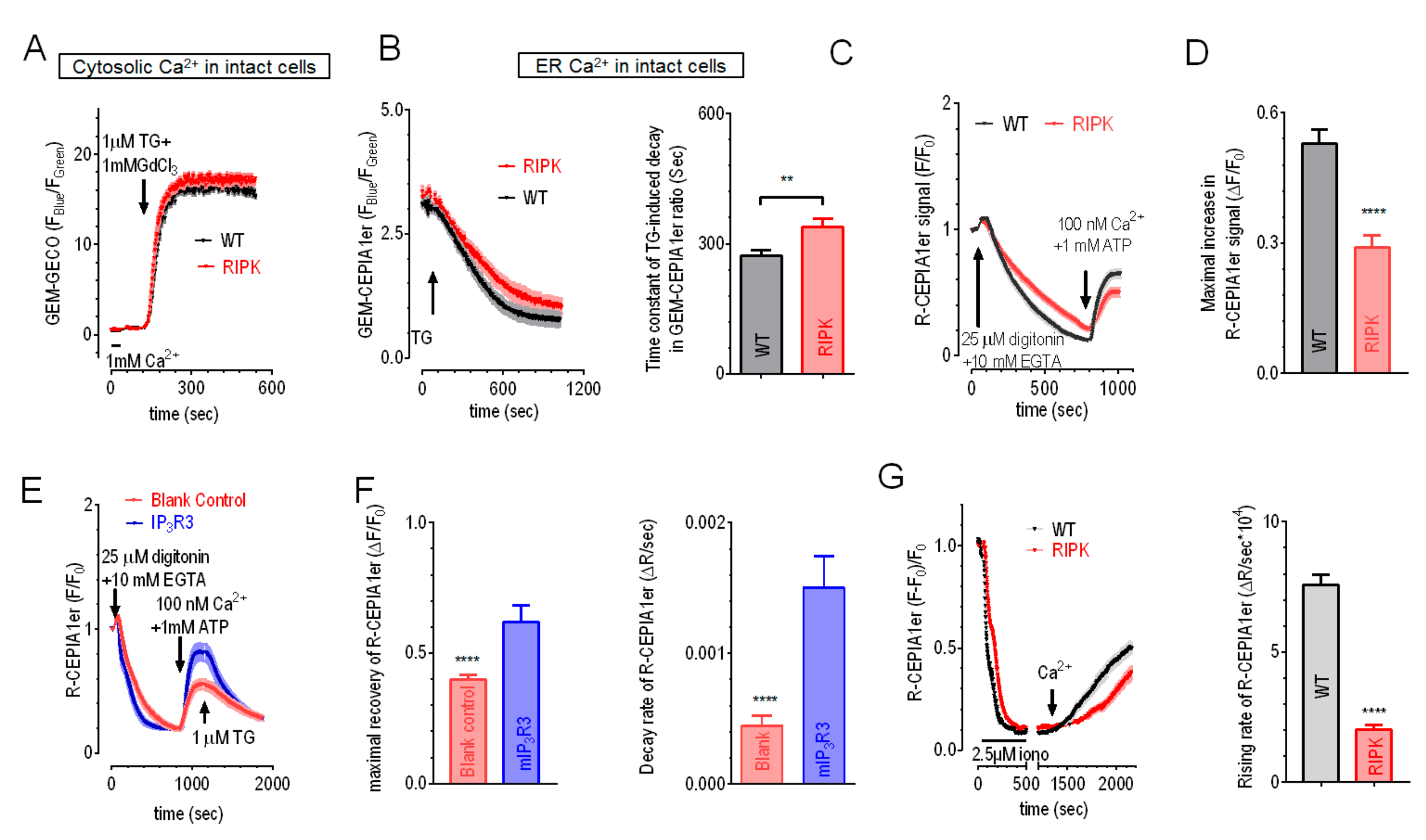
2.2. Overexpression of IP3R1 or IP3R3 Could Rescue the Impaired ER Ca2+ Leak and Refilling in IP3R-TKO Cells
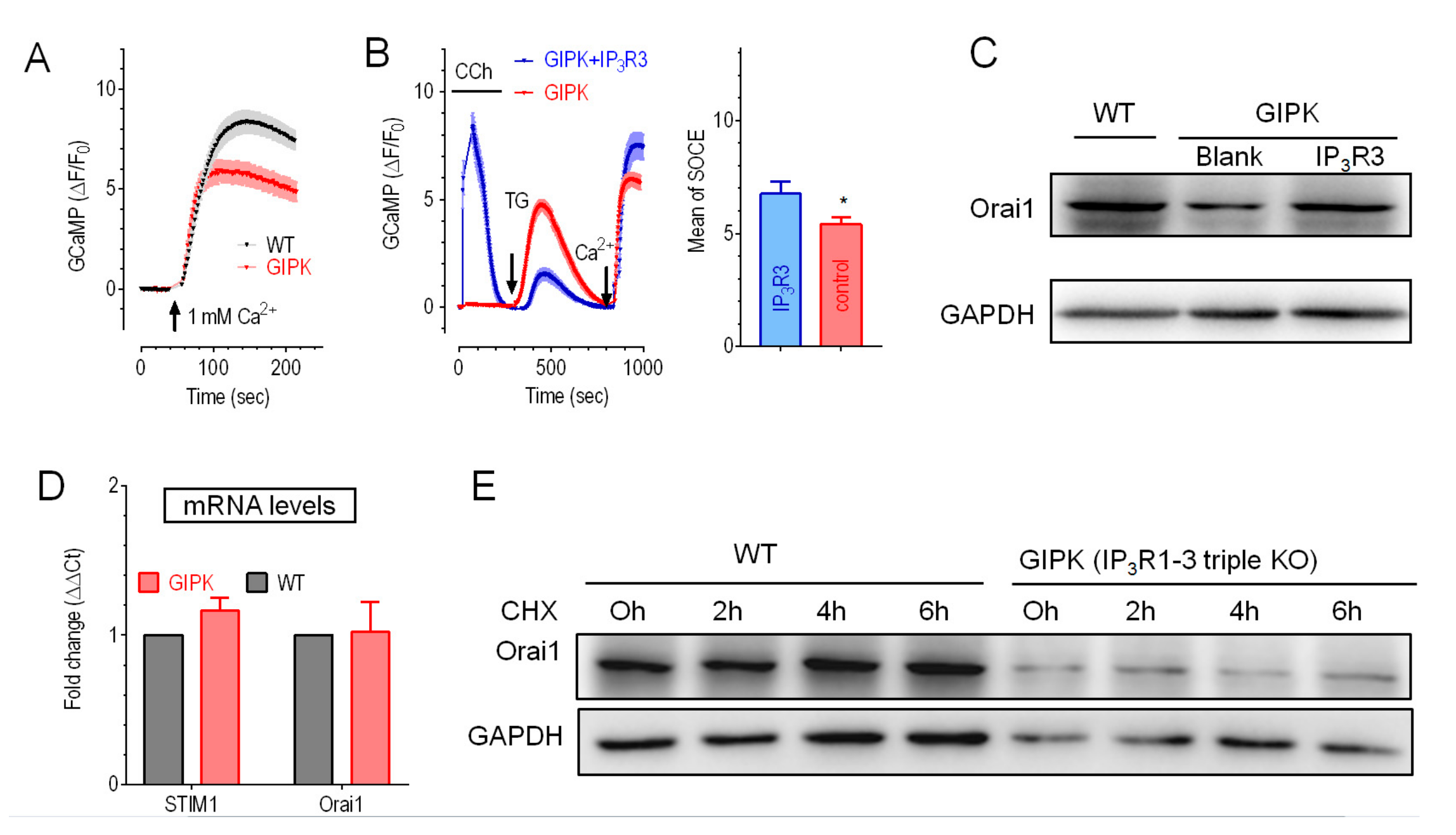
2.3. Expression of IP3R3 Restored the Reduced Orai1 Protein Level and SOCE in IP3Rs-TKO Cells
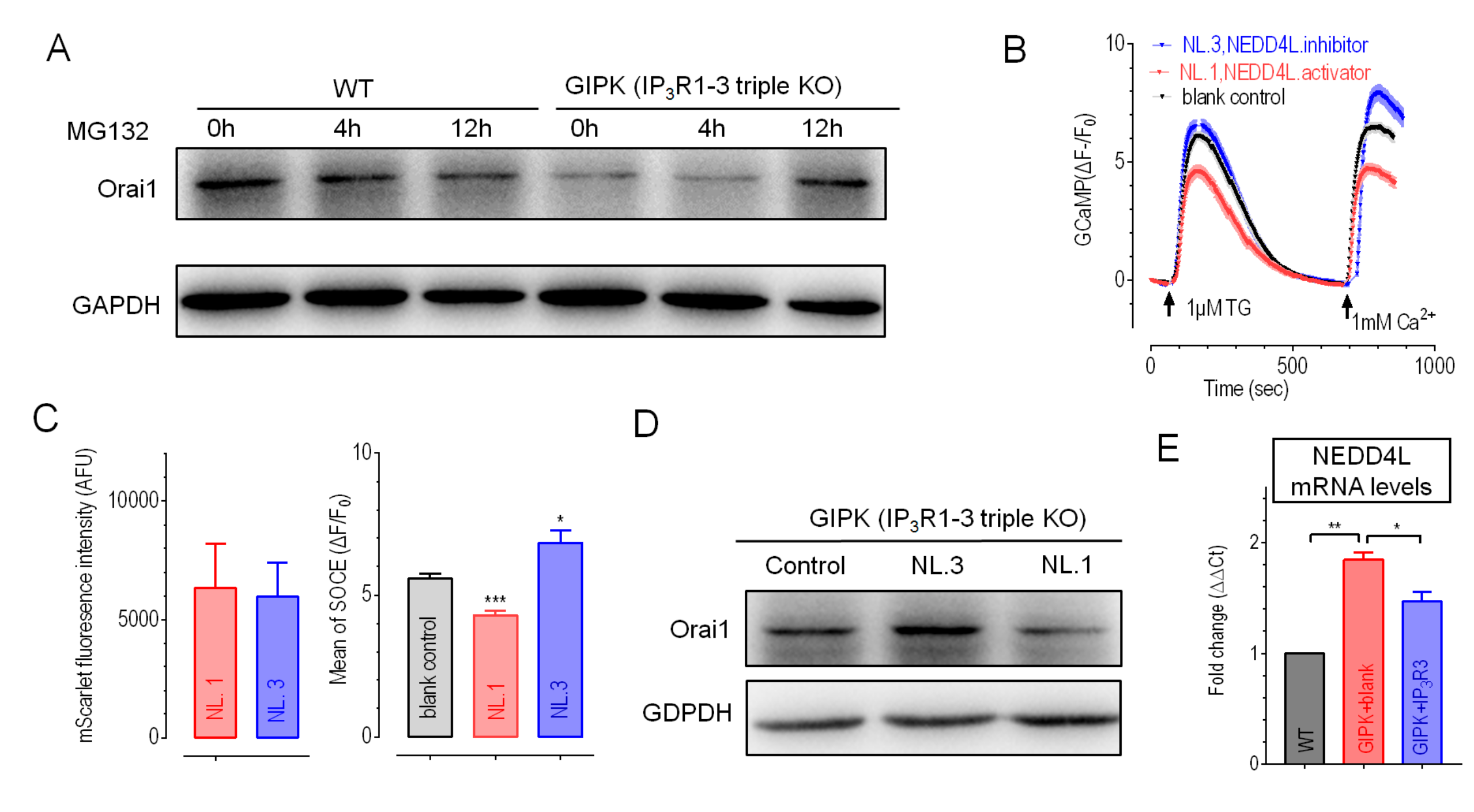
2.4. Overexpression of IP3R3 Restored SOCE by Inhibiting NEDD4L in GIPK Cells
3. Methods
3.1. Plasmids Construction
3.2. Cell Culture and Transfection
3.3. Construction of Knockout Cell Lines
3.4. Single-Cell Intracellular Ca2+ Measurements
3.5. Real-Time PCR
3.6. Cell Proliferation and Migration Essay
3.7. Western Blotting
3.8. Proteasomal Inhibition Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soboloff, J.; Rothberg, B.S.; Madesh, M.; Gill, D.L. STIM proteins: Dynamic calcium signal transducers. Nat. Rev. Mol. Cell Biol. 2012, 13, 549–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foskett, J.K.; White, C.; Cheung, K.H.; Mak, D.O.D. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 2007, 87, 593–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikoshiba, K. IP3 receptor/Ca2+ channel: From discovery to new signaling concepts. J. Neurochem. 2007, 102, 1426–1446. [Google Scholar] [CrossRef]
- Mikoshiba, K. Role of IP3 receptor signaling in cell functions and diseases. Adv. Biol. Regul. 2015, 57, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Hamada, K.; Mikoshiba, K. IP3 Receptor Plasticity Underlying Diverse Functions. Annu. Rev. Physiol. 2019, 82, 19.1–19.26. [Google Scholar] [CrossRef] [Green Version]
- Clapham, D.E. Calcium signaling. Cell 1995, 80, 259–268. [Google Scholar] [CrossRef] [Green Version]
- Prole, D.L.; Taylor, C.W. Inositol 1,4,5-trisphosphate receptors and their protein partners as signalling hubs. J. Physiol. 2016, 594, 2849–2866. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.M.; Zhao, X.S.; Zeng, W.; Mozhayeva, M.; Muallem, S. The mammalian Sec6/8 complex interacts with Ca(2+) signaling complexes and regulates their activity. J. Cell Biol. 2000, 150, 1101–1112. [Google Scholar] [CrossRef] [Green Version]
- Huang, G.; Yao, J.; Zeng, W.; Mizuno, Y.; Kamm, K.E.; Stull, J.T.; Harding, H.P.; Ron, D.; Muallem, S. ER stress disrupts Ca2+-signaling complexes and Ca2+ regulation in secretory and muscle cells from PERK-knockout mice. J. Cell Sci. 2006, 119, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Redondo, P.C.; Jardin, I.; Lopez, J.J.; Salido, G.M.; Rosado, J.A. Intracellular Ca2+ store depletion induces the formation of macromolecular complexes involving hTRPC1, hTRPC6, the type II IP3 receptor and SERCA3 in human platelets. Biochim. Biophys. Acta 2008, 1783, 1163–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoso, N.G.; Cebotaru, L.; Guggino, W.B. Polycystin-1, 2, and STIM1 interact with IP(3)R to modulate ER Ca release through the PI3K/Akt pathway. Cell. Physiol. Biochem. 2011, 27, 715–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodard, G.E.; Lopez, J.J.; Jardin, I.; Salido, G.M.; Rosado, J.A. TRPC3 regulates agonist-stimulated Ca2+ mobilization by mediating the interaction between type I inositol 1,4,5-trisphosphate receptor, RACK1, and Orai1. J. Biol. Chem. 2010, 285, 8045–8053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lur, G.; Sherwood, M.W.; Ebisui, E.; Haynes, L.; Feske, S.; Sutton, R.; Burgoyne, R.D.; Mikoshiba, K.; Petersen, O.H.; Tepikin, A.V. InsP(3)receptors and Orai channels in pancreatic acinar cells: Co-localization and its consequences. Biochem. J. 2011, 436, 231–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartok, A.; Weaver, D.; Golenar, T.; Nichtova, Z.; Katona, M.; Bansaghi, S.; Alzayady, K.J.; Thomas, V.K.; Ando, H.; Mikoshiba, K.; et al. IP3 receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer. Nat. Commun. 2019, 10, 3726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berridge, M.J. The Inositol Trisphosphate/Calcium Signaling Pathway in Health and Disease. Physiol. Rev. 2016, 96, 1261–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, S.; Zhou, L.; Ma, G.; Zhang, T.; Liu, J.; Li, J.; Nguyen, N.T.; Zhang, X.; Li, W.; Nwokonko, R.; et al. Calcium store refilling and STIM activation in STIM- and Orai-deficient cell lines. Pflügers Archiv Eur. J. Physiol. 2018, 470, 1555–1567. [Google Scholar] [CrossRef] [Green Version]
- Putney, J.W.; Steinckwich-Besancon, N.; Numaga-Tomita, T.; Davis, F.M.; Desai, P.N.; D’Agostin, D.M.; Wu, S.; Bird, G.S. The functions of store-operated calcium channels. Biochim. Biophys. Acta 2017, 1864, 900–906. [Google Scholar] [CrossRef]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [Green Version]
- Hogan, P.G.; Rao, A. Store-operated calcium entry: Mechanisms and modulation. Biochem. Biophys. Res. Commun. 2015, 460, 40–49. [Google Scholar] [CrossRef] [Green Version]
- Thillaiappan, N.B.; Chavda, A.P.; Tovey, S.C.; Prole, D.L.; Taylor, C.W. Ca(2+) signals initiate at immobile IP3 receptors adjacent to ER-plasma membrane junctions. Nat. Commun. 2017, 8, 1505. [Google Scholar] [CrossRef] [PubMed]
- Thillaiappan, N.B.; Chakraborty, P.; Hasan, G.; Taylor, C.W. IP3 receptors and Ca(2+) entry. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Kar, P.; Mirams, G.R.; Christian, H.C.; Parekh, A.B. Control of NFAT Isoform Activation and NFAT-Dependent Gene Expression through Two Coincident and Spatially Segregated Intracellular Ca(2+) Signals. Mol. Cell 2016, 64, 746–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courjaret, R.; Dib, M.; Machaca, K. Store-Operated Ca(2+) Entry in Oocytes Modulate the Dynamics of IP3 -Dependent Ca(2+) Release From Oscillatory to Tonic. J. Cell. Physiol. 2017, 232, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Courjaret, R.; Dib, M.; Machaca, K. Spatially restricted subcellular Ca(2+) signaling downstream of store-operated calcium entry encoded by a cortical tunneling mechanism. Sci. Rep. 2018, 8, 11214. [Google Scholar] [CrossRef]
- Sampieri, A.; Santoyo, K.; Asanov, A.; Vaca, L. Association of the IP3R to STIM1 provides a reduced intraluminal calcium microenvironment, resulting in enhanced store-operated calcium entry. Sci. Rep. 2018, 8, 13252. [Google Scholar] [CrossRef]
- Chakraborty, S.; Deb, B.K.; Chorna, T.; Konieczny, V.; Taylor, C.W.; Hasan, G. Mutant IP3 receptors attenuate store-operated Ca2+ entry by destabilizing STIM-Orai interactions in Drosophila neurons. J. Cell Sci. 2016, 129, 3903–3910. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, J.; Kanemaru, K.; Ishii, K.; Ohkura, M.; Okubo, Y.; Iino, M. Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat. Commun. 2014, 5, 4153. [Google Scholar] [CrossRef]
- Chen, T.W.; Wardill, T.J.; Sun, Y.; Pulver, S.R.; Renninger, S.L.; Baohan, A.; Schreiter, E.R.; Kerr, R.A.; Orger, M.B.; Jayaraman, V.; et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 2013, 499, 295–300. [Google Scholar] [CrossRef] [Green Version]
- Lock, J.T.; Alzayady, K.J.; Yule, D.I.; Parker, I. All three IP3 receptor isoforms generate Ca(2+) puffs that display similar characteristics. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Wedel, B.; Boyles, R.R.; Putney, J.W., Jr.; Bird, G.S. Role of the store-operated calcium entry proteins Stim1 and Orai1 in muscarinic cholinergic receptor-stimulated calcium oscillations in human embryonic kidney cells. J. Physiol. 2007, 579, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Inositol trisphosphate and calcium signalling mechanisms. Biochim. Biophys. Acta 2009, 1793, 933–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Araki, S.; Wu, J.; Teramoto, T.; Chang, Y.F.; Nakano, M.; Abdelfattah, A.S.; Fujiwara, M.; Ishihara, T.; Nagai, T.; et al. An expanded palette of genetically encoded Ca(2)(+) indicators. Science 2011, 333, 1888–1891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgo, J.; Muller, M.; Vais, H.; Cheung, K.H.; Yang, J.; Parker, I.; et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckstein, M.; Vaeth, M.; Aulestia, F.J.; Costiniti, V.; Kassam, S.N.; Bromage, T.G.; Pedersen, P.; Issekutz, T.; Idaghdour, Y.; Moursi, A.M.; et al. Differential regulation of Ca(2+) influx by ORAI channels mediates enamel mineralization. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef]
- Groenendyk, J.; Lynch, J.; Michalak, M. Calreticulin, Ca2+, and calcineurin—signaling from the endoplasmic reticulum. Mol. Cells 2004, 17, 383–389. [Google Scholar]
- Bastianutto, C.; Clementi, E.; Codazzi, F.; Podini, P.; De Giorgi, F.; Rizzuto, R.; Meldolesi, J.; Pozzan, T. Overexpression of calreticulin increases the Ca2+ capacity of rapidly exchanging Ca2+ stores and reveals aspects of their lumenal microenvironment and function. J. Cell Biol. 1995, 130, 847–855. [Google Scholar] [CrossRef]
- Llewelyn Roderick, H.; Llewellyn, D.H.; Campbell, A.K.; Kendall, J.M. Role of calreticulin in regulating intracellular Ca2+ storage and capacitative Ca2+ entry in HeLa cells. Cell Calcium 1998, 24, 253–262. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Longo, F.J.; Wintermantel, M.R.; Jiang, X.; Clark, R.A.; DeLisle, S. Calreticulin modulates capacitative Ca2+ influx by controlling the extent of inositol 1,4,5-trisphosphate-induced Ca2+ store depletion. J. Biol. Chem. 2000, 275, 36676–36682. [Google Scholar] [CrossRef] [Green Version]
- Fasolato, C.; Pizzo, P.; Pozzan, T. Delayed activation of the store-operated calcium current induced by calreticulin overexpression in RBL-1 cells. Mol. Biol. Cell 1998, 9, 1513–1522. [Google Scholar] [CrossRef] [Green Version]
- Courjaret, R.; Machaca, K. Mid-range Ca2+ signalling mediated by functional coupling between store-operated Ca2+ entry and IP3-dependent Ca2+ release. Nat. Commun. 2014, 5, 3916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.T.; Venkatachalam, K.; Li, H.S.; Montell, C.; Kurosaki, T.; Patterson, R.L.; Gill, D.L. Assessment of the role of the inositol 1,4,5-trisphosphate receptor in the activation of transient receptor potential channels and store-operated Ca2+ entry channels. J. Biol. Chem. 2001, 276, 18888–18896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, O.H.; Courjaret, R.; Machaca, K. Ca(2+) tunnelling through the ER lumen as a mechanism for delivering Ca(2+) entering via store-operated Ca(2+) channels to specific target sites. J. Physiol. 2017, 595, 2999–3014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, S.; Hasan, G. Spontaneous Ca(2+) Influx in Drosophila Pupal Neurons Is Modulated by IP3-Receptor Function and Influences Maturation of the Flight Circuit. Front. Mol. Neurosci. 2017, 10, 111. [Google Scholar] [CrossRef]
- Eylenstein, A.; Gehring, E.M.; Heise, N.; Shumilina, E.; Schmidt, S.; Szteyn, K.; Munzer, P.; Nurbaeva, M.K.; Eichenmuller, M.; Tyan, L.; et al. Stimulation of Ca2+-channel Orai1/STIM1 by serum- and glucocorticoid-inducible kinase 1 (SGK1). FASEB J. 2011, 25, 2012–2021. [Google Scholar] [CrossRef] [Green Version]
- Mocciaro, A.; Rape, M. Emerging regulatory mechanisms in ubiquitin-dependent cell cycle control. J. Cell Sci. 2012, 125, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Lang, F.; Pelzl, L.; Hauser, S.; Hermann, A.; Stournaras, C.; Schols, L. To die or not to die SGK1-sensitive ORAI/STIM in cell survival. Cell Calcium 2018, 74, 29–34. [Google Scholar] [CrossRef]
- Zhang, W.; Wu, K.P.; Sartori, M.A.; Kamadurai, H.B.; Ordureau, A.; Jiang, C.; Mercredi, P.Y.; Murchie, R.; Hu, J.; Persaud, A.; et al. System-Wide Modulation of HECT E3 Ligases with Selective Ubiquitin Variant Probes. Mol. Cell 2016, 62, 121–136. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; Ma, G.; He, L.; Zhang, T.; Li, J.; Yuan, X.; Nguyen, N.T.; Huang, Y.; Zhang, X.; Gao, P.; et al. Identification of molecular determinants that govern distinct STIM2 activation dynamics. PLoS Biol. 2018, 16, e2006898. [Google Scholar] [CrossRef] [Green Version]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [Green Version]
- Ma, G.; Wei, M.; He, L.; Liu, C.; Wu, B.; Zhang, S.L.; Jing, J.; Liang, X.; Senes, A.; Tan, P.; et al. Inside-out Ca2+ signalling prompted by STIM1 conformational switch. Nat. Commun. 2015, 6, 7826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Yuan, F.; Wen, Z.; Li, Y.; Wang, F.; Zhu, T.; Zhuo, W.; Jin, X.; Wang, Y.; Zhao, H.; et al. Genome-wide survey and expression analysis of the OSCA gene family in rice. BMC Plant Biol. 2015, 15, 261. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | sgRNA Sequence (5′→3′) | Target CDs Site | Primers (5′→3′) |
|---|---|---|---|
| ITPR1 | CAAAGACGACATATTAAAGG | 687–706 | F: TGCTGTGATTTTAGTGGCGT R: TCTCCACCCTACCCTTACCT |
| ITPR2 | ACACGATGTCCCCTATGTAG | 46–27 | F: TCTTGGCCGCTGTAGTCC R: CCAGGGAAAACAAGCACACA |
| ITPR3 | ACCCCCCTTCTCACGGAACG | 1593–1574 | F: GGGTTGTGGTCCAGCTTAGA R: CATCTAACCCAGTGCAAGGC |
| Gene | Forward Primer | Reverse Primer | NCBI Reference Sequence |
|---|---|---|---|
| ITPR1 | ATGACAGCTCTGAGGAGAA | GCAGAAGAGACAGGAGATTTAG | NM_001099952.2 |
| ITPR2 | CTGTGTGGAAGAGCAACTAC | ATGGGTCTGTAGGTAGGAATAG | NM_002223.3 |
| ITPR3 | CAAGCCCTCCAAAGATGAG | CGAAGGTGTCGATGATTACC | NM_002224.3 |
| STIM1 | GGATCTCAGAGGTTGGTAGA | GATGGAAGAGGAGCAAGAAG | NM_001277961.1 |
| Orai1 | GACGCTGACCACGACTA | CCTTGACCGAGTTGAGATTG | NM_032790.3 |
| GAPDH | AACTGCTTAGCACCCCTGGC | ATGACCTTGCCCACAGCCTT | NM_001256799.2 |
| NEDD4L | CCTAGATTGTCACGGTTTCC | GACTAAACTCTCACCTCCTTTC | NM_001144964.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yue, L.; Wang, L.; Du, Y.; Zhang, W.; Hamada, K.; Matsumoto, Y.; Jin, X.; Zhou, Y.; Mikoshiba, K.; Gill, D.L.; et al. Type 3 Inositol 1,4,5-Trisphosphate Receptor is a Crucial Regulator of Calcium Dynamics Mediated by Endoplasmic Reticulum in HEK Cells. Cells 2020, 9, 275. https://doi.org/10.3390/cells9020275
Yue L, Wang L, Du Y, Zhang W, Hamada K, Matsumoto Y, Jin X, Zhou Y, Mikoshiba K, Gill DL, et al. Type 3 Inositol 1,4,5-Trisphosphate Receptor is a Crucial Regulator of Calcium Dynamics Mediated by Endoplasmic Reticulum in HEK Cells. Cells. 2020; 9(2):275. https://doi.org/10.3390/cells9020275
Chicago/Turabian StyleYue, Lili, Liuqing Wang, Yangchun Du, Wei Zhang, Kozo Hamada, Yoshifumi Matsumoto, Xi Jin, Yandong Zhou, Katsuhiko Mikoshiba, Donald L. Gill, and et al. 2020. "Type 3 Inositol 1,4,5-Trisphosphate Receptor is a Crucial Regulator of Calcium Dynamics Mediated by Endoplasmic Reticulum in HEK Cells" Cells 9, no. 2: 275. https://doi.org/10.3390/cells9020275
APA StyleYue, L., Wang, L., Du, Y., Zhang, W., Hamada, K., Matsumoto, Y., Jin, X., Zhou, Y., Mikoshiba, K., Gill, D. L., Han, S., & Wang, Y. (2020). Type 3 Inositol 1,4,5-Trisphosphate Receptor is a Crucial Regulator of Calcium Dynamics Mediated by Endoplasmic Reticulum in HEK Cells. Cells, 9(2), 275. https://doi.org/10.3390/cells9020275