Design and Synthesis of Arf1-Targeting γ-Dipeptides as Potential Agents against Head and Neck Squamous Cell Carcinoma



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of Compounds 8a–d, 9a–c, 10a–d
2.2. NMR Experiments
2.3. Cell Culture and Cell Viability
2.4. Three-Dimensional (3D) Cell Cultures
2.5. Western Blotting and Determination of Arf1 Activation
2.6. Statistics
3. Results
3.1. Rational Design of γ-Dipeptides
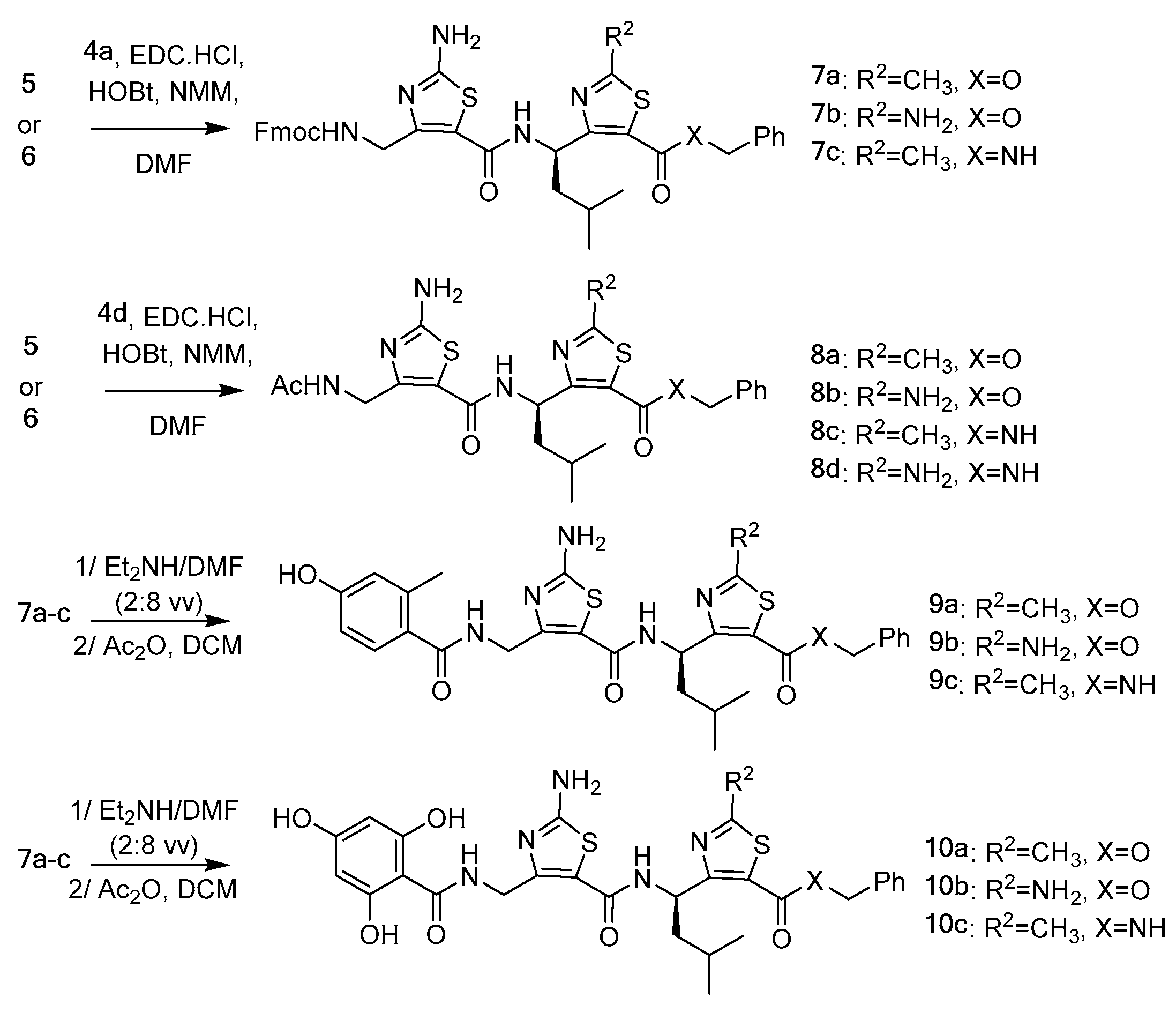
3.2. Synthesis of γ-Dipeptides
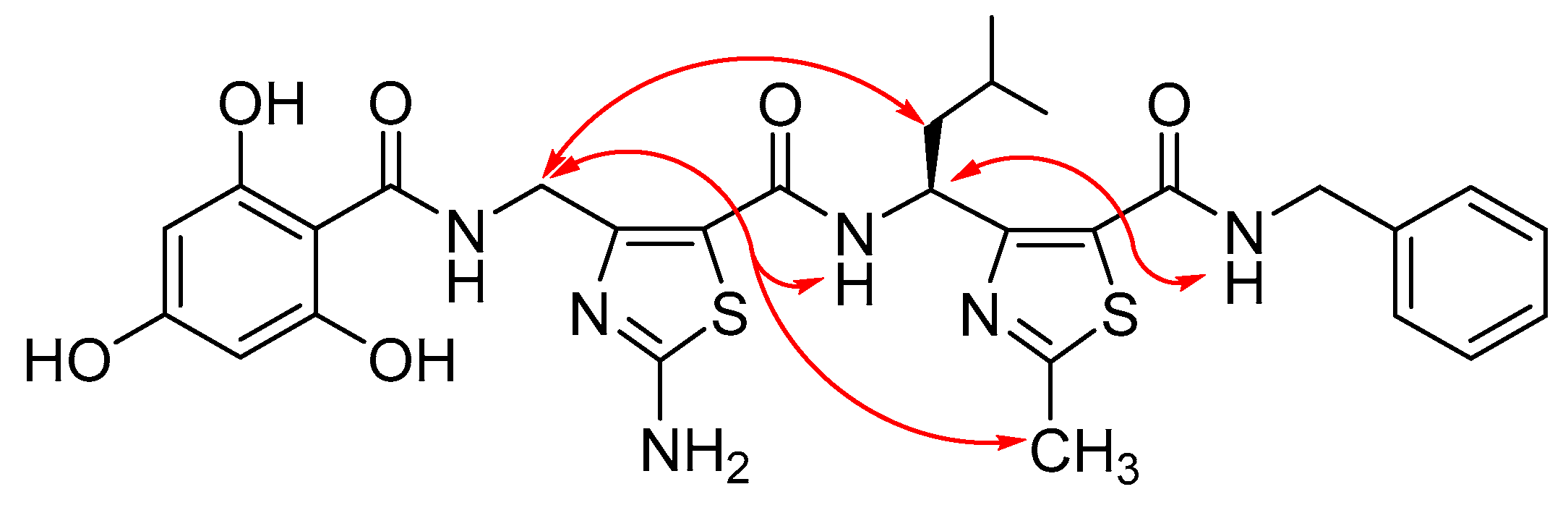
3.3. Structure Analysis
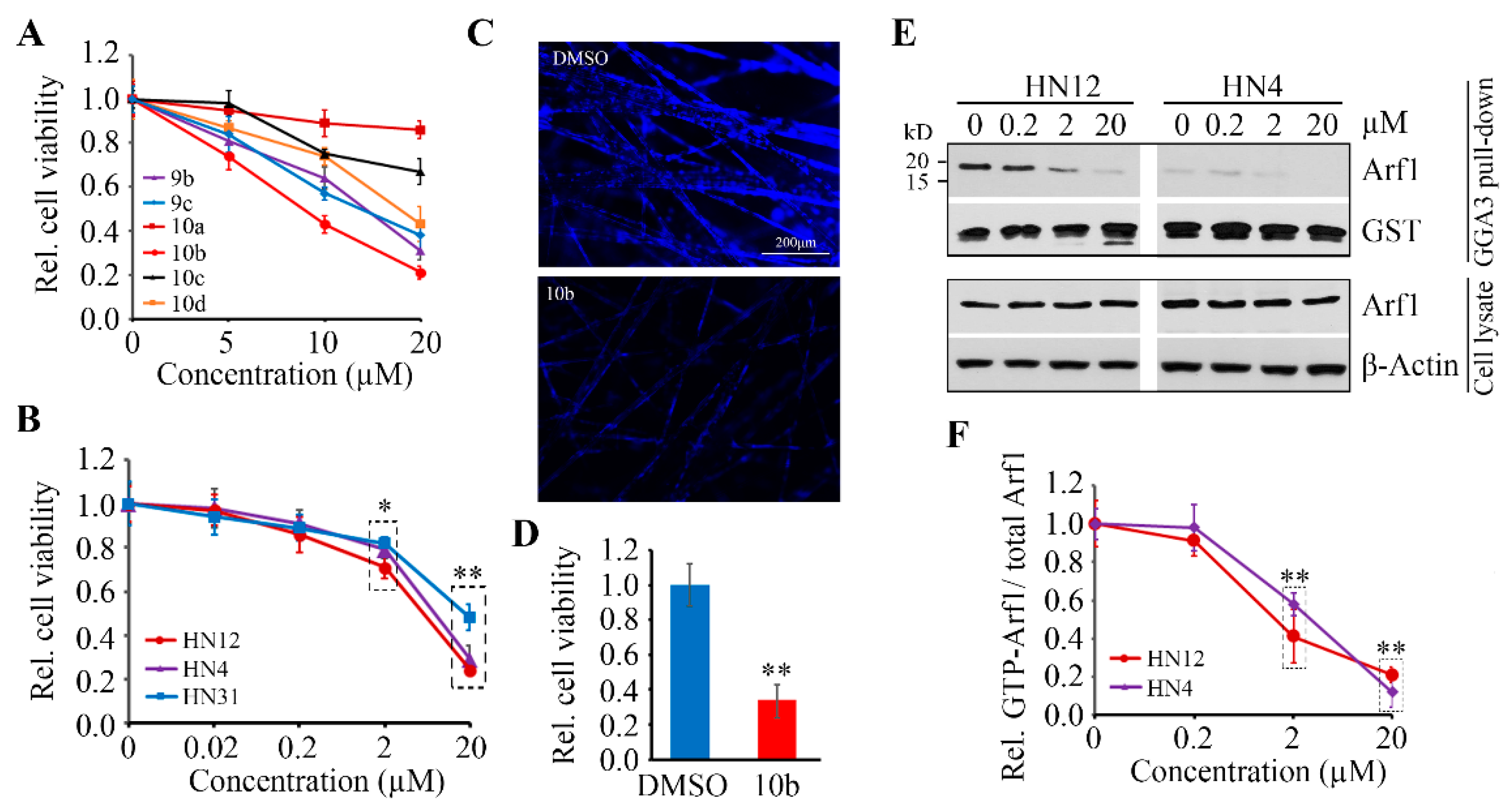
3.4. Compound 10b Exhibits Superior Anticancer Effect in HNSCC Cells by Inactivating Arf1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigneswaran, N.; Williams, M.D. Epidemiologic trends in head and neck cancer and aids in diagnosis. Oral Maxillofac. Surg. Clin. 2014, 26, 123–141. [Google Scholar] [CrossRef]
- Marur, S.; Forastiere, A.A. Head and Neck Squamous Cell Carcinoma: Update on Epidemiology, Diagnosis, and Treatment. Mayo Clin. Proc. 2016, 91, 386–396. [Google Scholar] [CrossRef]
- Machiels, J.-P.; Lambrecht, M.; Hanin, F.-X.; Duprez, T.; Gregoire, V.; Schmitz, S.; Hamoir, M. Advances in the management of squamous cell carcinoma of the head and neck. F1000Prime Rep. 2014, 6, 44. [Google Scholar] [CrossRef]
- Magnes, T.; Egle, A.; Greil, R.; Melchardt, T. Update on squamous cell carcinoma of the head and neck: ASCO annual meeting 2017. Mag. Eur. Med. Oncol. 2017, 10, 220–223. [Google Scholar] [CrossRef] [Green Version]
- Donaldson, J.G.; Jackson, C.L. ARF family G proteins and their regulators: Roles in membrane transport, development and disease. Nat. Rev. Mol. Cell Biol. 2011, 12, 362–375. [Google Scholar] [CrossRef] [PubMed]
- D’Souza-Schorey, C.; Chavrier, P. ARF proteins: Roles in membrane traffic and beyond. Nat. Rev. Mol. Cell Biol. 2006, 7, 347–358. [Google Scholar] [CrossRef]
- Jackson, C.L.; Casanova, J.E. Turning on ARF: The Sec7 family of guanine-nucleotide-exchange factors. Trends Cell Biol. 2000, 10, 60–67. [Google Scholar] [CrossRef]
- Casalou, C.; Faustino, A.; Barral, D.C. Arf proteins in cancer cell migration. Small GTPases 2016, 7, 270–282. [Google Scholar] [CrossRef]
- Lang, L.; Shay, C.; Zhao, X.; Teng, Y. Combined targeting of Arf1 and Ras potentiates anticancer activity for prostate cancer therapeutics. J. Exp. Clin. Cancer Res. 2017, 36, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieto-Dominguez, N.; Parnell, C.; Teng, Y. Drugging the Small GTPase Pathways in Cancer Treatment: Promises and Challenges. Cells 2019, 8, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigil, D.; Cherfils, J.; Rossman, K.L.; Der, C.J. Ras superfamily GEFs and GAPs: Validated and tractable targets for cancer therapy? Nat. Rev. Cancer 2010, 10, 842–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Gao, L.; Shay, C.; Lang, L.; Lv, F.; Teng, Y. Histone deacetylase inhibitors suppress aggressiveness of head and neck squamous cell carcinoma via histone acetylation-independent blockade of the EGFR-Arf1 axis. J. Exp. Clin. Cancer Res. 2019, 38, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singleton, V.L.; Bohonos, N.; Ullstrup, A.J. Decumbin, a new compound from a species of Penicillium. Nature 1958, 181, 1072–1073. [Google Scholar] [CrossRef] [PubMed]
- Peyroche, A.; Antonny, B.; Robineau, S.; Acker, J.; Cherfils, J.; Jackson, C.L. Brefeldin A acts to stabilize an abortive ARF-GDP-Sec7 domain protein complex: Involvement of specific residues of the Sec7 domain. Mol. Cell 1999, 3, 275–285. [Google Scholar] [CrossRef]
- Viaud, J.; Zeghouf, M.; Barelli, H.; Zeeh, J.-C.; Padilla, A.; Guibert, B.; Chardin, P.; Royer, C.A.; Cherfils, J.; Chavanieu, A. Structure-based discovery of an inhibitor of Arf activation by Sec7 domains through targeting of protein-protein complexes. Proc. Natl. Acad. Sci. USA 2007, 104, 10370–10375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, X.; Tang, S.-C.; Cai, Y.; Pi, W.; Deng, L.; Wu, G.; Chavanieu, A.; Teng, Y. Suppression of breast cancer metastasis through the inactivation of. Oncotarget 2016, 7, 58111–58120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, R.G.; Shimizu, T.; Pommier, Y. Brefeldin A is a potent inducer of apoptosis in human cancer cells independently of p53. Exp. Cell Res. 1996, 227, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, Y.; Iijima, H.; Yamaotsu, N.; Yamazaki, K.; Sato, S.; Okamura, M.; Sugimoto, K.; Dan, S.; Hirono, S.; Yamori, T. AMF-26, a novel inhibitor of the Golgi system, targeting ADP-ribosylation factor 1 (Arf1) with potential for cancer therapy. J. Biol. Chem. 2012, 287, 3885–3897. [Google Scholar] [CrossRef] [Green Version]
- Hafner, M.; Schmitz, A.; Grune, I.; Srivatsan, S.G.; Paul, B.; Kolanus, W.; Quast, T.; Kremmer, E.; Bauer, I.; Famulok, M. Inhibition of cytohesins by SecinH3 leads to hepatic insulin resistance. Nature 2006, 444, 941–944. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Sun, J.; Hu, J.; Hu, Y.; Zhou, J.; Chen, Z.; Xu, D.; Xu, W.; Zheng, S.; Zhang, S. Cytohesins/ARNO: The function in colorectal cancer cells. PLoS ONE 2014, 9, e90997. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Jadhav, A.P.; Rodighiero, C.; Fujinaga, Y.; Kirchhausen, T.; Lencer, W.I. Retrograde transport of cholera toxin from the plasma membrane to the endoplasmic reticulum requires the trans-Golgi network but not the Golgi apparatus in Exo2-treated cells. EMBO Rep. 2004, 5, 596–601. [Google Scholar] [CrossRef] [Green Version]
- Rouhana, J.; Hoh, F.; Estaran, S.; Henriquet, C.; Boublik, Y.; Kerkour, A.; Trouillard, R.; Martinez, J.; Pugniere, M.; Padilla, A.; et al. Fragment-based identification of a locus in the Sec7 domain of Arno for the design of protein-protein interaction inhibitors. J. Med. Chem. 2013, 56, 8497–8511. [Google Scholar] [CrossRef]
- DiNitto, J.P.; Delprato, A.; Gabe Lee, M.-T.; Cronin, T.C.; Huang, S.; Guilherme, A.; Czech, M.P.; Lambright, D.G. Structural basis and mechanism of autoregulation in 3-phosphoinositide-dependent Grp1 family Arf GTPase exchange factors. Mol. Cell 2007, 28, 569–583. [Google Scholar] [CrossRef] [Green Version]
- UniProtKB—Q99418 (CYH2_HUMAN). Available online: https://www.uniprot.org/uniprot/Q99418 (accessed on 20 September 2019).
- Mathieu, L.; Legrand, B.; Deng, C.; Vezenkov, L.; Wenger, E.; Didierjean, C.; Amblard, M.; Averlant-Petit, M.-C.; Masurier, N.; Lisowski, V.; et al. Helical oligomers of thiazole-based amino γ-acids: Synthesis and structural studies. Angew. Chem. Int. Ed. 2013, 52, 6006–6010. [Google Scholar] [CrossRef]
- Mathieu, L.; Bonnel, C.; Masurier, N.; Maillard, L.T.; Martinez, J.; Lisowski, V. Cross-Claisen condensation of N-Fmoc-amino acids—A short route to heterocyclic γ-amino acids. Eur. J. Org. Chem. 2015, 2015, 2262–2270. [Google Scholar] [CrossRef]
- Simon, M.; Ali, L.M.A.; El Cheikh, K.; Aguesseau, J.; Gary-Bobo, M.; Garcia, M.; Morere, A.; Maillard, L.T. Can Heterocyclic γ-Peptides Provide Polyfunctional Platforms for Synthetic Glycocluster Construction? Chem. Eur. J. 2018, 24, 11426–11432. [Google Scholar] [CrossRef]
- Chardin, P.; Paris, S.; Antonny, B.; Robineau, S.; Beraud-Dufour, S.; Jackson, C.L.; Chabre, M. A human exchange factor for ARF contains Sec7- and pleckstrin-homology domains. Nature 1996, 384, 481–484. [Google Scholar] [CrossRef]
- Klarlund, J.K.; Guilherme, A.; Holik, J.J.; Virbasius, J.V.; Chawla, A.; Czech, M.P. Signaling by phosphoinositide-3,4,5-trisphosphate through proteins containing pleckstrin and Sec7 homology domains. Science 1997, 275, 1927–1930. [Google Scholar] [CrossRef]
- Vezenkov, L.L.; Martin, V.; Bettache, N.; Simon, M.; Messerschmitt, A.; Legrand, B.; Bantignies, J.-L.; Subra, G.; Maynadier, M.; Bellet, V.; et al. Ribbon-like Foldamers for Cellular Uptake and Drug Delivery. ChemBioChem 2017, 18, 2110–2114. [Google Scholar] [CrossRef] [PubMed]
- Fivush, A.M.; Willson, T.M. AMEBA: An acid sensitive aldehyde resin for solid phase synthesis. Tetrahedron Lett. 1997, 38, 7151–7154. [Google Scholar] [CrossRef]
- Bonnel, C.; Legrand, B.; Bantignies, J.-L.; Petitjean, H.; Martinez, J.; Masurier, N.; Maillard, L.T. FT-IR and NMR structural markers for thiazole-based γ-peptide foldamers. Org. Biomol. Chem. 2016, 14, 8664–8669. [Google Scholar] [CrossRef] [PubMed]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef] [Green Version]
- Alsahafi, E.; Begg, K.; Amelio, I.; Raulf, N.; Lucarelli, P.; Sauter, T.; Tavassoli, M. Clinical update on head and neck cancer: Molecular biology and ongoing challenges. Cell Death Dis. 2019, 10, 540. [Google Scholar] [CrossRef] [Green Version]
- Ferris, R.L.; Licitra, L.; Fayette, J.; Even, C.; Blumenschein, G.; Harrington, K.J.; Guigay, J.; Vokes, E.E.; Saba, N.F.; Haddad, R.; et al. Nivolumab in Patients with Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck: Efficacy and Safety in CheckMate 141 by Prior Cetuximab Use. Clin. Cancer Res. 2019, 25, 5221–5230. [Google Scholar] [CrossRef] [Green Version]
- Seiwert, T.Y.; Burtness, B.; Mehra, R.; Weiss, J.; Berger, R.; Eder, J.P.; Heath, K.; McClanahan, T.; Lunceford, J.; Gause, C.; et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): An open-label, multicentre, phase 1b trial. Lancet Oncol. 2016, 17, 956–965. [Google Scholar] [CrossRef]
- Troiano, G.; Caponio, V.C.A.; Zhurakivska, K.; Arena, C.; Pannone, G.; Mascitti, M.; Santarelli, A.; Lo Muzio, L. High PD-L1 expression in the tumour cells did not correlate with poor prognosis of patients suffering for oral squamous cells carcinoma: A meta-analysis of the literature. Cell Prolif. 2019, 52, e12537. [Google Scholar] [CrossRef] [Green Version]
- Lindemann, A.; Patel, A.A.; Silver, N.L.; Tang, L.; Liu, Z.; Wang, L.; Tanaka, N.; Rao, X.; Takahashi, H.; Maduka, N.K.; et al. COTI-2, A Novel Thiosemicarbazone Derivative, Exhibits Antitumor Activity in HNSCC through p53-dependent and -independent Mechanisms. Clin. Cancer Res. 2019, 25, 5650–5662. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.E.; Xie, X.; Guo, J.; Huang, W.; Chu, W.-M.; Huang, S.; Teng, Y.; Wu, G. ARF1 promotes prostate tumorigenesis via targeting oncogenic MAPK signaling. Oncotarget 2016, 7, 39834–39845. [Google Scholar] [CrossRef] [Green Version]
- Boulay, P.-L.; Schlienger, S.; Lewis-Saravalli, S.; Vitale, N.; Ferbeyre, G.; Claing, A. ARF1 controls proliferation of breast cancer cells by regulating the retinoblastoma protein. Oncogene 2011, 30, 3846–3861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulay, P.-L.; Cotton, M.; Melançon, P.; Claing, A. ADP-ribosylation factor 1 controls the activation of the phosphatidylinositol 3-kinase pathway to regulate epidermal growth factor-dependent growth and migration of breast cancer cells. J. Biol. Chem. 2008, 283, 36425–36434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anadu, N.O.; Davisson, V.J.; Cushman, M. Synthesis and anticancer activity of brefeldin A ester derivatives. J. Med. Chem. 2006, 49, 3897–3905. [Google Scholar] [CrossRef]
- Rouhana, J.; Padilla, A.; Estaran, S.; Bakari, S.; Delbecq, S.; Boublik, Y.; Chopineau, J.; Pugniere, M.; Chavanieu, A. Kinetics of interaction between ADP-ribosylation factor-1 (Arf1) and the Sec7 domain of Arno guanine nucleotide exchange factor, modulation by allosteric factors, and the uncompetitive inhibitor brefeldin A. J. Biol. Chem. 2013, 288, 4659–4672. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, Y.; Okamura, M.; Katayama, R.; Fang, S.; Tsutsui, S.; Akatsuka, A.; Shan, M.; Choi, H.-W.; Fujita, N.; Yoshimatsu, K.; et al. Targeting the Golgi apparatus to overcome acquired resistance of non-small cell lung cancer cells to EGFR tyrosine kinase inhibitors. Oncotarget 2018, 9, 1641–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haines, E.; Schlienger, S.; Claing, A. The small GTPase ADP-Ribosylation Factor 1 mediates the sensitivity of triple negative breast cancer cells to EGFR tyrosine kinase inhibitors. Cancer Biol. Ther. 2015, 16, 1535–1547. [Google Scholar] [CrossRef] [Green Version]
- Schlienger, S.; Campbell, S.; Claing, A. ARF1 regulates the Rho/MLC pathway to control EGF-dependent breast cancer cell invasion. Mol. Biol. Cell 2014, 25, 17–29. [Google Scholar] [CrossRef]
- Haines, E.; Saucier, C.; Claing, A. The adaptor proteins p66Shc and Grb2 regulate the activation of the GTPases ARF1 and ARF6 in invasive breast cancer cells. J. Biol. Chem. 2014, 289, 5687–5703. [Google Scholar] [CrossRef] [Green Version]
- Lang, L.; Shay, C.; Zhao, X.; Xiong, Y.; Wang, X.; Teng, Y. Simultaneously inactivating Src and AKT by saracatinib/capivasertib co-delivery nanoparticles to improve the efficacy of anti-Src therapy in head and neck squamous cell carcinoma. J. Hematol. Oncol. 2019, 12, 132. [Google Scholar] [CrossRef] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vo-Hoang, Y.; Paiva, S.; He, L.; Estaran, S.; Teng, Y. Design and Synthesis of Arf1-Targeting γ-Dipeptides as Potential Agents against Head and Neck Squamous Cell Carcinoma. Cells 2020, 9, 286. https://doi.org/10.3390/cells9020286
Vo-Hoang Y, Paiva S, He L, Estaran S, Teng Y. Design and Synthesis of Arf1-Targeting γ-Dipeptides as Potential Agents against Head and Neck Squamous Cell Carcinoma. Cells. 2020; 9(2):286. https://doi.org/10.3390/cells9020286
Chicago/Turabian StyleVo-Hoang, Yen, Sergio Paiva, Leilei He, Sébastien Estaran, and Yong Teng. 2020. "Design and Synthesis of Arf1-Targeting γ-Dipeptides as Potential Agents against Head and Neck Squamous Cell Carcinoma" Cells 9, no. 2: 286. https://doi.org/10.3390/cells9020286
APA StyleVo-Hoang, Y., Paiva, S., He, L., Estaran, S., & Teng, Y. (2020). Design and Synthesis of Arf1-Targeting γ-Dipeptides as Potential Agents against Head and Neck Squamous Cell Carcinoma. Cells, 9(2), 286. https://doi.org/10.3390/cells9020286