Targeting CD38-Expressing Multiple Myeloma and Burkitt Lymphoma Cells In Vitro with Nanobody-Based Chimeric Antigen Receptors (Nb-CARs)

 ,
, 
 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Generation of Nb-CARs
2.3. Stable Transduction of NK-92 Cells with Nb-CARs
2.4. Production of Alexa Fluor 647-Labeled CD38 and ARTC2.2
2.5. Luminescence CARDCC Assays
2.6. Flow Cytometric CARDCC Assays
2.7. CARDCC Assays with Primary Human Bone Marrow Samples
3. Results
3.1. Generation of CD38-Deficient Cell Lines and Lentiviral Transduction of CD38+ and CD38− Cell Lines with Luciferase
3.2. Generation of Nanobody-Based Chimeric Antigen Receptors (Nb-CARs) and Transduction of NK-92 Cells with Nb-CARs
3.3. Luminescence-Based CAR-Dependent Cellular Cytotoxicity Assay (CARDCC)
3.4. A Flow-Cytometric CARDCC
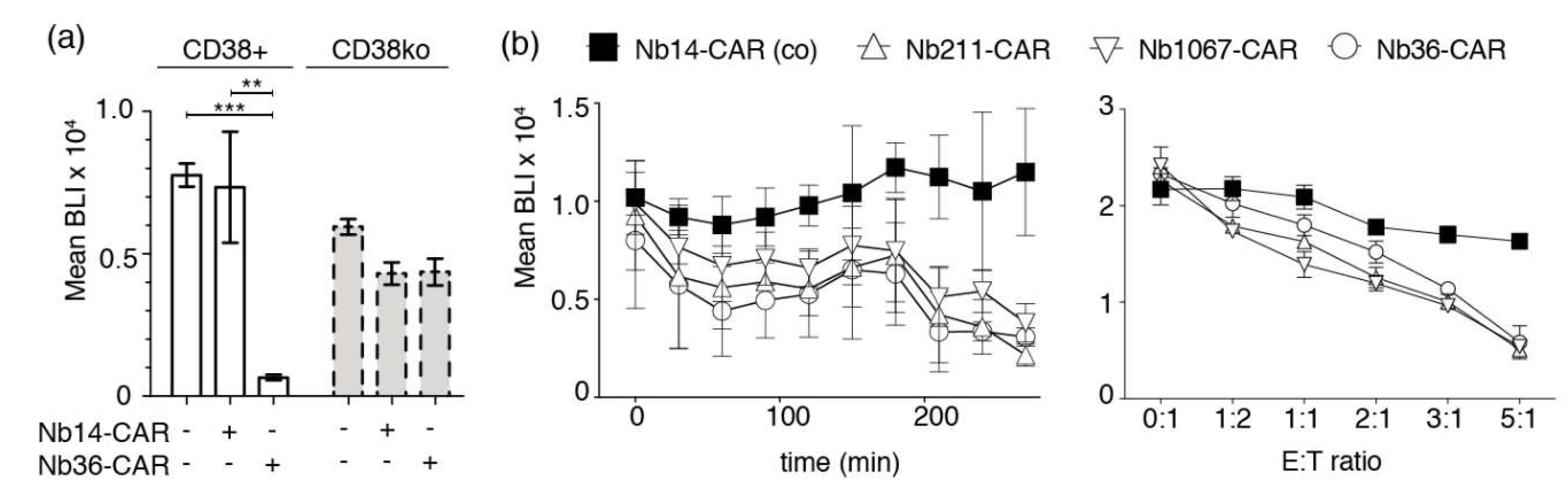
3.5. CD38-Directed Nb-CAR-NKs Specifically Deplete CD38+/CD56+ Myeloma Cells from Primary Human Bone Marrow Samples
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Deaglio, S.; Aydin, S.; Vaisitti, T.; Bergui, L.; Malavasi, F. CD38 at the junction between prognostic marker and therapeutic target. Trends Mol. Med. 2008, 14, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Horenstein, A.L.; Bracci, C.; Morandi, F.; Malavasi, F. CD38 in adenosinergic pathways and metabolic re-programming in human multiple myeloma cells: In-tandem insights from basic science to therapy. Front. Immunol. 2019, 10, 760. [Google Scholar] [CrossRef] [PubMed]
- Morandi, F.; Horenstein, A.L.; Costa, F.; Giuliani, N.; Pistoia, V.; Malavasi, F. CD38: A target for immunotherapeutic approaches in multiple myeloma. Front. Immunol. 2018, 9, 2722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonial, S.; Weiss, B.M.; Usmani, S.Z.; Singhal, S.; Chari, A.; Bahlis, N.J.; Belch, A.; Krishnan, A.; Vescio, R.A.; Mateos, M.V.; et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): An open-label, randomised, phase 2 trial. Lancet 2016, 387, 1551–1560. [Google Scholar] [CrossRef]
- van de Donk, N.; Richardson, P.G.; Malavasi, F. CD38 antibodies in multiple myeloma: Back to the future. Blood 2018, 131, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.W.; Comenzo, R.L. CD38 Monoclonal Antibody Therapies for Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2015, 15, 635–645. [Google Scholar] [CrossRef]
- Muyldermans, S. Nanobodies: Natural single-domain antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [Green Version]
- Wesolowski, J.; Alzogaray, V.; Reyelt, J.; Unger, M.; Juarez, K.; Urrutia, M.; Cauerhff, A.; Danquah, W.; Rissiek, B.; Scheuplein, F.; et al. Single domain antibodies: Promising experimental and therapeutic tools in infection and immunity. Med. Microbiol. Immunol. 2009, 198, 157–174. [Google Scholar] [CrossRef] [Green Version]
- Conrath, K.E.; Wernery, U.; Muyldermans, S.; Nguyen, V.K. Emergence and evolution of functional heavy-chain antibodies in Camelidae. Dev. Comp. Immunol. 2003, 27, 87–103. [Google Scholar] [CrossRef]
- Bannas, P.; Hambach, J.; Koch-Nolte, F. Nanobodies and Nanobody-Based Human Heavy Chain Antibodies As Antitumor Therapeutics. Front. Immunol. 2017, 8, 1603. [Google Scholar] [CrossRef]
- Ingram, J.R.; Schmidt, F.I.; Ploegh, H.L. Exploiting Nanobodies’ Singular Traits. Annu. Rev. Immunol. 2018, 36, 695–715. [Google Scholar] [CrossRef] [PubMed]
- Bannas, P.; Koch-Nolte, F. Perspectives for the Development of CD38-Specific Heavy Chain Antibodies as Therapeutics for Multiple Myeloma. Front. Immunol. 2018, 9, 2559. [Google Scholar] [CrossRef] [PubMed]
- Fumey, W.; Koenigsdorf, J.; Kunick, V.; Menzel, S.; Schutze, K.; Unger, M.; Schriewer, L.; Haag, F.; Adam, G.; Oberle, A.; et al. Nanobodies effectively modulate the enzymatic activity of CD38 and allow specific imaging of CD38(+) tumors in mouse models in vivo. Sci. Rep. 2017, 7, 14289. [Google Scholar] [CrossRef] [PubMed]
- Schriewer, L.; Schutze, K.; Petry, K.; Hambach, J.; Fumey, W.; Koenigsdorf, J.; Baum, N.; Menzel, S.; Rissiek, B.; Riecken, K.; et al. Nanobody-based CD38-specific heavy chain antibodies induce killing of multiple myeloma and other hematological malignancies. Theranostics 2019. (In press) [Google Scholar] [CrossRef]
- Schutze, K.; Petry, K.; Hambach, J.; Schuster, N.; Fumey, W.; Schriewer, L.; Rockendorf, J.; Menzel, S.; Albrecht, B.; Haag, F.; et al. CD38-Specific Biparatopic Heavy Chain Antibodies Display Potent Complement-Dependent Cytotoxicity Against Multiple Myeloma Cells. Front. Immunol. 2018, 9, 2553. [Google Scholar] [CrossRef]
- Klingemann, H.; Boissel, L.; Toneguzzo, F. Natural Killer Cells for Immunotherapy - Advantages of the NK-92 Cell Line over Blood NK Cells. Front. Immunol. 2016, 7, 91. [Google Scholar] [CrossRef] [Green Version]
- Weber, K.; Bartsch, U.; Stocking, C.; Fehse, B. A multicolor panel of novel lentiviral “gene ontology” (LeGO) vectors for functional gene analysis. Mol. Ther. 2008, 16, 698–706. [Google Scholar] [CrossRef]
- Weber, K.; Mock, U.; Petrowitz, B.; Bartsch, U.; Fehse, B. Lentiviral gene ontology (LeGO) vectors equipped with novel drug-selectable fluorescent proteins: New building blocks for cell marking and multi-gene analysis. Gene Ther. 2010, 17, 511–520. [Google Scholar] [CrossRef] [Green Version]
- Koch-Nolte, F.; Reyelt, J.; Schossow, B.; Schwarz, N.; Scheuplein, F.; Rothenburg, S.; Haag, F.; Alzogaray, V.; Cauerhff, A.; Goldbaum, F.A. Single domain antibodies from llama effectively and specifically block T cell ecto-ADP-ribosyltransferase ART2.2 in vivo. FASEB J. 2007, 21, 3490–3498. [Google Scholar] [CrossRef]
- Schambach, A.; Mueller, D.; Galla, M.; Verstegen, M.M.; Wagemaker, G.; Loew, R.; Baum, C.; Bohne, J. Overcoming promoter competition in packaging cells improves production of self-inactivating retroviral vectors. Gene Ther. 2006, 13, 1524–1533. [Google Scholar] [CrossRef] [Green Version]
- Schambach, A.; Bohne, J.; Chandra, S.; Will, E.; Margison, G.P.; Williams, D.A.; Baum, C. Equal potency of gammaretroviral and lentiviral SIN vectors for expression of O6-methylguanine-DNA methyltransferase in hematopoietic cells. Mol. Ther. 2006, 13, 391–400. [Google Scholar] [CrossRef]
- Frigyesi, I.; Adolfsson, J.; Ali, M.; Christophersen, M.K.; Johnsson, E.; Turesson, I.; Gullberg, U.; Hansson, M.; Nilsson, B. Robust isolation of malignant plasma cells in multiple myeloma. Blood 2014, 123, 1336–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodgins, J.J.; Khan, S.T.; Park, M.M.; Auer, R.C.; Ardolino, M. Killers 2.0: NK cell therapies at the forefront of cancer control. J. Clin. Invest. 2019, 129, 3499–3510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, J.H.; Maki, G.; Klingemann, H.G. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia 1994, 8, 652–658. [Google Scholar] [PubMed]
- Arai, S.; Meagher, R.; Swearingen, M.; Myint, H.; Rich, E.; Martinson, J.; Klingemann, H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: A phase I trial. Cytotherapy 2008, 10, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Tonn, T.; Schwabe, D.; Klingemann, H.G.; Becker, S.; Esser, R.; Koehl, U.; Suttorp, M.; Seifried, E.; Ottmann, O.G.; Bug, G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy 2013, 15, 1563–1570. [Google Scholar] [CrossRef]
- An, N.; Hou, Y.N.; Zhang, Q.X.; Li, T.; Zhang, Q.L.; Fang, C.; Chen, H.; Lee, H.C.; Zhao, Y.J.; Du, X. Anti-Multiple Myeloma Activity of Nanobody-Based Anti-CD38 Chimeric Antigen Receptor T Cells. Mol. Pharm. 2018, 15, 4577–4588. [Google Scholar] [CrossRef]
- Bakhtiari, S.H.; Rahbarizadeh, F.; Hasannia, S.; Ahmadvand, D.; Iri-Sofla, F.J.; Rasaee, M.J. Anti-MUC1 nanobody can redirect T-body cytotoxic effector function. Hybrid 2009, 28, 85–92. [Google Scholar] [CrossRef]
- Hassani, M.; Hajari Taheri, F.; Sharifzadeh, Z.; Arashkia, A.; Hadjati, J.; van Weerden, W.M.; Modarressi, M.H.; Abolhassani, M. Construction of a chimeric antigen receptor bearing a nanobody against prostate a specific membrane antigen in prostate cancer. J. Cell Biochem. 2019, 120, 10787–10795. [Google Scholar] [CrossRef]
- Xie, Y.J.; Dougan, M.; Jailkhani, N.; Ingram, J.; Fang, T.; Kummer, L.; Momin, N.; Pishesha, N.; Rickelt, S.; Hynes, R.O.; et al. Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc. Natl. Acad. Sci. USA 2019, 116, 7624–7631. [Google Scholar] [CrossRef] [Green Version]
- Drent, E.; Groen, R.W.; Noort, W.A.; Themeli, M.; Lammerts van Bueren, J.J.; Parren, P.W.; Kuball, J.; Sebestyen, Z.; Yuan, H.; de Bruijn, J.; et al. Pre-clinical evaluation of CD38 chimeric antigen receptor engineered T cells for the treatment of multiple myeloma. Haematologica 2016, 101, 616–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihara, K.; Yanagihara, K.; Takigahira, M.; Imai, C.; Kitanaka, A.; Takihara, Y.; Kimura, A. Activated T-cell-mediated immunotherapy with a chimeric receptor against CD38 in B-cell non-Hodgkin lymphoma. J. Immunother. 2009, 32, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Drent, E.; Poels, R.; Ruiter, R.; van de Donk, N.; Zweegman, S.; Yuan, H.; de Bruijn, J.; Sadelain, M.; Lokhorst, H.M.; Groen, R.W.J.; et al. Combined CD28 and 4-1BB Costimulation Potentiates Affinity-tuned Chimeric Antigen Receptor-engineered T Cells. Clin. Cancer Res. 2019, 25, 4014–4025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chmielewski, M.; Hombach, A.; Heuser, C.; Adams, G.P.; Abken, H. T cell activation by antibody-like immunoreceptors: Increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. J. Immunol. 2004, 173, 7647–7653. [Google Scholar] [CrossRef]
- Drent, E.; Themeli, M.; Poels, R.; de Jong-Korlaar, R.; Yuan, H.; de Bruijn, J.; Martens, A.C.M.; Zweegman, S.; van de Donk, N.; Groen, R.W.J.; et al. A Rational Strategy for Reducing On-Target Off-Tumor Effects of CD38-Chimeric Antigen Receptors by Affinity Optimization. Mol. Ther. 2017, 25, 1946–1958. [Google Scholar] [CrossRef]
- Hudecek, M.; Lupo-Stanghellini, M.T.; Kosasih, P.L.; Sommermeyer, D.; Jensen, M.C.; Rader, C.; Riddell, S.R. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin. Cancer Res. 2013, 19, 3153–3164. [Google Scholar] [CrossRef] [Green Version]
- Perna, F.; Berman, S.H.; Soni, R.K.; Mansilla-Soto, J.; Eyquem, J.; Hamieh, M.; Hendrickson, R.C.; Brennan, C.W.; Sadelain, M. Integrating Proteomics and Transcriptomics for Systematic Combinatorial Chimeric Antigen Receptor Therapy of AML. Cancer Cell 2017, 32, 506–519. [Google Scholar] [CrossRef] [Green Version]
- De Munter, S.; Ingels, J.; Goetgeluk, G.; Bonte, S.; Pille, M.; Weening, K.; Kerre, T.; Abken, H.; Vandekerckhove, B. Nanobody Based Dual Specific CARs. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hambach, J.; Riecken, K.; Cichutek, S.; Schütze, K.; Albrecht, B.; Petry, K.; Röckendorf, J.L.; Baum, N.; Kröger, N.; Hansen, T.; et al. Targeting CD38-Expressing Multiple Myeloma and Burkitt Lymphoma Cells In Vitro with Nanobody-Based Chimeric Antigen Receptors (Nb-CARs). Cells 2020, 9, 321. https://doi.org/10.3390/cells9020321
Hambach J, Riecken K, Cichutek S, Schütze K, Albrecht B, Petry K, Röckendorf JL, Baum N, Kröger N, Hansen T, et al. Targeting CD38-Expressing Multiple Myeloma and Burkitt Lymphoma Cells In Vitro with Nanobody-Based Chimeric Antigen Receptors (Nb-CARs). Cells. 2020; 9(2):321. https://doi.org/10.3390/cells9020321
Chicago/Turabian StyleHambach, Julia, Kristoffer Riecken, Sophia Cichutek, Kerstin Schütze, Birte Albrecht, Katharina Petry, Jana Larissa Röckendorf, Natalie Baum, Nicolaus Kröger, Timon Hansen, and et al. 2020. "Targeting CD38-Expressing Multiple Myeloma and Burkitt Lymphoma Cells In Vitro with Nanobody-Based Chimeric Antigen Receptors (Nb-CARs)" Cells 9, no. 2: 321. https://doi.org/10.3390/cells9020321
APA StyleHambach, J., Riecken, K., Cichutek, S., Schütze, K., Albrecht, B., Petry, K., Röckendorf, J. L., Baum, N., Kröger, N., Hansen, T., Schuch, G., Haag, F., Adam, G., Fehse, B., Bannas, P., & Koch-Nolte, F. (2020). Targeting CD38-Expressing Multiple Myeloma and Burkitt Lymphoma Cells In Vitro with Nanobody-Based Chimeric Antigen Receptors (Nb-CARs). Cells, 9(2), 321. https://doi.org/10.3390/cells9020321