Metabolic Dysfunction in Hutchinson–Gilford Progeria Syndrome


Abstract
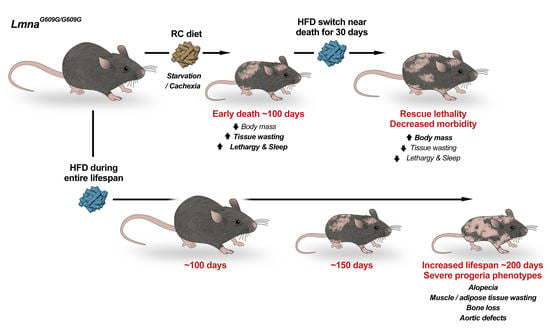
:
1. Introduction
2. The Progeria Patient
3. The Progeria Cell and Energy
4. The Progeria Mice
5. Metabolic Approaches in Progeria
6. Current Therapeutic Strategies
7. A Case for Metabolism
Funding
Acknowledgments
Conflicts of Interest
References
- Merideth, M.A.; Gordon, L.B.; Clauss, S.; Sachdev, V.; Smith, A.C.; Perry, M.B.; Brewer, C.C.; Zalewski, C.; Kim, H.J.; Solomon, B.; et al. Phenotype and course of hutchinson-gilford progeria syndrome. N. Engl. J. Med. 2008, 358, 592–604. [Google Scholar] [CrossRef] [Green Version]
- Ullrich, N.J.; Gordon, L.B. Hutchinson-gilford progeria syndrome. Handb. Clin. Neurol. 2015, 132, 249–264. [Google Scholar]
- Gordon, L.B.; Massaro, J.; D’Agostino, R.B., Sr.; Campbell, S.E.; Brazier, J.; Brown, W.T.; Kleinman, M.E.; Kieran, M.W.; Progeria Clinical Trials, C. Impact of farnesylation inhibitors on survival in hutchinson-gilford progeria syndrome. Circulation 2014, 130, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Gordon, L.B.; Rothman, F.G.; Lopez-Otin, C.; Misteli, T. Progeria: A paradigm for translational medicine. Cell 2014, 156, 400–407. [Google Scholar] [CrossRef] [Green Version]
- Prakash, A.; Gordon, L.B.; Kleinman, M.E.; Gurary, E.B.; Massaro, J.; D’Agostino, R., Sr.; Kieran, M.W.; Gerhard-Herman, M.; Smoot, L. Cardiac abnormalities in patients with hutchinson-gilford progeria syndrome. JAMA Cardiol. 2018, 3, 326–334. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin a cause hutchinson-gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef] [Green Version]
- De Sandre-Giovannoli, A.; Bernard, R.; Cau, P.; Navarro, C.; Amiel, J.; Boccaccio, I.; Lyonnet, S.; Stewart, C.L.; Munnich, A.; Le Merrer, M.; et al. Lamin a truncation in hutchinson-gilford progeria. Science 2003, 300, 2055. [Google Scholar] [CrossRef]
- Burke, B.; Stewart, C.L. Functional architecture of the cell’s nucleus in development, aging, and disease. Curr. Top. Dev. Biol. 2014, 109, 1–52. [Google Scholar]
- Goldman, R.D.; Gruenbaum, Y.; Moir, R.D.; Shumaker, D.K.; Spann, T.P. Nuclear lamins: Building blocks of nuclear architecture. Genes Dev. 2002, 16, 533–547. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Suarez, I.; Redwood, A.B.; Perkins, S.M.; Vermolen, B.; Lichtensztejin, D.; Grotsky, D.A.; Morgado-Palacin, L.; Gapud, E.J.; Sleckman, B.P.; Sullivan, T.; et al. Novel roles for a-type lamins in telomere biology and the DNA damage response pathway. EMBO J. 2009, 28, 2414–2427. [Google Scholar] [CrossRef]
- Gruenbaum, Y.; Goldman, R.D.; Meyuhas, R.; Mills, E.; Margalit, A.; Fridkin, A.; Dayani, Y.; Prokocimer, M.; Enosh, A. The nuclear lamina and its functions in the nucleus. Int. Rev. Cytol. 2003, 226, 1–62. [Google Scholar] [PubMed]
- Goldman, R.D.; Shumaker, D.K.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Gordon, L.B.; Gruenbaum, Y.; Khuon, S.; Mendez, M.; Varga, R.; et al. Accumulation of mutant lamin a causes progressive changes in nuclear architecture in hutchinson-gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 8963–8968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaffidi, P.; Misteli, T. Lamin a-dependent nuclear defects in human aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constantinescu, D.; Csoka, A.B.; Navara, C.S.; Schatten, G.P. Defective dsb repair correlates with abnormal nuclear morphology and is improved with fti treatment in hutchinson-gilford progeria syndrome fibroblasts. Exp. Cell Res. 2010, 316, 2747–2759. [Google Scholar] [CrossRef] [PubMed]
- Decker, M.L.; Chavez, E.; Vulto, I.; Lansdorp, P.M. Telomere length in hutchinson-gilford progeria syndrome. Mech. Ageing Dev. 2009, 130, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Kreienkamp, R.; Graziano, S.; Coll-Bonfill, N.; Bedia-Diaz, G.; Cybulla, E.; Vindigni, A.; Dorsett, D.; Kubben, N.; Batista, L.F.Z.; Gonzalo, S. A cell-intrinsic interferon-like response links replication stress to cellular aging caused by progerin. Cell Rep. 2018, 22, 2006–2015. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Rusinol, A.; Sinensky, M.; Wang, Y.; Zou, Y. DNA damage responses in progeroid syndromes arise from defective maturation of prelamin a. J. Cell Sci. 2006, 119, 4644–4649. [Google Scholar] [CrossRef] [Green Version]
- McCord, R.P.; Nazario-Toole, A.; Zhang, H.; Chines, P.S.; Zhan, Y.; Erdos, M.R.; Collins, F.S.; Dekker, J.; Cao, K. Correlated alterations in genome organization, histone methylation, and DNA-lamin a/c interactions in hutchinson-gilford progeria syndrome. Genome Res. 2013, 23, 260–269. [Google Scholar] [CrossRef] [Green Version]
- Wheaton, K.; Campuzano, D.; Ma, W.; Sheinis, M.; Ho, B.; Brown, G.W.; Benchimol, S. Progerin-induced replication stress facilitates premature senescence in hutchinson-gilford progeria syndrome. Mol. Cell Biol. 2017, 37. [Google Scholar] [CrossRef] [Green Version]
- Hennekam, R.C. Hutchinson-gilford progeria syndrome: Review of the phenotype. Am. J. Med. Genet. 2006, 140, 2603–2624. [Google Scholar] [CrossRef] [Green Version]
- McClintock, D.; Ratner, D.; Lokuge, M.; Owens, D.M.; Gordon, L.B.; Collins, F.S.; Djabali, K. The mutant form of lamin a that causes hutchinson-gilford progeria is a biomarker of cellular aging in human skin. PLoS ONE 2007, 2, e1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lattanzi, G.; Ortolani, M.; Columbaro, M.; Prencipe, S.; Mattioli, E.; Lanzarini, C.; Maraldi, N.M.; Cenni, V.; Garagnani, P.; Salvioli, S.; et al. Lamins are rapamycin targets that impact human longevity: A study in centenarians. J. Cell Sci. 2014, 127, 147–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barzilai, N.; Huffman, D.M.; Muzumdar, R.H.; Bartke, A. The critical role of metabolic pathways in aging. Diabetes 2012, 61, 1315–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, B.K.; Berger, S.L.; Brunet, A.; Campisi, J.; Cuervo, A.M.; Epel, E.S.; Franceschi, C.; Lithgow, G.J.; Morimoto, R.I.; Pessin, J.E.; et al. Geroscience: Linking aging to chronic disease. Cell 2014, 159, 709–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rork, J.F.; Huang, J.T.; Gordon, L.B.; Kleinman, M.; Kieran, M.W.; Liang, M.G. Initial cutaneous manifestations of hutchinson-gilford progeria syndrome. Pediatr. Dermatol. 2014, 31, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.M.; Gordon, L.B.; Snyder, B.D.; Nazarian, A.; Quinn, N.; Huh, S.; Giobbie-Hurder, A.; Neuberg, D.; Cleveland, R.; Kleinman, M.; et al. Hutchinson-gilford progeria is a skeletal dysplasia. J. Bone Miner. Res. 2011, 26, 1670–1679. [Google Scholar] [CrossRef] [Green Version]
- Olive, M.; Harten, I.; Mitchell, R.; Beers, J.K.; Djabali, K.; Cao, K.; Erdos, M.R.; Blair, C.; Funke, B.; Smoot, L.; et al. Cardiovascular pathology in hutchinson-gilford progeria: Correlation with the vascular pathology of aging. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2301–2309. [Google Scholar] [CrossRef] [Green Version]
- Stehbens, W.E.; Wakefield, S.J.; Gilbert-Barness, E.; Olson, R.E.; Ackerman, J. Histological and ultrastructural features of atherosclerosis in progeria. Cardiovasc. Pathol. 1999, 8, 29–39. [Google Scholar] [CrossRef]
- Gerhard-Herman, M.; Smoot, L.B.; Wake, N.; Kieran, M.W.; Kleinman, M.E.; Miller, D.T.; Schwartzman, A.; Giobbie-Hurder, A.; Neuberg, D.; Gordon, L.B. Mechanisms of premature vascular aging in children with hutchinson-gilford progeria syndrome. Hypertension 2012, 59, 92–97. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.J.; Coffinier, C.; Choe, Y.; Beigneux, A.P.; Davies, B.S.; Yang, S.H.; Barnes, R.H., 2nd; Hong, J.; Sun, T.; Pleasure, S.J.; et al. Regulation of prelamin a but not lamin c by mir-9, a brain-specific microrna. Proc. Natl. Acad. Sci. USA 2012, 109, E423–E431. [Google Scholar] [CrossRef] [Green Version]
- Kieran, M.W.; Gordon, L.; Kleinman, M. New approaches to progeria. Pediatrics 2007, 120, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Gordon, L.B.; McCarten, K.M.; Giobbie-Hurder, A.; Machan, J.T.; Campbell, S.E.; Berns, S.D.; Kieran, M.W. Disease progression in hutchinson-gilford progeria syndrome: Impact on growth and development. Pediatrics 2007, 120, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Gordon, L.B.; Campbell, S.E.; Massaro, J.M.; D’Agostino, R.B., Sr.; Kleinman, M.E.; Kieran, M.W.; Moses, M.A. Survey of plasma proteins in children with progeria pre-therapy and on-therapy with lonafarnib. Pediatr. Res. 2018, 83, 982–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, K.K.; Park, S.M.; Quon, M.J. Leptin and cardiovascular disease: Response to therapeutic interventions. Circulation 2008, 117, 3238–3249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piemonti, L.; Calori, G.; Mercalli, A.; Lattuada, G.; Monti, P.; Garancini, M.P.; Costantino, F.; Ruotolo, G.; Luzi, L.; Perseghin, G. Fasting plasma leptin, tumor necrosis factor-alpha receptor 2, and monocyte chemoattracting protein 1 concentration in a population of glucose-tolerant and glucose-intolerant women: Impact on cardiovascular mortality. Diabetes Care 2003, 26, 2883–2889. [Google Scholar] [CrossRef] [Green Version]
- Gordon, L.B.; Brown, W.T.; Collins, F.S. Hutchinson-gilford progeria syndrome. In Genereviews((r)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Gast, K.B.; Tjeerdema, N.; Stijnen, T.; Smit, J.W.; Dekkers, O.M. Insulin resistance and risk of incident cardiovascular events in adults without diabetes: Meta-analysis. PLoS ONE 2012, 7, e52036. [Google Scholar] [CrossRef] [Green Version]
- Ormazabal, V.; Nair, S.; Elfeky, O.; Aguayo, C.; Salomon, C.; Zuniga, F.A. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc. Diabetol. 2018, 17, 122. [Google Scholar] [CrossRef]
- Greer, M.M.; Kleinman, M.E.; Gordon, L.B.; Massaro, J.; D’Agostino, R.B., Sr.; Baltrusaitis, K.; Kieran, M.W.; Gordon, C.M. Pubertal progression in female adolescents with progeria. J. Pediatr. Adolesc. Gynecol. 2018, 31, 238–241. [Google Scholar] [CrossRef]
- Iorga, A.; Cunningham, C.M.; Moazeni, S.; Ruffenach, G.; Umar, S.; Eghbali, M. The protective role of estrogen and estrogen receptors in cardiovascular disease and the controversial use of estrogen therapy. Biol. Sex Differ. 2017, 8, 33. [Google Scholar] [CrossRef]
- Rivera-Torres, J.; Acin-Perez, R.; Cabezas-Sanchez, P.; Osorio, F.G.; Gonzalez-Gomez, C.; Megias, D.; Camara, C.; Lopez-Otin, C.; Enriquez, J.A.; Luque-Garcia, J.L.; et al. Identification of mitochondrial dysfunction in hutchinson-gilford progeria syndrome through use of stable isotope labeling with amino acids in cell culture. J. Proteom. 2013, 91, 466–477. [Google Scholar] [CrossRef]
- Gabriel, D.; Gordon, L.B.; Djabali, K. Temsirolimus partially rescues the hutchinson-gilford progeria cellular phenotype. PLoS ONE 2016, 11, e0168988. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, F.; Evangelisti, C.; Cenni, V.; Fazio, A.; Paganelli, F.; Martelli, A.M.; Lattanzi, G. The cutting edge: The role of mtor signaling in laminopathies. Int. J. Mol. Sci. 2019, 20, 847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimobayashi, M.; Hall, M.N. Making new contacts: The mtor network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014, 15, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. Mtor signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.W.; Guo, Z.J.; Chu, A.L.; Zhang, Y.; Liang, B.; Guo, X.; Chai, T.; Song, R.; Hou, G.; Yuan, J.J. High incidence of coding gene mutations in mitochondrial DNA in esophageal cancer. Mol. Med. Rep. 2017, 16, 8537–8541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, K.; Graziotto, J.J.; Blair, C.D.; Mazzulli, J.R.; Erdos, M.R.; Krainc, D.; Collins, F.S. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in hutchinson-gilford progeria syndrome cells. Sci. Transl. Med. 2011, 3, 89ra58. [Google Scholar] [CrossRef]
- Pellegrini, C.; Columbaro, M.; Capanni, C.; D’Apice, M.R.; Cavallo, C.; Murdocca, M.; Lattanzi, G.; Squarzoni, S. All-trans retinoic acid and rapamycin normalize hutchinson gilford progeria fibroblast phenotype. Oncotarget 2015, 6, 29914–29928. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, Y.; Hambright, W.S.; Takayama, K.; Mu, X.; Lu, A.; Cummins, J.H.; Matsumoto, T.; Yurube, T.; Kuroda, R.; Kurosaka, M.; et al. Rapamycin rescues age-related changes in muscle-derived stem/progenitor cells from progeroid mice. Mol. Ther. Methods Clin. Dev. 2019, 14, 64–76. [Google Scholar] [CrossRef] [Green Version]
- Takayama, K.; Kawakami, Y.; Lavasani, M.; Mu, X.; Cummins, J.H.; Yurube, T.; Kuroda, R.; Kurosaka, M.; Fu, F.H.; Robbins, P.D.; et al. Mtor signaling plays a critical role in the defects observed in muscle-derived stem/progenitor cells isolated from a murine model of accelerated aging. J. Orthop. Res. 2017, 35, 1375–1382. [Google Scholar] [CrossRef] [Green Version]
- Ramos, F.J.; Chen, S.C.; Garelick, M.G.; Dai, D.F.; Liao, C.Y.; Schreiber, K.H.; MacKay, V.L.; An, E.H.; Strong, R.; Ladiges, W.C.; et al. Rapamycin reverses elevated mtorc1 signaling in lamin a/c-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci. Transl. Med. 2012, 4, 144ra103. [Google Scholar] [CrossRef] [Green Version]
- Buchwalter, A.; Hetzer, M.W. Nucleolar expansion and elevated protein translation in premature aging. Nat. Commun. 2017, 8, 328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evangelisti, C.; Cenni, V.; Lattanzi, G. Potential therapeutic effects of the mtor inhibitors for preventing ageing and progeria-related disorders. Br. J. Clin. Pharmacol. 2016, 82, 1229–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa-Bellosta, R.; Rivera-Torres, J.; Osorio, F.G.; Acin-Perez, R.; Enriquez, J.A.; Lopez-Otin, C.; Andres, V. Defective extracellular pyrophosphate metabolism promotes vascular calcification in a mouse model of hutchinson-gilford progeria syndrome that is ameliorated on pyrophosphate treatment. Circulation 2013, 127, 2442–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Mejia, I.C.; de Toledo, M.; Chavey, C.; Lapasset, L.; Cavelier, P.; Lopez-Herrera, C.; Chebli, K.; Fort, P.; Beranger, G.; Fajas, L.; et al. Antagonistic functions of lmna isoforms in energy expenditure and lifespan. EMBO Rep. 2014, 15, 529–539. [Google Scholar] [CrossRef] [Green Version]
- Viteri, G.; Chung, Y.W.; Stadtman, E.R. Effect of progerin on the accumulation of oxidized proteins in fibroblasts from hutchinson gilford progeria patients. Mech. Ageing Dev. 2010, 131, 2–8. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Z.M.; Choi, J.Y.; Wang, K.; Zhang, H.; Tariq, Z.; Wu, D.; Ko, E.; LaDana, C.; Sesaki, H.; Cao, K. Methylene blue alleviates nuclear and mitochondrial abnormalities in progeria. Aging Cell 2016, 15, 279–290. [Google Scholar] [CrossRef]
- Kubben, N.; Zhang, W.; Wang, L.; Voss, T.C.; Yang, J.; Qu, J.; Liu, G.H.; Misteli, T. Repression of the antioxidant nrf2 pathway in premature aging. Cell 2016, 165, 1361–1374. [Google Scholar] [CrossRef] [Green Version]
- Kubben, N.; Misteli, T. Shared molecular and cellular mechanisms of premature ageing and ageing-associated diseases. Nat. Rev. Mol. Cell Biol. 2017, 18, 595–609. [Google Scholar] [CrossRef]
- Nogueiras, R.; Habegger, K.M.; Chaudhary, N.; Finan, B.; Banks, A.S.; Dietrich, M.O.; Horvath, T.L.; Sinclair, D.A.; Pfluger, P.T.; Tschop, M.H. Sirtuin 1 and sirtuin 3: Physiological modulators of metabolism. Physiol. Rev. 2012, 92, 1479–1514. [Google Scholar] [CrossRef] [Green Version]
- Zhong, L.; D’Urso, A.; Toiber, D.; Sebastian, C.; Henry, R.E.; Vadysirisack, D.D.; Guimaraes, A.; Marinelli, B.; Wikstrom, J.D.; Nir, T.; et al. The histone deacetylase sirt6 regulates glucose homeostasis via HIF1alpha. Cell 2010, 140, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Ghosh, S.; Yang, X.; Zheng, H.; Liu, X.; Wang, Z.; Jin, G.; Zheng, B.; Kennedy, B.K.; Suh, Y.; et al. Resveratrol rescues sirt1-dependent adult stem cell decline and alleviates progeroid features in laminopathy-based progeria. Cell Metab. 2012, 16, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Mostoslavsky, R.; Chua, K.F.; Lombard, D.B.; Pang, W.W.; Fischer, M.R.; Gellon, L.; Liu, P.; Mostoslavsky, G.; Franco, S.; Murphy, M.M.; et al. Genomic instability and aging-like phenotype in the absence of mammalian sirt6. Cell 2006, 124, 315–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endisha, H.; Merrill-Schools, J.; Zhao, M.; Bristol, M.; Wang, X.; Kubben, N.; Elmore, L.W. Restoring sirt6 expression in hutchinson-gilford progeria syndrome cells impedes premature senescence and formation of dysmorphic nuclei. Pathobiology 2015, 82, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barlow, C.; Hirotsune, S.; Paylor, R.; Liyanage, M.; Eckhaus, M.; Collins, F.; Shiloh, Y.; Crawley, J.N.; Ried, T.; Tagle, D.; et al. Atm-deficient mice: A paradigm of ataxia telangiectasia. Cell 1996, 86, 159–171. [Google Scholar] [CrossRef] [Green Version]
- Lenaz, G.; D’Aurelio, M.; Merlo Pich, M.; Genova, M.L.; Ventura, B.; Bovina, C.; Formiggini, G.; Parenti Castelli, G. Mitochondrial bioenergetics in aging. Biochim. Biophys. Acta 2000, 1459, 397–404. [Google Scholar] [CrossRef] [Green Version]
- Qian, M.; Liu, Z.; Peng, L.; Tang, X.; Meng, F.; Ao, Y.; Zhou, M.; Wang, M.; Cao, X.; Qin, B.; et al. Boosting atm activity alleviates aging and extends lifespan in a mouse model of progeria. Elife 2018, 7. [Google Scholar] [CrossRef]
- Sullivan, T.; Escalante-Alcalde, D.; Bhatt, H.; Anver, M.; Bhat, N.; Nagashima, K.; Stewart, C.L.; Burke, B. Loss of a-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol. 1999, 147, 913–920. [Google Scholar] [CrossRef] [Green Version]
- Pendas, A.M.; Zhou, Z.; Cadinanos, J.; Freije, J.M.; Wang, J.; Hultenby, K.; Astudillo, A.; Wernerson, A.; Rodriguez, F.; Tryggvason, K.; et al. Defective prelamin a processing and muscular and adipocyte alterations in zmpste24 metalloproteinase-deficient mice. Nat. Genet. 2002, 31, 94–99. [Google Scholar] [CrossRef]
- Bergo, M.O.; Gavino, B.; Ross, J.; Schmidt, W.K.; Hong, C.; Kendall, L.V.; Mohr, A.; Meta, M.; Genant, H.; Jiang, Y.; et al. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin a processing defect. Proc. Natl. Acad. Sci. USA 2002, 99, 13049–13054. [Google Scholar] [CrossRef] [Green Version]
- Varga, R.; Eriksson, M.; Erdos, M.R.; Olive, M.; Harten, I.; Kolodgie, F.; Capell, B.C.; Cheng, J.; Faddah, D.; Perkins, S.; et al. Progressive vascular smooth muscle cell defects in a mouse model of hutchinson-gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 3250–3255. [Google Scholar] [CrossRef] [Green Version]
- Osorio, F.G.; Navarro, C.L.; Cadinanos, J.; Lopez-Mejia, I.C.; Quiros, P.M.; Bartoli, C.; Rivera, J.; Tazi, J.; Guzman, G.; Varela, I.; et al. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci. Transl. Med. 2011, 3, 106ra107. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, L.; Sanchez-Lopez, A.; Salaices, M.; von Kleeck, R.A.; Exposito, E.; Gonzalez-Gomez, C.; Cusso, L.; Guzman-Martinez, G.; Ruiz-Cabello, J.; Desco, M.; et al. Vascular smooth muscle cell-specific progerin expression in a mouse model of hutchinson-gilford progeria syndrome promotes arterial stiffness: Therapeutic effect of dietary nitrite. Aging Cell 2019, 18, e12936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emini Veseli, B.; Perrotta, P.; De Meyer, G.R.A.; Roth, L.; Van der Donckt, C.; Martinet, W.; De Meyer, G.R.Y. Animal models of atherosclerosis. Eur. J. Pharmacol. 2017, 816, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Hamczyk, M.R.; Villa-Bellosta, R.; Quesada, V.; Gonzalo, P.; Vidak, S.; Nevado, R.M.; Andres-Manzano, M.J.; Misteli, T.; Lopez-Otin, C.; Andres, V. Progerin accelerates atherosclerosis by inducing endoplasmic reticulum stress in vascular smooth muscle cells. EMBO Mol. Med. 2019, 11. [Google Scholar] [CrossRef]
- Hamczyk, M.R.; Villa-Bellosta, R.; Gonzalo, P.; Andres-Manzano, M.J.; Nogales, P.; Bentzon, J.F.; Lopez-Otin, C.; Andres, V. Vascular smooth muscle-specific progerin expression accelerates atherosclerosis and death in a mouse model of hutchinson-gilford progeria syndrome. Circulation 2018, 138, 266–282. [Google Scholar] [CrossRef]
- Osmanagic-Myers, S.; Kiss, A.; Manakanatas, C.; Hamza, O.; Sedlmayer, F.; Szabo, P.L.; Fischer, I.; Fichtinger, P.; Podesser, B.K.; Eriksson, M.; et al. Endothelial progerin expression causes cardiovascular pathology through an impaired mechanoresponse. J. Clin. Investig. 2019, 129, 531–545. [Google Scholar] [CrossRef] [Green Version]
- Chow, E.; Bernjak, A.; Williams, S.; Fawdry, R.A.; Hibbert, S.; Freeman, J.; Sheridan, P.J.; Heller, S.R. Risk of cardiac arrhythmias during hypoglycemia in patients with type 2 diabetes and cardiovascular risk. Diabetes 2014, 63, 1738–1747. [Google Scholar] [CrossRef] [Green Version]
- Marques, J.L.; George, E.; Peacey, S.R.; Harris, N.D.; Macdonald, I.A.; Cochrane, T.; Heller, S.R. Altered ventricular repolarization during hypoglycaemia in patients with diabetes. Diabet. Med. 1997, 14, 648–654. [Google Scholar] [CrossRef]
- Kreienkamp, R.; Gonzalo, S. Hutchinson-gilford progeria syndrome: Challenges at bench and bedside. Subcell Biochem. 2019, 91, 435–451. [Google Scholar]
- Kreienkamp, R.; Billon, C.; Bedia-Diaz, G.; Albert, C.J.; Toth, Z.; Butler, A.A.; McBride-Gagyi, S.; Ford, D.A.; Baldan, A.; Burris, T.P.; et al. Doubled lifespan and patient-like pathologies in progeria mice fed high-fat diet. Aging Cell 2018, e12852. [Google Scholar] [CrossRef] [Green Version]
- Ocampo, A.; Reddy, P.; Martinez-Redondo, P.; Platero-Luengo, A.; Hatanaka, F.; Hishida, T.; Li, M.; Lam, D.; Kurita, M.; Beyret, E.; et al. In vivo amelioration of age-associated hallmarks by partial reprogramming. Cell 2016, 167, 1719–1733 e1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balmus, G.; Larrieu, D.; Barros, A.C.; Collins, C.; Abrudan, M.; Demir, M.; Geisler, N.J.; Lelliott, C.J.; White, J.K.; Karp, N.A.; et al. Targeting of nat10 enhances healthspan in a mouse model of human accelerated aging syndrome. Nat. Commun. 2018, 9, 1700. [Google Scholar] [CrossRef] [PubMed]
- Barcena, C.; Quiros, P.M.; Durand, S.; Mayoral, P.; Rodriguez, F.; Caravia, X.M.; Marino, G.; Garabaya, C.; Fernandez-Garcia, M.T.; Kroemer, G.; et al. Methionine restriction extends lifespan in progeroid mice and alters lipid and bile acid metabolism. Cell Rep. 2018, 24, 2392–2403. [Google Scholar] [CrossRef] [Green Version]
- Beyret, E.; Liao, H.K.; Yamamoto, M.; Hernandez-Benitez, R.; Fu, Y.; Erikson, G.; Reddy, P.; Izpisua Belmonte, J.C. Single-dose crispr-cas9 therapy extends lifespan of mice with hutchinson-gilford progeria syndrome. Nat. Med. 2019, 25, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Fernandez, O.; Osorio, F.G.; Quesada, V.; Rodriguez, F.; Basso, S.; Maeso, D.; Rolas, L.; Barkaway, A.; Nourshargh, S.; Folgueras, A.R.; et al. Development of a crispr/cas9-based therapy for hutchinson-gilford progeria syndrome. Nat. Med. 2019, 25, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Barcena, C.; Valdes-Mas, R.; Mayoral, P.; Garabaya, C.; Durand, S.; Rodriguez, F.; Fernandez-Garcia, M.T.; Salazar, N.; Nogacka, A.M.; Garatachea, N.; et al. Healthspan and lifespan extension by fecal microbiota transplantation into progeroid mice. Nat. Med. 2019, 25, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.A.; Buehner, G.; Chang, Y.; Harper, J.M.; Sigler, R.; Smith-Wheelock, M. Methionine-deficient diet extends mouse lifespan, slows immune and lens aging, alters glucose, t4, igf-i and insulin levels, and increases hepatocyte mif levels and stress resistance. Aging Cell 2005, 4, 119–125. [Google Scholar] [CrossRef]
- Goodrick, C.L.; Ingram, D.K.; Reynolds, M.A.; Freeman, J.R.; Cider, N. Effects of intermittent feeding upon body weight and lifespan in inbred mice: Interaction of genotype and age. Mech. Ageing Dev. 1990, 55, 69–87. [Google Scholar] [CrossRef]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a tool to target aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef] [Green Version]
- Zi, F.; Zi, H.; Li, Y.; He, J.; Shi, Q.; Cai, Z. Metformin and cancer: An existing drug for cancer prevention and therapy. Oncol. Lett. 2018, 15, 683–690. [Google Scholar] [CrossRef] [Green Version]
- Park, S.K.; Shin, O.S. Metformin alleviates ageing cellular phenotypes in hutchinson-gilford progeria syndrome dermal fibroblasts. Exp. Dermatol. 2017, 26, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Egesipe, A.L.; Blondel, S.; Lo Cicero, A.; Jaskowiak, A.L.; Navarro, C.; Sandre-Giovannoli, A.; Levy, N.; Peschanski, M.; Nissan, X. Metformin decreases progerin expression and alleviates pathological defects of hutchinson-gilford progeria syndrome cells. NPJ Aging Mech. Dis. 2016, 2, 16026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, L.B.; Kleinman, M.E.; Massaro, J.; D’Agostino, R.B., Sr.; Shappell, H.; Gerhard-Herman, M.; Smoot, L.B.; Gordon, C.M.; Cleveland, R.H.; Nazarian, A.; et al. Clinical trial of the protein farnesylation inhibitors lonafarnib, pravastatin, and zoledronic acid in children with hutchinson-gilford progeria syndrome. Circulation 2016, 134, 114–125. [Google Scholar] [CrossRef] [Green Version]
- Capell, B.C.; Erdos, M.R.; Madigan, J.P.; Fiordalisi, J.J.; Varga, R.; Conneely, K.N.; Gordon, L.B.; Der, C.J.; Cox, A.D.; Collins, F.S. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of hutchinson-gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 12879–12884. [Google Scholar] [CrossRef] [Green Version]
- Capell, B.C.; Olive, M.; Erdos, M.R.; Cao, K.; Faddah, D.A.; Tavarez, U.L.; Conneely, K.N.; Qu, X.; San, H.; Ganesh, S.K.; et al. A farnesyltransferase inhibitor prevents both the onset and late progression of cardiovascular disease in a progeria mouse model. Proc. Natl. Acad. Sci. USA 2008, 105, 15902–15907. [Google Scholar] [CrossRef] [Green Version]
- Mallampalli, M.P.; Huyer, G.; Bendale, P.; Gelb, M.H.; Michaelis, S. Inhibiting farnesylation reverses the nuclear morphology defect in a hela cell model for hutchinson-gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 14416–14421. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.H.; Bergo, M.O.; Toth, J.I.; Qiao, X.; Hu, Y.; Sandoval, S.; Meta, M.; Bendale, P.; Gelb, M.H.; Young, S.G.; et al. Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted hutchinson-gilford progeria syndrome mutation. Proc. Natl. Acad. Sci. USA 2005, 102, 10291–10296. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.H.; Meta, M.; Qiao, X.; Frost, D.; Bauch, J.; Coffinier, C.; Majumdar, S.; Bergo, M.O.; Young, S.G.; Fong, L.G. A farnesyltransferase inhibitor improves disease phenotypes in mice with a hutchinson-gilford progeria syndrome mutation. J. Clin. Investig. 2006, 116, 2115–2121. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, M.X.; Sayin, V.I.; Akula, M.K.; Liu, M.; Fong, L.G.; Young, S.G.; Bergo, M.O. Targeting isoprenylcysteine methylation ameliorates disease in a mouse model of progeria. Science 2013, 340, 1330–1333. [Google Scholar] [CrossRef] [Green Version]
- DuBose, A.J.; Lichtenstein, S.T.; Petrash, N.M.; Erdos, M.R.; Gordon, L.B.; Collins, F.S. Everolimus rescues multiple cellular defects in laminopathy-patient fibroblasts. Proc. Natl. Acad. Sci. USA 2018, 115, 4206–4211. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, D.; Roedl, D.; Gordon, L.B.; Djabali, K. Sulforaphane enhances progerin clearance in hutchinson-gilford progeria fibroblasts. Aging Cell 2015, 14, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, D.; Shafry, D.D.; Gordon, L.B.; Djabali, K. Intermittent treatment with farnesyltransferase inhibitor and sulforaphane improves cellular homeostasis in hutchinson-gilford progeria fibroblasts. Oncotarget 2017, 8, 64809–64826. [Google Scholar] [CrossRef] [PubMed]
- Kreienkamp, R.; Croke, M.; Neumann, M.A.; Bedia-Diaz, G.; Graziano, S.; Dusso, A.; Dorsett, D.; Carlberg, C.; Gonzalo, S. Vitamin d receptor signaling improves hutchinson-gilford progeria syndrome cellular phenotypes. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.M.; Nobumori, C.; Tu, Y.; Choi, C.; Yang, S.H.; Jung, H.J.; Vickers, T.A.; Rigo, F.; Bennett, C.F.; Young, S.G.; et al. Modulation of lmna splicing as a strategy to treat prelamin a diseases. J. Clin. Investig. 2016, 126, 1592–1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larrieu, D.; Britton, S.; Demir, M.; Rodriguez, R.; Jackson, S.P. Chemical inhibition of nat10 corrects defects of laminopathic cells. Science 2014, 344, 527–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, R.E.; Sears, D.D. Metabolic effects of intermittent fasting. Annu. Rev. Nutr. 2017, 37, 371–393. [Google Scholar] [CrossRef] [Green Version]
- Roberts, M.N.; Wallace, M.A.; Tomilov, A.A.; Zhou, Z.; Marcotte, G.R.; Tran, D.; Perez, G.; Gutierrez-Casado, E.; Koike, S.; Knotts, T.A.; et al. A ketogenic diet extends longevity and healthspan in adult mice. Cell Metab. 2017, 26, 539–546. [Google Scholar] [CrossRef] [Green Version]
- Perlman, R.L. Mouse models of human disease: An evolutionary perspective. Evol. Med. Public Health 2016, 2016, 170–176. [Google Scholar] [CrossRef] [Green Version]
- Ballinger, S.W.; Patterson, C.; Yan, C.N.; Doan, R.; Burow, D.L.; Young, C.G.; Yakes, F.M.; Van Houten, B.; Ballinger, C.A.; Freeman, B.A.; et al. Hydrogen peroxide- and peroxynitrite-induced mitochondrial DNA damage and dysfunction in vascular endothelial and smooth muscle cells. Circ. Res. 2000, 86, 960–966. [Google Scholar] [CrossRef]
- Docherty, C.K.; Carswell, A.; Friel, E.; Mercer, J.R. Impaired mitochondrial respiration in human carotid plaque atherosclerosis: A potential role for pink1 in vascular smooth muscle cell energetics. Atherosclerosis 2018, 268, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, P.; Scaffidi, P.; Markert, E.; Lee, J.H.; Rane, S.; Misteli, T. Transformation resistance in a premature aging disorder identifies a tumor-protective function of brd4. Cell Rep. 2014, 9, 248–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Treatment | Median Lifespan Untreated | Median Lifespan Treated | Percentage Increase | Max Lifespan | Citation |
|---|---|---|---|---|---|
| Antisense Oligonucleotides Against Progerin | 111 (Mean) | 155 (Mean) | 39.6% | 190 | (Osorio et al., 2011) [72] |
| Partial Reprogramming | 126 * | 168 * | 33.3% * | 205 * | (Ocampo et al., 2016) [82] |
| Remodelin | 80 * | 110 * | 37.5% * | 120 * | (Balmus et al., 2018) [83] |
| Methionine Restriction | 155 * | 180 * | 16.1% * | 225 * | (Bárcena et al., 2018) [84] |
| High-Fat Diet | 113 | 194 | 71.7% | 229 | (Kreienkamp et al., 2019) [80] |
| CRISPR-Cas9 | 128 (Mean) | 167 (Mean) | 30.4% | 212 | (Beyret et al., 2019) [85] |
| CRISPR-Cas9 | 140 * | 177 * | 26.4% | 224 * | (Santiago-fernández et al., 2019) [86] |
| Fecal Microbiota Transplantation | 141 | 160 | 13.5% | 184 | (Bárcena et al., 2019) [87] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kreienkamp, R.; Gonzalo, S. Metabolic Dysfunction in Hutchinson–Gilford Progeria Syndrome. Cells 2020, 9, 395. https://doi.org/10.3390/cells9020395
Kreienkamp R, Gonzalo S. Metabolic Dysfunction in Hutchinson–Gilford Progeria Syndrome. Cells. 2020; 9(2):395. https://doi.org/10.3390/cells9020395
Chicago/Turabian StyleKreienkamp, Ray, and Susana Gonzalo. 2020. "Metabolic Dysfunction in Hutchinson–Gilford Progeria Syndrome" Cells 9, no. 2: 395. https://doi.org/10.3390/cells9020395
APA StyleKreienkamp, R., & Gonzalo, S. (2020). Metabolic Dysfunction in Hutchinson–Gilford Progeria Syndrome. Cells, 9(2), 395. https://doi.org/10.3390/cells9020395