Oncolytic Virus Encoding a Master Pro-Inflammatory Cytokine Interleukin 12 in Cancer Immunotherapy


Abstract
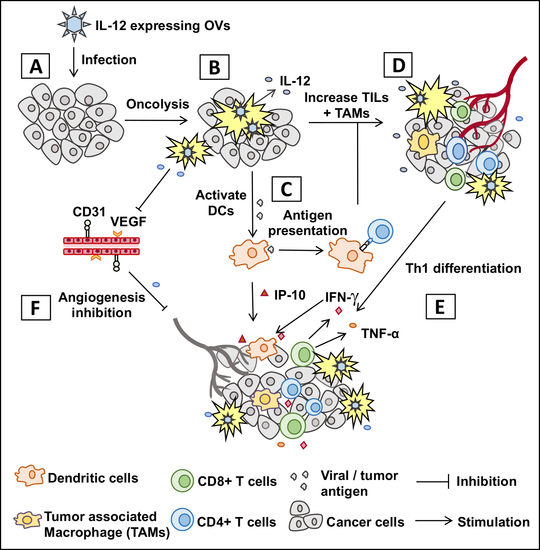
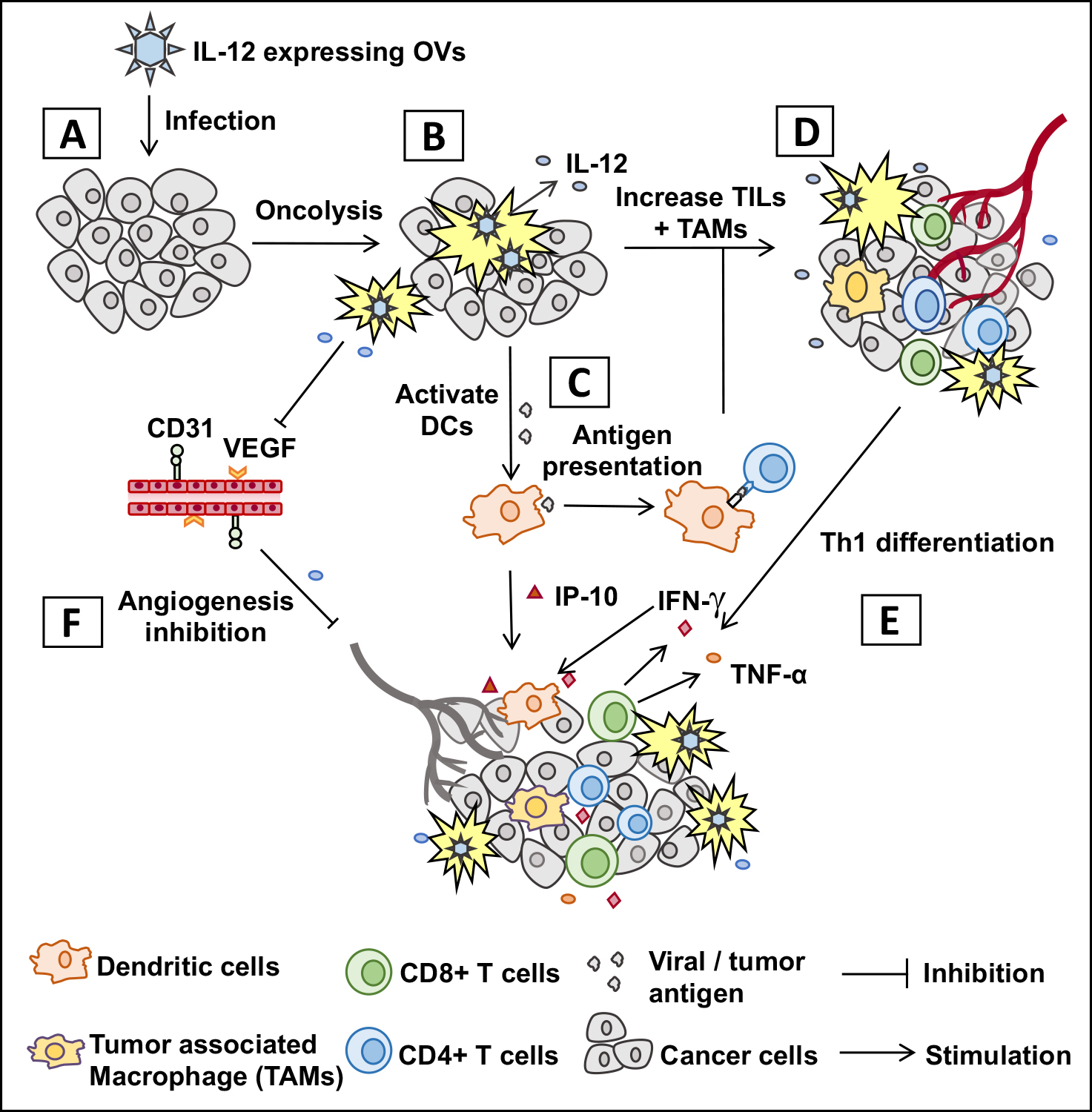
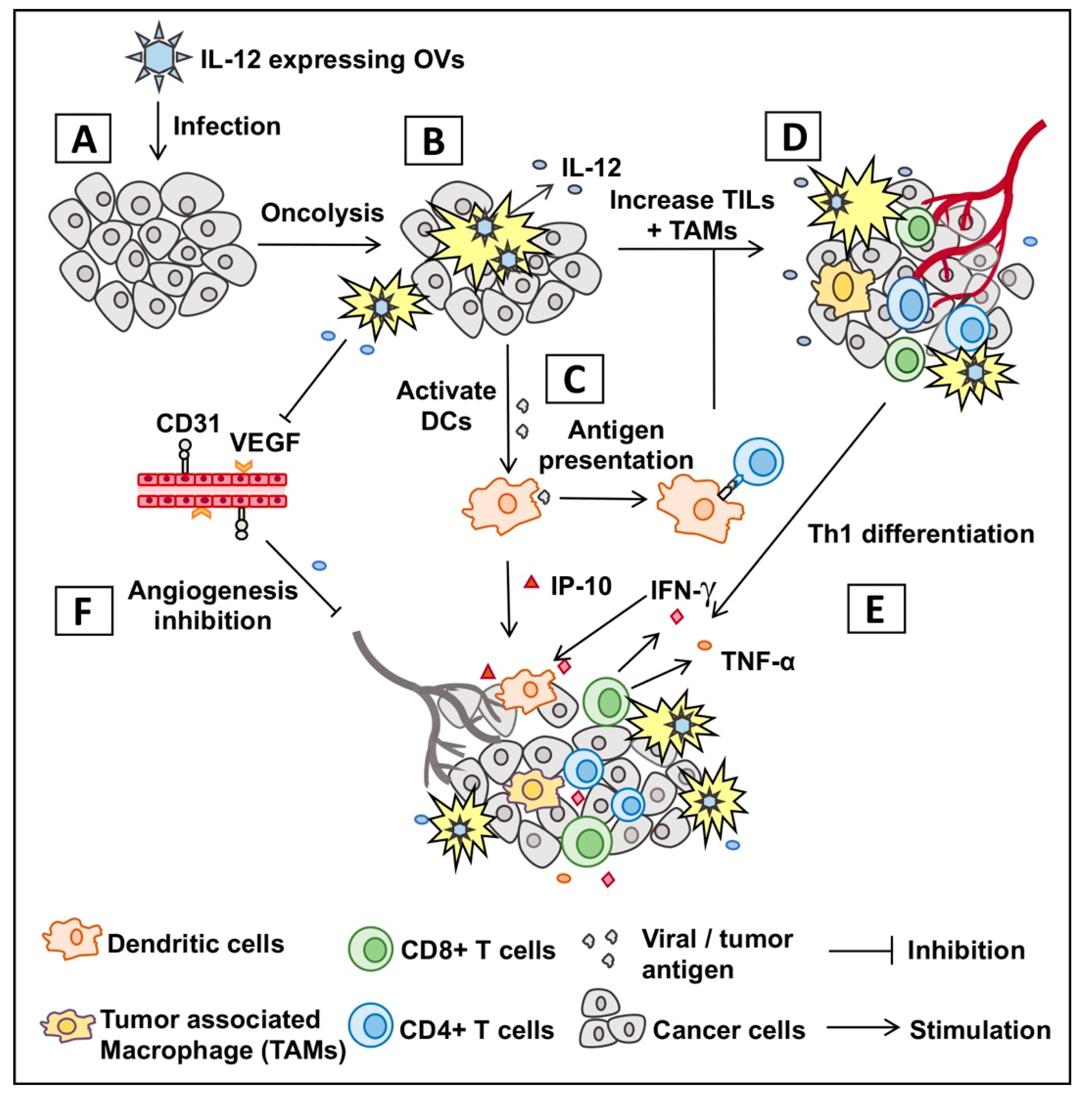
:
1. Introduction
2. Genetically Engineered IL-12 Expressing OHSVs and Their Therapeutic Efficacy
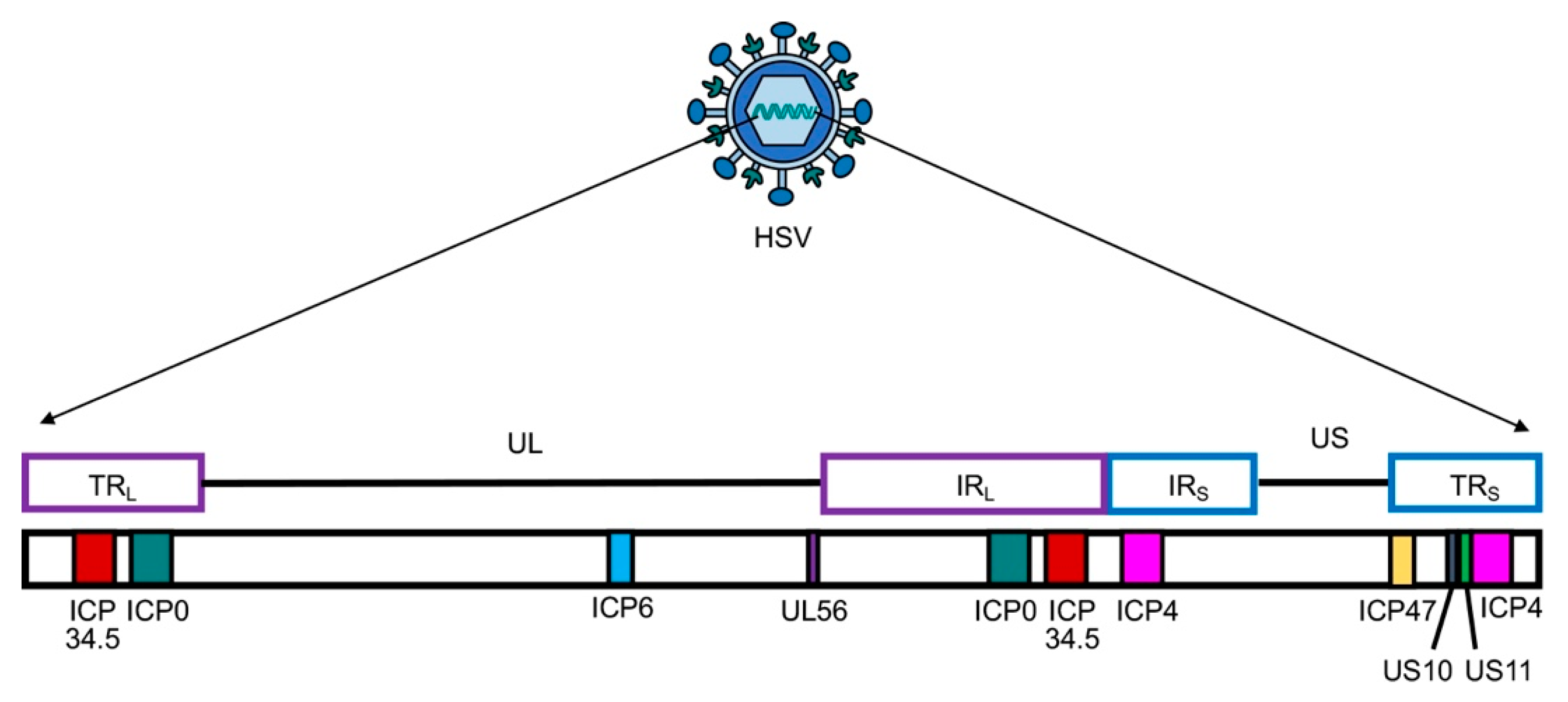
2.1. IL-12 Insertion Does Not Compromise Safety and Tumor Specificity of Genetically Modified OHSV-IL12 Viruses
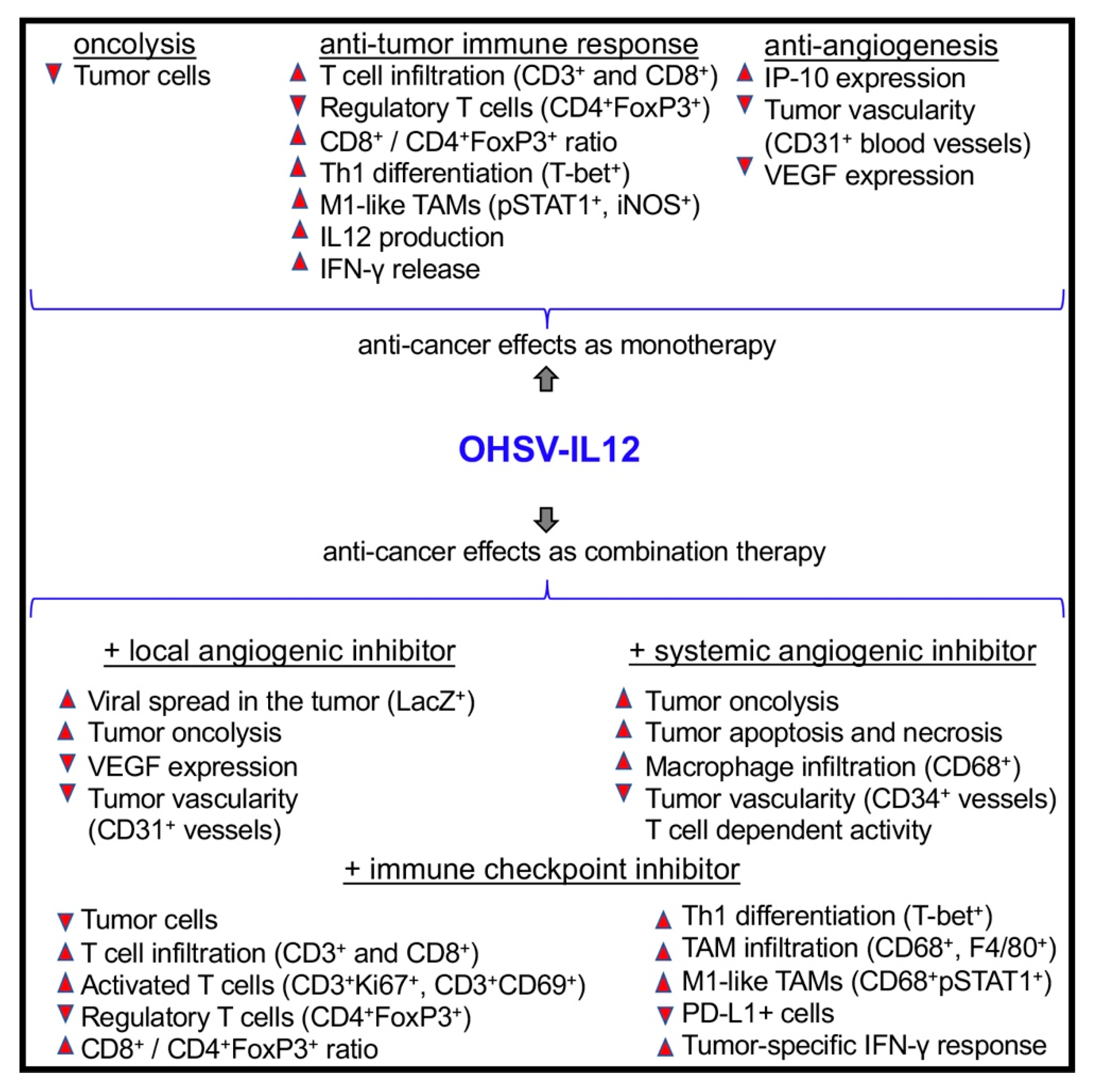
2.2. IL-12 Expressing OHSVs Produce Superior Anti-Tumor Immunity/Efficacy than Non-IL12 OHSVs
2.3. Anti-Tumor Anti-Vascular Effects of OHSV-IL12
2.4. Inhibition of Tumor Angiogenesis Enhances Anti-Tumor Potential of OHSV-IL12 Treatment
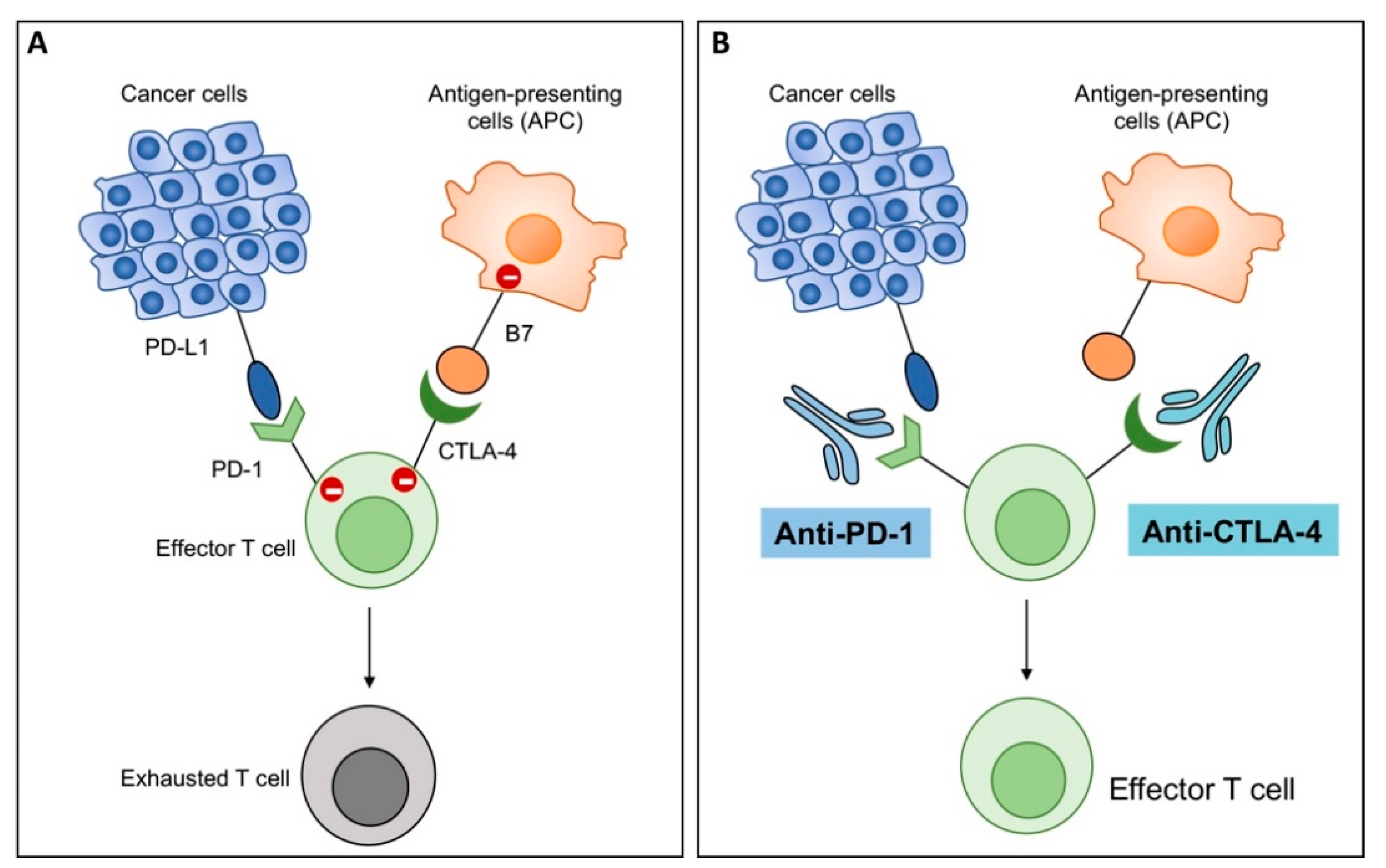
2.5. Immune Checkpoint Inhibition Enhances OHSV-IL12 Treatment-Induced Anti-Tumor Immunity
3. Anti-Cancer Potential of other OVs Encoding IL-12
4. Clinical Perspectives
5. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Berraondo, P.; Etxeberria, I.; Ponz-Sarvise, M.; Melero, I. Revisiting Interleukin-12 as a Cancer Immunotherapy Agent. Clin. Cancer Res. 2018, 24, 2716–2718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X. Impact of IL-12 in Cancer. Curr. Cancer Drug Targets 2017, 17, 682–697. [Google Scholar] [CrossRef] [PubMed]
- Zeh, H.J., 3rd; Hurd, S.; Storkus, W.J.; Lotze, M.T. Interleukin-12 promotes the proliferation and cytolytic maturation of immune effectors: Implications for the immunotherapy of cancer. J. Immunother. Emphasis Tumor Immunol. 1993, 14, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Lasek, W.; Zagozdzon, R.; Jakobisiak, M. Interleukin 12: Still a promising candidate for tumor immunotherapy? Cancer Immunol. Immunother. 2014, 63, 419–435. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, D.; Spanholtz, J.; Sturtzel, C.; Tordoir, M.; Schlechta, B.; Groenewegen, D.; Hofer, E. IL-12 directs further maturation of ex vivo differentiated NK cells with improved therapeutic potential. PLoS ONE 2014, 9, e87131. [Google Scholar] [CrossRef] [Green Version]
- Trinchieri, G.; Wysocka, M.; D’Andrea, A.; Rengaraju, M.; Aste-Amezaga, M.; Kubin, M.; Valiante, N.M.; Chehimi, J. Natural killer cell stimulatory factor (NKSF) or interleukin-12 is a key regulator of immune response and inflammation. Prog. Growth Factor Res. 1992, 4, 355–368. [Google Scholar] [CrossRef]
- Trinchieri, G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 2003, 3, 133–146. [Google Scholar] [CrossRef]
- Otani, T.; Nakamura, S.; Toki, M.; Motoda, R.; Kurimoto, M.; Orita, K. Identification of IFN-gamma-producing cells in IL-12/IL-18-treated mice. Cell Immunol. 1999, 198, 111–119. [Google Scholar] [CrossRef]
- Tugues, S.; Burkhard, S.H.; Ohs, I.; Vrohlings, M.; Nussbaum, K.; Vom Berg, J.; Kulig, P.; Becher, B. New insights into IL-12-mediated tumor suppression. Cell Death Differ. 2015, 22, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Angiolillo, A.L.; Sgadari, C.; Tosato, G. A role for the interferon-inducible protein 10 in inhibition of angiogenesis by interleukin-12. Ann. N. Y. Acad. Sci. 1996, 795, 158–167. [Google Scholar] [CrossRef]
- Lamont, A.G.; Adorini, L. IL-12: A key cytokine in immune regulation. Immunol. Today 1996, 17, 214–217. [Google Scholar] [CrossRef]
- Jenks, S. After initial setback, IL-12 regaining popularity. J. Natl. Cancer Inst. 1996, 88, 576–577. [Google Scholar] [CrossRef] [Green Version]
- Duvic, M.; Sherman, M.L.; Wood, G.S.; Kuzel, T.M.; Olsen, E.; Foss, F.; Laliberte, R.J.; Ryan, J.L.; Zonno, K.; Rook, A.H. A phase II open-label study of recombinant human interleukin-12 in patients with stage IA, IB, or IIA mycosis fungoides. J. Am. Acad. Dermatol. 2006, 55, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Rook, A.H.; Wood, G.S.; Yoo, E.K.; Elenitsas, R.; Kao, D.M.; Sherman, M.L.; Witmer, W.K.; Rockwell, K.A.; Shane, R.B.; Lessin, S.R.; et al. Interleukin-12 therapy of cutaneous T-cell lymphoma induces lesion regression and cytotoxic T-cell responses. Blood 1999, 94, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Little, R.F.; Pluda, J.M.; Wyvill, K.M.; Rodriguez-Chavez, I.R.; Tosato, G.; Catanzaro, A.T.; Steinberg, S.M.; Yarchoan, R. Activity of subcutaneous interleukin-12 in AIDS-related Kaposi sarcoma. Blood 2006, 107, 4650–4657. [Google Scholar] [CrossRef] [Green Version]
- Younes, A.; Pro, B.; Robertson, M.J.; Flinn, I.W.; Romaguera, J.E.; Hagemeister, F.; Dang, N.H.; Fiumara, P.; Loyer, E.M.; Cabanillas, F.F.; et al. Phase II clinical trial of interleukin-12 in patients with relapsed and refractory non-Hodgkin’s lymphoma and Hodgkin’s disease. Clin. Cancer Res. 2004, 10, 5432–5438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Vecchio, M.; Bajetta, E.; Canova, S.; Lotze, M.T.; Wesa, A.; Parmiani, G.; Anichini, A. Interleukin-12: Biological properties and clinical application. Clin. Cancer Res. 2007, 13, 4677–4685. [Google Scholar] [CrossRef] [Green Version]
- Poutou, J.; Bunuales, M.; Gonzalez-Aparicio, M.; Garcia-Aragoncillo, E.; Quetglas, J.I.; Casado, R.; Bravo-Perez, C.; Alzuguren, P.; Hernandez-Alcoceba, R. Safety and antitumor effect of oncolytic and helper-dependent adenoviruses expressing interleukin-12 variants in a hamster pancreatic cancer model. Gene Ther. 2015, 22, 696–706. [Google Scholar] [CrossRef] [Green Version]
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell 2017, 32, 253–267.e255. [Google Scholar] [CrossRef] [Green Version]
- Veinalde, R.; Grossardt, C.; Hartmann, L.; Bourgeois-Daigneault, M.C.; Bell, J.C.; Jager, D.; von Kalle, C.; Ungerechts, G.; Engeland, C.E. Oncolytic measles virus encoding interleukin-12 mediates potent antitumor effects through T cell activation. Oncoimmunology 2017, 6, e1285992. [Google Scholar] [CrossRef] [Green Version]
- Roth, J.C.; Cassady, K.A.; Cody, J.J.; Parker, J.N.; Price, K.H.; Coleman, J.M.; Peggins, J.O.; Noker, P.E.; Powers, N.W.; Grimes, S.D.; et al. Evaluation of the safety and biodistribution of M032, an attenuated herpes simplex virus type 1 expressing hIL-12, after intracerebral administration to aotus nonhuman primates. Hum. Gene Ther. Clin Dev. 2014, 25, 16–27. [Google Scholar] [CrossRef] [Green Version]
- Saha, D.; Ahmed, S.S.; Rabkin, S.D. Exploring the Antitumor Effect of Virus in Malignant Glioma. Drugs Future 2015, 40, 739–749. [Google Scholar] [CrossRef] [Green Version]
- Saha, D.; Martuza, R.L.; Curry, W.T. Viral oncolysis of glioblastoma. In Neurotropic viral infections, 2nd ed.; Reiss, C.S., Ed.; Springer: New York, NY, USA, 2016; Volume 2, pp. 481–517. [Google Scholar]
- Saha, D.; Wakimoto, H.; Rabkin, S.D. Oncolytic herpes simplex virus interactions with the host immune system. Curr. Opin. Virol. 2016, 21, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Oncolytic herpes simplex virus immunovirotherapy in combination with immune checkpoint blockade to treat glioblastoma. Immunotherapy 2018, 10, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Curing glioblastoma: Oncolytic HSV-IL12 and checkpoint blockade. Oncoscience 2017, 4, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Bommareddy, P.K. Two roads for oncolytic immunotherapy development. J. Immunother. Cancer 2019, 7, 26. [Google Scholar] [CrossRef] [Green Version]
- Raja, J.; Ludwig, J.M.; Gettinger, S.N.; Schalper, K.A.; Kim, H.S. Oncolytic virus immunotherapy: Future prospects for oncology. J. Immunother. Cancer 2018, 6, 140. [Google Scholar] [CrossRef]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Cheema, T.A.; Wakimoto, H.; Fecci, P.E.; Ning, J.; Kuroda, T.; Jeyaretna, D.S.; Martuza, R.L.; Rabkin, S.D. Multifaceted oncolytic virus therapy for glioblastoma in an immunocompetent cancer stem cell model. Proc. Natl. Acad. Sci. USA 2013, 110, 12006–12011. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.; Ino, Y.; Fukuhara, H.; Todo, T. Antitumor Efficacy of Intravenous Administration of Oncolytic Herpes Simplex Virus Expressing Interleukin 12. Mol. Ther. 2006, 13, S108. [Google Scholar] [CrossRef]
- Wong, R.J.; Patel, S.G.; Kim, S.; DeMatteo, R.P.; Malhotra, S.; Bennett, J.J.; St-Louis, M.; Shah, J.P.; Johnson, P.A.; Fong, Y. Cytokine gene transfer enhances herpes oncolytic therapy in murine squamous cell carcinoma. Hum. Gene. Ther. 2001, 12, 253–265. [Google Scholar] [CrossRef]
- Wong, R.J.; Chan, M.K.; Yu, Z.; Kim, T.H.; Bhargava, A.; Stiles, B.M.; Horsburgh, B.C.; Shah, J.P.; Ghossein, R.A.; Singh, B.; et al. Effective intravenous therapy of murine pulmonary metastases with an oncolytic herpes virus expressing interleukin 12. Clin. Cancer Res. 2004, 10, 251–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, G.K.; Bernstock, J.D.; Chen, D.; Nan, L.; Moore, B.P.; Kelly, V.M.; Youngblood, S.L.; Langford, C.P.; Han, X.; Ring, E.K.; et al. Enhanced Sensitivity of Patient-Derived Pediatric High-Grade Brain Tumor Xenografts to Oncolytic HSV-1 Virotherapy Correlates with Nectin-1 Expression. Sci. Rep. 2018, 8, 13930. [Google Scholar] [CrossRef] [PubMed]
- Cody, J.J.; Scaturro, P.; Cantor, A.B.; Yancey Gillespie, G.; Parker, J.N.; Markert, J.M. Preclinical evaluation of oncolytic deltagamma(1)34.5 herpes simplex virus expressing interleukin-12 for therapy of breast cancer brain metastases. Int. J. Breast Cancer 2012, 2012, 628697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillory, L.A.; Megison, M.L.; Stewart, J.E.; Mroczek-Musulman, E.; Nabers, H.C.; Waters, A.M.; Kelly, V.; Coleman, J.M.; Markert, J.M.; Gillespie, G.Y.; et al. Preclinical evaluation of engineered oncolytic herpes simplex virus for the treatment of neuroblastoma. PLoS ONE 2013, 8, e77753. [Google Scholar] [CrossRef] [PubMed]
- Megison, M.L.; Gillory, L.A.; Stewart, J.E.; Nabers, H.C.; Mroczek-Musulman, E.; Waters, A.M.; Coleman, J.M.; Kelly, V.; Markert, J.M.; Gillespie, G.Y.; et al. Preclinical evaluation of engineered oncolytic herpes simplex virus for the treatment of pediatric solid tumors. PLoS ONE 2014, 9, e86843. [Google Scholar] [CrossRef] [PubMed]
- Leoni, V.; Vannini, A.; Gatta, V.; Rambaldi, J.; Sanapo, M.; Barboni, C.; Zaghini, A.; Nanni, P.; Lollini, P.L.; Casiraghi, C.; et al. A fully-virulent retargeted oncolytic HSV armed with IL-12 elicits local immunity and vaccine therapy towards distant tumors. PLoS Pathog 2018, 14, e1007209. [Google Scholar] [CrossRef] [Green Version]
- Alessandrini, F.; Menotti, L.; Avitabile, E.; Appolloni, I.; Ceresa, D.; Marubbi, D.; Campadelli-Fiume, G.; Malatesta, P. Eradication of glioblastoma by immuno-virotherapy with a retargeted oncolytic HSV in a preclinical model. Oncogene 2019, 38, 4467–4479. [Google Scholar] [CrossRef]
- Ino, Y.; Saeki, Y.; Fukuhara, H.; Todo, T. Triple combination of oncolytic herpes simplex virus-1 vectors armed with interleukin-12, interleukin-18, or soluble B7-1 results in enhanced antitumor efficacy. Clin. Cancer Res. 2006, 12, 643–652. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Zhou, X.; Chen, X.; Liu, X.; Ma, J.; Wang, L.; Liu, Z.; Zhan, B.; Chen, H.; Wang, J.; et al. Enhancement of Oncolytic Activity of oHSV Expressing IL-12 and Anti PD-1 Antibody by Concurrent Administration of Exosomes Carrying CTLA-4 miRNA. Immunotherapy 2019, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Freytag, S.O.; Barton, K.N.; Zhang, Y. Efficacy of oncolytic adenovirus expressing suicide genes and interleukin-12 in preclinical model of prostate cancer. Gene Ther. 2013, 20, 1131–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freytag, S.O.; Zhang, Y.; Siddiqui, F. Preclinical toxicology of oncolytic adenovirus-mediated cytotoxic and interleukin-12 gene therapy for prostate cancer. Mol. Ther. Oncolytics. 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, X.; Wang, J.; Gao, D.; Li, Y.; Li, H.; Chu, Y.; Zhang, Z.; Liu, H.; Jiang, G.; et al. Re-designing Interleukin-12 to enhance its safety and potential as an anti-tumor immunotherapeutic agent. Nat. Commun. 2017, 8, 1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, K.J.; Zhang, S.N.; Choi, I.K.; Kim, J.S.; Yun, C.O. Strengthening of antitumor immune memory and prevention of thymic atrophy mediated by adenovirus expressing IL-12 and GM-CSF. Gene Ther. 2012, 19, 711–723. [Google Scholar] [CrossRef] [Green Version]
- Choi, I.K.; Lee, J.S.; Zhang, S.N.; Park, J.; Sonn, C.H.; Lee, K.M.; Yun, C.O. Oncolytic adenovirus co-expressing IL-12 and IL-18 improves tumor-specific immunity via differentiation of T cells expressing IL-12Rbeta2 or IL-18Ralpha. Gene Ther. 2011, 18, 898–909. [Google Scholar] [CrossRef] [Green Version]
- Oh, E.; Choi, I.K.; Hong, J.; Yun, C.O. Oncolytic adenovirus coexpressing interleukin-12 and decorin overcomes Treg-mediated immunosuppression inducing potent antitumor effects in a weakly immunogenic tumor model. Oncotarget 2017, 8, 4730–4746. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, J.H.; Choi, K.J.; Choi, I.K.; Kim, H.; Cho, S.; Cho, B.C.; Yun, C.O. Enhanced antitumor effect of oncolytic adenovirus expressing interleukin-12 and B7-1 in an immunocompetent murine model. Clin. Cancer Res. 2006, 12, 5859–5868. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.H.; Zhang, S.N.; Choi, K.J.; Choi, I.K.; Kim, J.H.; Lee, M.G.; Kim, H.; Yun, C.O. Therapeutic and tumor-specific immunity induced by combination of dendritic cells and oncolytic adenovirus expressing IL-12 and 4-1BBL. Mol. Ther. 2010, 18, 264–274. [Google Scholar] [CrossRef]
- Backhaus, P.S.; Veinalde, R.; Hartmann, L.; Dunder, J.E.; Jeworowski, L.M.; Albert, J.; Hoyler, B.; Poth, T.; Jager, D.; Ungerechts, G.; et al. Immunological Effects and Viral Gene Expression Determine the Efficacy of Oncolytic Measles Vaccines Encoding IL-12 or IL-15 Agonists. Viruses 2019, 11, 914. [Google Scholar] [CrossRef] [Green Version]
- Alkayyal, A.A.; Tai, L.H.; Kennedy, M.A.; de Souza, C.T.; Zhang, J.; Lefebvre, C.; Sahi, S.; Ananth, A.A.; Mahmoud, A.B.; Makrigiannis, A.P.; et al. NK-Cell Recruitment Is Necessary for Eradication of Peritoneal Carcinomatosis with an IL12-Expressing Maraba Virus Cellular Vaccine. Cancer Immunol. Res. 2017, 5, 211–221. [Google Scholar] [CrossRef] [Green Version]
- Ren, G.; Tian, G.; Liu, Y.; He, J.; Gao, X.; Yu, Y.; Liu, X.; Zhang, X.; Sun, T.; Liu, S.; et al. Recombinant Newcastle Disease Virus Encoding IL-12 and/or IL-2 as Potential Candidate for Hepatoma Carcinoma Therapy. Technol. Cancer Res. Treat. 2016, 15, NP83-94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asselin-Paturel, C.; Lassau, N.; Guinebretiere, J.M.; Zhang, J.; Gay, F.; Bex, F.; Hallez, S.; Leclere, J.; Peronneau, P.; Mami-Chouaib, F.; et al. Transfer of the murine interleukin-12 gene in vivo by a Semliki Forest virus vector induces B16 tumor regression through inhibition of tumor blood vessel formation monitored by Doppler ultrasonography. Gene Ther. 1999, 6, 606–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colmenero, P.; Chen, M.; Castanos-Velez, E.; Liljestrom, P.; Jondal, M. Immunotherapy with recombinant SFV-replicons expressing the P815A tumor antigen or IL-12 induces tumor regression. Int. J. Cancer 2002, 98, 554–560. [Google Scholar] [CrossRef] [Green Version]
- Melero, I.; Quetglas, J.I.; Reboredo, M.; Dubrot, J.; Rodriguez-Madoz, J.R.; Mancheno, U.; Casales, E.; Riezu-Boj, J.I.; Ruiz-Guillen, M.; Ochoa, M.C.; et al. Strict requirement for vector-induced type I interferon in efficacious antitumor responses to virally encoded IL12. Cancer Res. 2015, 75, 497–507. [Google Scholar] [CrossRef] [Green Version]
- Roche, F.P.; Sheahan, B.J.; O’Mara, S.M.; Atkins, G.J. Semliki Forest virus-mediated gene therapy of the RG2 rat glioma. Neuropathol. Appl. Neurobiol. 2010, 36, 648–660. [Google Scholar] [CrossRef]
- Shin, E.J.; Wanna, G.B.; Choi, B.; Aguila, D., 3rd; Ebert, O.; Genden, E.M.; Woo, S.L. Interleukin-12 expression enhances vesicular stomatitis virus oncolytic therapy in murine squamous cell carcinoma. Laryngoscope 2007, 117, 210–214. [Google Scholar] [CrossRef] [Green Version]
- Granot, T.; Venticinque, L.; Tseng, J.C.; Meruelo, D. Activation of cytotoxic and regulatory functions of NK cells by Sindbis viral vectors. PLoS ONE 2011, 6, e20598. [Google Scholar] [CrossRef] [Green Version]
- Peters, C.; Rabkin, S.D. Designing Herpes Viruses as Oncolytics. Mol Ther Oncolytics 2015, 2. [Google Scholar] [CrossRef]
- Todo, T.; Martuza, R.L.; Rabkin, S.D.; Johnson, P.A. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc. Natl. Acad. Sci. USA 2001, 98, 6396–6401. [Google Scholar] [CrossRef] [Green Version]
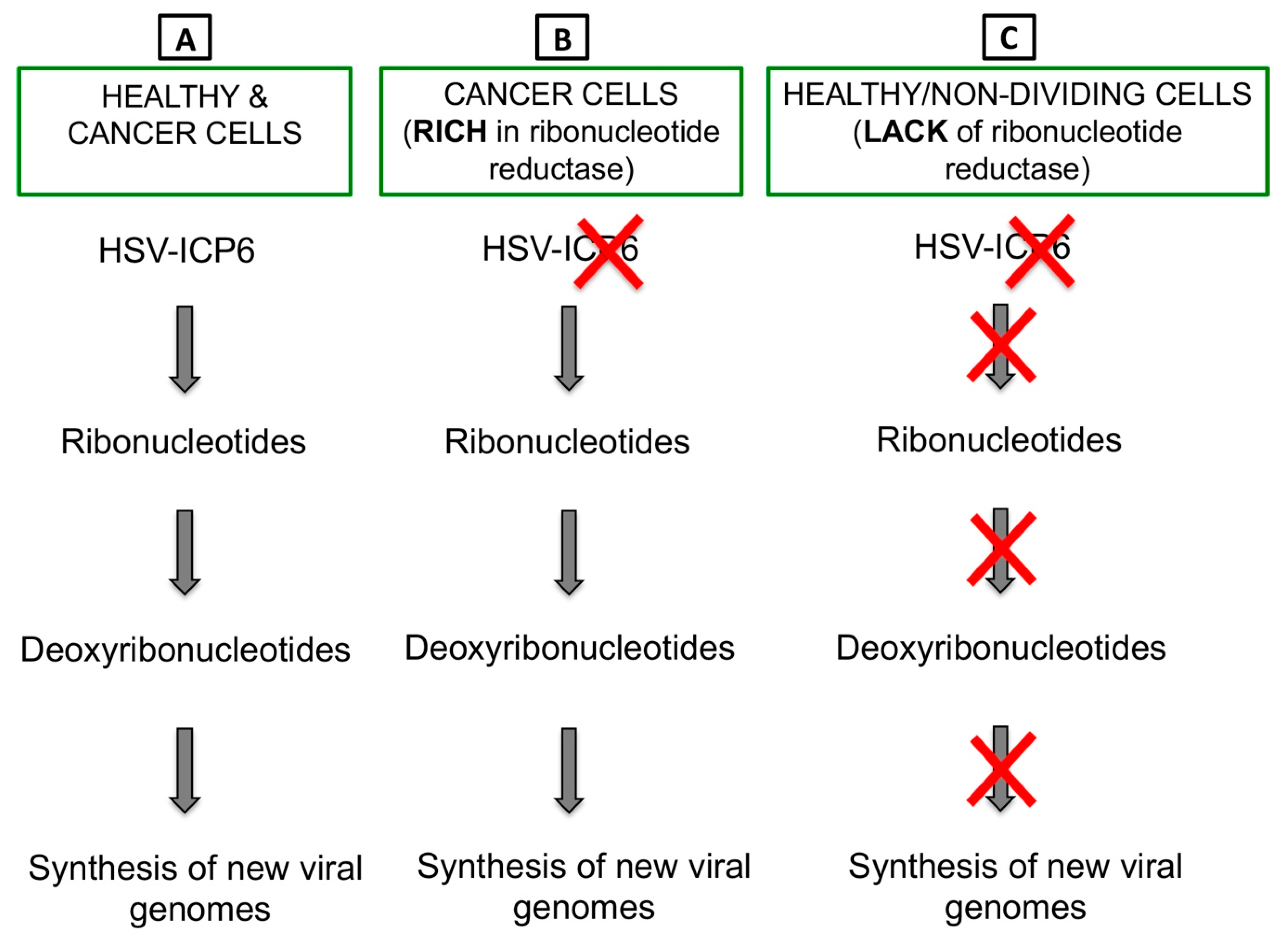
- Goldstein, D.J.; Weller, S.K. Factor(s) present in herpes simplex virus type 1-infected cells can compensate for the loss of the large subunit of the viral ribonucleotide reductase: characterization of an ICP6 deletion mutant. Virology 1988, 166, 41–51. [Google Scholar] [CrossRef]
- Aghi, M.; Visted, T.; Depinho, R.A.; Chiocca, E.A. Oncolytic herpes virus with defective ICP6 specifically replicates in quiescent cells with homozygous genetic mutations in p16. Oncogene 2008, 27, 4249–4254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, J.M.; McDougall, I.; Marsden, H.S.; Preston, V.G.; Ryan, D.M.; Subak-Sharpe, J.H. Ribonucleotide reductase encoded by herpes simplex virus is a determinant of the pathogenicity of the virus in mice and a valid antiviral target. J. Gen. Virol. 1988, 69, 2607–2612. [Google Scholar] [CrossRef] [PubMed]
- Markert, J.M.; Razdan, S.N.; Kuo, H.C.; Cantor, A.; Knoll, A.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Agee, B.S.; Coleman, J.M.; et al. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol. Ther. 2014, 22, 1048–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
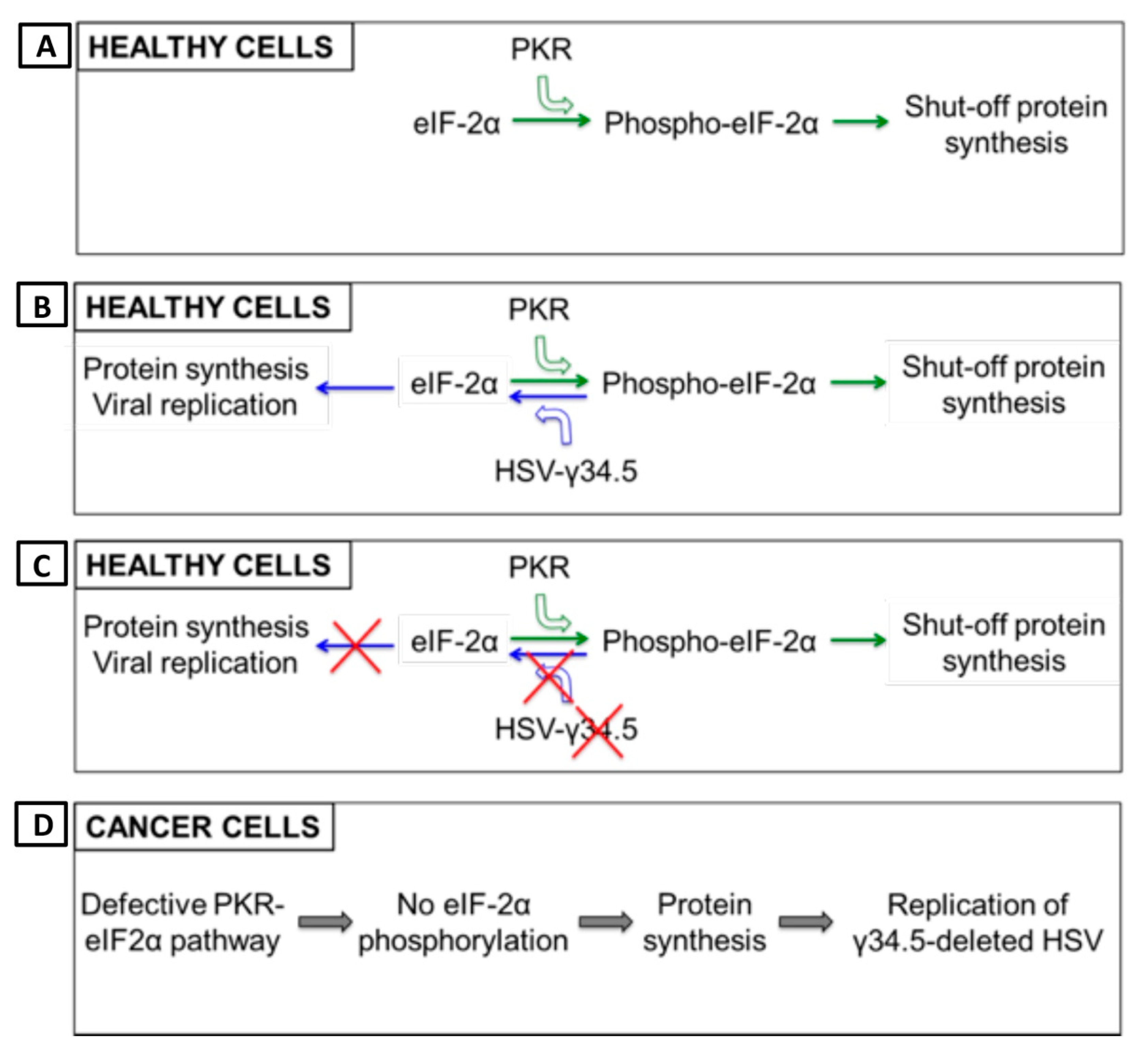
- Pasieka, T.J.; Baas, T.; Carter, V.S.; Proll, S.C.; Katze, M.G.; Leib, D.A. Functional genomic analysis of herpes simplex virus type 1 counteraction of the host innate response. J. Virol. 2006, 80, 7600–7612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wylie, K.M.; Schrimpf, J.E.; Morrison, L.A. Increased eIF2alpha phosphorylation attenuates replication of herpes simplex virus 2 vhs mutants in mouse embryonic fibroblasts and correlates with reduced accumulation of the PKR antagonist ICP34.5. J. Virol. 2009, 83, 9151–9162. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Gross, M.; Roizman, B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 843–848. [Google Scholar]
- Li, Y.; Zhang, C.; Chen, X.; Yu, J.; Wang, Y.; Yang, Y.; Du, M.; Jin, H.; Ma, Y.; He, B.; et al. ICP34.5 protein of herpes simplex virus facilitates the initiation of protein translation by bridging eukaryotic initiation factor 2alpha (eIF2alpha) and protein phosphatase 1. J. Biol. Chem. 2011, 286, 24785–24792. [Google Scholar] [CrossRef] [Green Version]
- Orr, M.T.; Edelmann, K.H.; Vieira, J.; Corey, L.; Raulet, D.H.; Wilson, C.B. Inhibition of MHC class I is a virulence factor in herpes simplex virus infection of mice. PLoS Pathog 2005, 1, e7. [Google Scholar] [CrossRef]
- Eggensperger, S.; Tampe, R. The transporter associated with antigen processing: A key player in adaptive immunity. Biol. Chem. 2015, 396, 1059–1072. [Google Scholar] [CrossRef]
- Goldsmith, K.; Chen, W.; Johnson, D.C.; Hendricks, R.L. Infected cell protein (ICP)47 enhances herpes simplex virus neurovirulence by blocking the CD8+ T cell response. J. Exp. Med. 1998, 187, 341–348. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.L.; Robinson, M.; Han, Z.Q.; Branston, R.H.; English, C.; Reay, P.; McGrath, Y.; Thomas, S.K.; Thornton, M.; Bullock, P.; et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003, 10, 292–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wollmann, G.; Ozduman, K.; van den Pol, A.N. Oncolytic virus therapy for glioblastoma multiforme: concepts and candidates. Cancer J. 2012, 18, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Mulvey, M.; Poppers, J.; Sternberg, D.; Mohr, I. Regulation of eIF2alpha phosphorylation by different functions that act during discrete phases in the herpes simplex virus type 1 life cycle. J. Virol. 2003, 77, 10917–10928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassady, K.A.; Gross, M.; Roizman, B. The herpes simplex virus US11 protein effectively compensates for the gamma1(34.5) gene if present before activation of protein kinase R by precluding its phosphorylation and that of the alpha subunit of eukaryotic translation initiation factor 2. J. Virol. 1998, 72, 8620–8626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, D.; Wakimoto, H.; Peters, C.W.; Antoszczyk, S.J.; Rabkin, S.D.; Martuza, R.L. Combinatorial effects of VEGFR kinase inhibitor axitinib and oncolytic virotherapy in mouse and human glioblastoma stem-like cell models. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todo, T. “Armed” oncolytic herpes simplex viruses for brain tumor therapy. Cell Adh. Migr. 2008, 2, 208–213. [Google Scholar] [CrossRef] [Green Version]
- Kelly, K.J.; Wong, J.; Fong, Y. Herpes simplex virus NV1020 as a novel and promising therapy for hepatic malignancy. Expert Opin. Investig. Drugs 2008, 17, 1105–1113. [Google Scholar] [CrossRef]
- Bennett, J.J.; Malhotra, S.; Wong, R.J.; Delman, K.; Zager, J.; St-Louis, M.; Johnson, P.; Fong, Y. Interleukin 12 secretion enhances antitumor efficacy of oncolytic herpes simplex viral therapy for colorectal cancer. Ann. Surg. 2001, 233, 819–826. [Google Scholar] [CrossRef]
- Smith, M.C.; Boutell, C.; Davido, D.J. HSV-1 ICP0: Paving the way for viral replication. Future Virol. 2011, 6, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Lanfranca, M.P.; Mostafa, H.H.; Davido, D.J. HSV-1 ICP0: An E3 Ubiquitin Ligase That Counteracts Host Intrinsic and Innate Immunity. Cells 2014, 3, 438–454. [Google Scholar] [CrossRef]
- Jacobs, A.; Breakefield, X.O.; Fraefel, C. HSV-1-based vectors for gene therapy of neurological diseases and brain tumors: Part I. HSV-1 structure, replication and pathogenesis. Neoplasia 1999, 1, 387–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, J.N.; Pfister, L.A.; Quenelle, D.; Gillespie, G.Y.; Markert, J.M.; Kern, E.R.; Whitley, R.J. Genetically engineered herpes simplex viruses that express IL-12 or GM-CSF as vaccine candidates. Vaccine 2006, 24, 1644–1652. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.N.; Gillespie, G.Y.; Love, C.E.; Randall, S.; Whitley, R.J.; Markert, J.M. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc. Natl. Acad. Sci. USA 2000, 97, 2208–2213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, D.F.; Pereboeva, L.; Gillespie, G.Y.; Cloud, G.A.; Elzafarany, O.; Langford, C.; Markert, J.M.; Lamb, L.S., Jr. Effect of HSV-IL12 Loaded Tumor Cell-Based Vaccination in a Mouse Model of High-Grade Neuroblastoma. J. Immunol. Res. 2016, 2016, 2568125. [Google Scholar] [CrossRef] [PubMed]
- Hellums, E.K.; Markert, J.M.; Parker, J.N.; He, B.; Perbal, B.; Roizman, B.; Whitley, R.J.; Langford, C.P.; Bharara, S.; Gillespie, G.Y. Increased efficacy of an interleukin-12-secreting herpes simplex virus in a syngeneic intracranial murine glioma model. Neuro. Oncol. 2005, 7, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Markert, J.M.; Cody, J.J.; Parker, J.N.; Coleman, J.M.; Price, K.H.; Kern, E.R.; Quenelle, D.C.; Lakeman, A.D.; Schoeb, T.R.; Palmer, C.A.; et al. Preclinical evaluation of a genetically engineered herpes simplex virus expressing interleukin-12. J. Virol. 2012, 86, 5304–5313. [Google Scholar] [CrossRef] [Green Version]
- Patel, D.M.; Foreman, P.M.; Nabors, L.B.; Riley, K.O.; Gillespie, G.Y.; Markert, J.M. Design of a Phase I Clinical Trial to Evaluate M032, a Genetically Engineered HSV-1 Expressing IL-12, in Patients with Recurrent/Progressive Glioblastoma Multiforme, Anaplastic Astrocytoma, or Gliosarcoma. Hum. Gene Ther. Clin. Dev. 2016, 27, 69–78. [Google Scholar] [CrossRef]
- Menotti, L.; Avitabile, E.; Gatta, V.; Malatesta, P.; Petrovic, B.; Campadelli-Fiume, G. HSV as A Platform for the Generation of Retargeted, Armed, and Reporter-Expressing Oncolytic Viruses. Viruses 2018, 10, 352. [Google Scholar] [CrossRef] [Green Version]
- Menotti, L.; Cerretani, A.; Hengel, H.; Campadelli-Fiume, G. Construction of a fully retargeted herpes simplex virus 1 recombinant capable of entering cells solely via human epidermal growth factor receptor 2. J. Virol. 2008, 82, 10153–10161. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.J.; Wang, X.Y.; Guo, J.X.; Zhu, H.J.; Zhang, C.R.; Ma, Z.H. Oncolytic property of HSV-1 recombinant viruses carrying the human IL-12. Zhonghua Yi Xue Za Zhi 2017, 97, 2135–2140. [Google Scholar] [CrossRef]
- Athie-Morales, V.; Smits, H.H.; Cantrell, D.A.; Hilkens, C.M. Sustained IL-12 signaling is required for Th1 development. J. Immunol. 2004, 172, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoszczyk, S.; Spyra, M.; Mautner, V.F.; Kurtz, A.; Stemmer-Rachamimov, A.O.; Martuza, R.L.; Rabkin, S.D. Treatment of orthotopic malignant peripheral nerve sheath tumors with oncolytic herpes simplex virus. Neuro. Oncol. 2014, 16, 1057–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varghese, S.; Rabkin, S.D.; Nielsen, G.P.; MacGarvey, U.; Liu, R.; Martuza, R.L. Systemic therapy of spontaneous prostate cancer in transgenic mice with oncolytic herpes simplex viruses. Cancer Res. 2007, 67, 9371–9379. [Google Scholar] [CrossRef] [Green Version]
- Ring, E.K.; Li, R.; Moore, B.P.; Nan, L.; Kelly, V.M.; Han, X.; Beierle, E.A.; Markert, J.M.; Leavenworth, J.W.; Gillespie, G.Y.; et al. Newly Characterized Murine Undifferentiated Sarcoma Models Sensitive to Virotherapy with Oncolytic HSV-1 M002. Mol. Ther. Oncolytics 2017, 7, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Thomas, E.D.; Meza-Perez, S.; Bevis, K.S.; Randall, T.D.; Gillespie, G.Y.; Langford, C.; Alvarez, R.D. IL-12 Expressing oncolytic herpes simplex virus promotes anti-tumor activity and immunologic control of metastatic ovarian cancer in mice. J. Ovarian Res. 2016, 9, 70. [Google Scholar] [CrossRef]
- Krist, L.F.; Kerremans, M.; Broekhuis-Fluitsma, D.M.; Eestermans, I.L.; Meyer, S.; Beelen, R.H. Milky spots in the greater omentum are predominant sites of local tumour cell proliferation and accumulation in the peritoneal cavity. Cancer Immunol. Immunother. 1998, 47, 205–212. [Google Scholar] [CrossRef]
- Sorensen, E.W.; Gerber, S.A.; Frelinger, J.G.; Lord, E.M. IL-12 suppresses vascular endothelial growth factor receptor 3 expression on tumor vessels by two distinct IFN-gamma-dependent mechanisms. J. Immunol. 2010, 184, 1858–1866. [Google Scholar] [CrossRef]
- Cicchelero, L.; Denies, S.; Haers, H.; Vanderperren, K.; Stock, E.; Van Brantegem, L.; de Rooster, H.; Sanders, N.N. Intratumoural interleukin 12 gene therapy stimulates the immune system and decreases angiogenesis in dogs with spontaneous cancer. Vet. Comp. Oncol. 2017, 15, 1187–1205. [Google Scholar] [CrossRef]
- Wong, R.J.; Chan, M.K.; Yu, Z.; Ghossein, R.A.; Ngai, I.; Adusumilli, P.S.; Stiles, B.M.; Shah, J.P.; Singh, B.; Fong, Y. Angiogenesis inhibition by an oncolytic herpes virus expressing interleukin 12. Clin. Cancer Res. 2004, 10, 4509–4516. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Marsden, P.A. Angiogenesis in glioblastoma. N Engl. J. Med. 2013, 369, 1561–1563. [Google Scholar] [CrossRef] [Green Version]
- Gatson, N.N.; Chiocca, E.A.; Kaur, B. Anti-angiogenic gene therapy in the treatment of malignant gliomas. Neurosci. Lett. 2012, 527, 62–70. [Google Scholar] [CrossRef] [Green Version]
- Hardee, M.E.; Zagzag, D. Mechanisms of glioma-associated neovascularization. Am. J. Pathol. 2012, 181, 1126–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, R.K.; di Tomaso, E.; Duda, D.G.; Loeffler, J.S.; Sorensen, A.G.; Batchelor, T.T. Angiogenesis in brain tumours. Nat. Rev. Neurosci. 2007, 8, 610–622. [Google Scholar] [CrossRef] [PubMed]
- Zirlik, K.; Duyster, J. Anti-Angiogenics: Current Situation and Future Perspectives. Oncol. Res. Treat. 2018, 41, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Ramjiawan, R.R.; Griffioen, A.W.; Duda, D.G. Anti-angiogenesis for cancer revisited: Is there a role for combinations with immunotherapy? Angiogenesis 2017, 20, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Rajabi, M.; Mousa, S.A. The Role of Angiogenesis in Cancer Treatment. Biomedicines 2017, 5, 34. [Google Scholar] [CrossRef] [Green Version]
- Melegh, Z.; Oltean, S. Targeting Angiogenesis in Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 2676. [Google Scholar] [CrossRef] [Green Version]
- van Moorselaar, R.J.; Voest, E.E. Angiogenesis in prostate cancer: Its role in disease progression and possible therapeutic approaches. Mol. Cell Endocrinol. 2002, 197, 239–250. [Google Scholar] [CrossRef]
- Batchelor, T.T.; Reardon, D.A.; de Groot, J.F.; Wick, W.; Weller, M. Antiangiogenic therapy for glioblastoma: Current status and future prospects. Clin. Cancer Res. 2014, 20, 5612–5619. [Google Scholar] [CrossRef] [Green Version]
- Wick, W.; Chinot, O.L.; Bendszus, M.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Revil, C.; Kerloeguen, Y.; Cloughesy, T. Evaluation of pseudoprogression rates and tumor progression patterns in a phase III trial of bevacizumab plus radiotherapy/temozolomide for newly diagnosed glioblastoma. Neuro. Oncol. 2016, 18, 1434–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardi, G.; Pambuku, A.; Bellu, L.; Farina, M.; Della Puppa, A.; Denaro, L.; Zagonel, V. Effectiveness of antiangiogenic drugs in glioblastoma patients: A systematic review and meta-analysis of randomized clinical trials. Crit. Rev. Oncol. Hematol. 2017, 111, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Kelly, W.K.; Halabi, S.; Carducci, M.; George, D.; Mahoney, J.F.; Stadler, W.M.; Morris, M.; Kantoff, P.; Monk, J.P.; Kaplan, E.; et al. Randomized, double-blind, placebo-controlled phase III trial comparing docetaxel and prednisone with or without bevacizumab in men with metastatic castration-resistant prostate cancer: CALGB 90401. J. Clin. Oncol. 2012, 30, 1534–1540. [Google Scholar] [CrossRef]
- Hu-Lowe, D.D.; Zou, H.Y.; Grazzini, M.L.; Hallin, M.E.; Wickman, G.R.; Amundson, K.; Chen, J.H.; Rewolinski, D.A.; Yamazaki, S.; Wu, E.Y.; et al. Nonclinical antiangiogenesis and antitumor activities of axitinib (AG-013736), an oral, potent, and selective inhibitor of vascular endothelial growth factor receptor tyrosine kinases 1, 2, 3. Clin. Cancer Res. 2008, 14, 7272–7283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, A.L.; Dunn, L.; Sherman, E.J.; Fury, M.G.; Baxi, S.S.; Chandramohan, R.; Dogan, S.; Morris, L.G.; Cullen, G.D.; Haque, S.; et al. A phase II study of axitinib (AG-013736) in patients with incurable adenoid cystic carcinoma. Ann. Oncol. 2016, 27, 1902–1908. [Google Scholar] [CrossRef] [PubMed]
- McNamara, M.G.; Le, L.W.; Horgan, A.M.; Aspinall, A.; Burak, K.W.; Dhani, N.; Chen, E.; Sinaei, M.; Lo, G.; Kim, T.K.; et al. A phase II trial of second-line axitinib following prior antiangiogenic therapy in advanced hepatocellular carcinoma. Cancer 2015, 121, 1620–1627. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.H.; Larson, T.; Ou, S.H.; Limentani, S.; Sandler, A.; Vokes, E.; Kim, S.; Liau, K.; Bycott, P.; Olszanski, A.J.; et al. Efficacy and safety of axitinib in patients with advanced non-small-cell lung cancer: Results from a phase II study. J. Clin. Oncol. 2009, 27, 3836–3841. [Google Scholar] [CrossRef]
- Du Four, S.; Maenhout, S.K.; Benteyn, D.; De Keersmaecker, B.; Duerinck, J.; Thielemans, K.; Neyns, B.; Aerts, J.L. Disease progression in recurrent glioblastoma patients treated with the VEGFR inhibitor axitinib is associated with increased regulatory T cell numbers and T cell exhaustion. Cancer Immunol. Immunother. 2016, 65, 727–740. [Google Scholar] [CrossRef]
- Du Four, S.; Maenhout, S.K.; De Pierre, K.; Renmans, D.; Niclou, S.P.; Thielemans, K.; Neyns, B.; Aerts, J.L. Axitinib increases the infiltration of immune cells and reduces the suppressive capacity of monocytic MDSCs in an intracranial mouse melanoma model. Oncoimmunology 2015, 4, e998107. [Google Scholar] [CrossRef] [Green Version]
- Laubli, H.; Muller, P.; D’Amico, L.; Buchi, M.; Kashyap, A.S.; Zippelius, A. The multi-receptor inhibitor axitinib reverses tumor-induced immunosuppression and potentiates treatment with immune-modulatory antibodies in preclinical murine models. Cancer Immunol. Immunother. 2018, 67, 815–824. [Google Scholar] [CrossRef]
- Wilmes, L.J.; Pallavicini, M.G.; Fleming, L.M.; Gibbs, J.; Wang, D.; Li, K.L.; Partridge, S.C.; Henry, R.G.; Shalinsky, D.R.; Hu-Lowe, D.; et al. AG-013736, a novel inhibitor of VEGF receptor tyrosine kinases, inhibits breast cancer growth and decreases vascular permeability as detected by dynamic contrast-enhanced magnetic resonance imaging. Magn Reson Imaging 2007, 25, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Rini, B.I.; Motzer, R.J.; Dutcher, J.P.; Rixe, O.; Wilding, G.; Stadler, W.M.; Tarazi, J.; Garrett, M.; Pithavala, Y.K. Effect of Renal Impairment on the Pharmacokinetics and Safety of Axitinib. Target Oncol. 2016, 11, 229–234. [Google Scholar] [CrossRef] [Green Version]
- Gross-Goupil, M.; Francois, L.; Quivy, A.; Ravaud, A. Axitinib: A review of its safety and efficacy in the treatment of adults with advanced renal cell carcinoma. Clin. Med. Insights Oncol. 2013, 7, 269–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Fulci, G.; Wakimoto, H.; Cheema, T.A.; Buhrman, J.S.; Jeyaretna, D.S.; Stemmer Rachamimov, A.O.; Rabkin, S.D.; Martuza, R.L. Combination of oncolytic herpes simplex viruses armed with angiostatin and IL-12 enhances antitumor efficacy in human glioblastoma models. Neoplasia 2013, 15, 591–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passer, B.J.; Cheema, T.; Wu, S.; Wu, C.L.; Rabkin, S.D.; Martuza, R.L. Combination of vinblastine and oncolytic herpes simplex virus vector expressing IL-12 therapy increases antitumor and antiangiogenic effects in prostate cancer models. Cancer Gene Ther. 2013, 20, 17–24. [Google Scholar] [CrossRef]
- Chen, C.Y.; Hutzen, B.; Wedekind, M.F.; Cripe, T.P. Oncolytic virus and PD-1/PD-L1 blockade combination therapy. Oncolytic Virother 2018, 7, 65–77. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Liu, F.; Liu, Z.; Tang, H.; Wu, H.; Gong, Q.; Chen, J. Immune Checkpoint in Glioblastoma: Promising and Challenging. Front. Pharmacol. 2017, 8, 242. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Twyman-Saint Victor, C.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373–377. [Google Scholar] [CrossRef] [Green Version]
- Curran, M.A.; Montalvo, W.; Yagita, H.; Allison, J.P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 4275–4280. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [Green Version]
- El-Shemi, A.G.; Ashshi, A.M.; Na, Y.; Li, Y.; Basalamah, M.; Al-Allaf, F.A.; Oh, E.; Jung, B.K.; Yun, C.O. Combined therapy with oncolytic adenoviruses encoding TRAIL and IL-12 genes markedly suppressed human hepatocellular carcinoma both in vitro and in an orthotopic transplanted mouse model. J. Exp. Clin. Cancer Res. 2016, 35, 74. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.N.; Choi, I.K.; Huang, J.H.; Yoo, J.Y.; Choi, K.J.; Yun, C.O. Optimizing DC vaccination by combination with oncolytic adenovirus coexpressing IL-12 and GM-CSF. Mol. Ther. 2011, 19, 1558–1568. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Seong, J.; Oh, H.J.; Koom, W.S.; Choi, K.J.; Yun, C.O. A novel combination treatment of armed oncolytic adenovirus expressing IL-12 and GM-CSF with radiotherapy in murine hepatocarcinoma. J. Radiat. Res. 2011, 52, 646–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, E.; Oh, J.E.; Hong, J.; Chung, Y.; Lee, Y.; Park, K.D.; Kim, S.; Yun, C.O. Optimized biodegradable polymeric reservoir-mediated local and sustained co-delivery of dendritic cells and oncolytic adenovirus co-expressing IL-12 and GM-CSF for cancer immunotherapy. J. Control. Release 2017, 259, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Bortolanza, S.; Bunuales, M.; Otano, I.; Gonzalez-Aseguinolaza, G.; Ortiz-de-Solorzano, C.; Perez, D.; Prieto, J.; Hernandez-Alcoceba, R. Treatment of pancreatic cancer with an oncolytic adenovirus expressing interleukin-12 in Syrian hamsters. Mol. Ther. 2009, 17, 614–622. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, Q.; Xu, K.; Shan, J.; Shen, J.; Liu, L.; Xu, Y.; Xia, F.; Bie, P.; Zhang, X.; et al. Combined therapy with cytokine-induced killer cells and oncolytic adenovirus expressing IL-12 induce enhanced antitumor activity in liver tumor model. PLoS ONE 2012, 7, e44802. [Google Scholar] [CrossRef] [Green Version]
- Quetglas, J.I.; Labiano, S.; Aznar, M.A.; Bolanos, E.; Azpilikueta, A.; Rodriguez, I.; Casales, E.; Sanchez-Paulete, A.R.; Segura, V.; Smerdou, C.; et al. Virotherapy with a Semliki Forest Virus-Based Vector Encoding IL12 Synergizes with PD-1/PD-L1 Blockade. Cancer Immunol. Res. 2015, 3, 449–454. [Google Scholar] [CrossRef] [Green Version]
- Schirmacher, V.; Forg, P.; Dalemans, W.; Chlichlia, K.; Zeng, Y.; Fournier, P.; von Hoegen, P. Intra-pinna anti-tumor vaccination with self-replicating infectious RNA or with DNA encoding a model tumor antigen and a cytokine. Gene Ther. 2000, 7, 1137–1147. [Google Scholar] [CrossRef] [Green Version]
- Quetglas, J.I.; Dubrot, J.; Bezunartea, J.; Sanmamed, M.F.; Hervas-Stubbs, S.; Smerdou, C.; Melero, I. Immunotherapeutic synergy between anti-CD137 mAb and intratumoral administration of a cytopathic Semliki Forest virus encoding IL-12. Mol. Ther. 2012, 20, 1664–1675. [Google Scholar] [CrossRef] [Green Version]
- Klas, S.D.; Robison, C.S.; Whitt, M.A.; Miller, M.A. Adjuvanticity of an IL-12 fusion protein expressed by recombinant deltaG-vesicular stomatitis virus. Cell Immunol. 2002, 218, 59–73. [Google Scholar] [CrossRef]
- Lawler, S.E.; Speranza, M.C.; Cho, C.F.; Chiocca, E.A. Oncolytic Viruses in Cancer Treatment: A Review. JAMA Oncol. 2017, 3, 841–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senzer, N.N.; Kaufman, H.L.; Amatruda, T.; Nemunaitis, M.; Reid, T.; Daniels, G.; Gonzalez, R.; Glaspy, J.; Whitman, E.; Harrington, K.; et al. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J. Clin. Oncol 2009, 27, 5763–5771. [Google Scholar] [CrossRef]
- Johnson, D.B.; Puzanov, I.; Kelley, M.C. Talimogene laherparepvec (T-VEC) for the treatment of advanced melanoma. Immunotherapy 2015, 7, 611–619. [Google Scholar] [CrossRef] [Green Version]
- Bommareddy, P.K.; Patel, A.; Hossain, S.; Kaufman, H.L. Talimogene Laherparepvec (T-VEC) and Other Oncolytic Viruses for the Treatment of Melanoma. Am. J. Clin. Dermatol 2017, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Vere Hodge, R.A.; Field, H.J. Antiviral agents for herpes simplex virus. Adv. Pharmacol. 2013, 67, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.N.; Meleth, S.; Hughes, K.B.; Gillespie, G.Y.; Whitley, R.J.; Markert, J.M. Enhanced inhibition of syngeneic murine tumors by combinatorial therapy with genetically engineered HSV-1 expressing CCL2 and IL-12. Cancer Gene Ther. 2005, 12, 359–368. [Google Scholar] [CrossRef] [Green Version]
- Ghouse, S.M.; Bommareddy, P.K.; Nguyen, H.M.; Guz-Montgomery, K.; Saha, D. Oncolytic herpes simplex virus encoding IL12 controls triple-negative breast cancer growth and metastasis in CD8-dependent manner. In Proceedings of the International Oncolytic Virus Conference, Rochester, MN, USA, 9–12 October 2019. [Google Scholar]
- Esaki, S.; Nigim, F.; Moon, E.; Luk, S.; Kiyokawa, J.; Curry, W., Jr.; Cahill, D.P.; Chi, A.S.; Iafrate, A.J.; Martuza, R.L.; et al. Blockade of transforming growth factor-beta signaling enhances oncolytic herpes simplex virus efficacy in patient-derived recurrent glioblastoma models. Int. J. Cancer 2017, 141, 2348–2358. [Google Scholar] [CrossRef] [Green Version]
- Ning, J.; Wakimoto, H.; Peters, C.; Martuza, R.L.; Rabkin, S.D. Rad51 Degradation: Role in Oncolytic Virus-Poly(ADP-Ribose) Polymerase Inhibitor Combination Therapy in Glioblastoma. J. Natl. Cancer Inst. 2017, 109, 1–13. [Google Scholar] [CrossRef]
- Johnson, D.B.; Chandra, S.; Sosman, J.A. Immune Checkpoint Inhibitor Toxicity in 2018. JAMA 2018, 320, 1702–1703. [Google Scholar] [CrossRef]
- Lyon, A.R.; Yousaf, N.; Battisti, N.M.L.; Moslehi, J.; Larkin, J. Immune checkpoint inhibitors and cardiovascular toxicity. Lancet Oncol. 2018, 19, e447–e458. [Google Scholar] [CrossRef]
- Pai, C.S.; Simons, D.M.; Lu, X.; Evans, M.; Wei, J.; Wang, Y.H.; Chen, M.; Huang, J.; Park, C.; Chang, A.; et al. Tumor-conditional anti-CTLA4 uncouples antitumor efficacy from immunotherapy-related toxicity. J. Clin. Investig. 2019, 129, 349–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmieri, D.J.; Carlino, M.S. Immune Checkpoint Inhibitor Toxicity. Curr. Oncol Rep. 2018, 20, 72. [Google Scholar] [CrossRef] [PubMed]
- Rota, E.; Varese, P.; Agosti, S.; Celli, L.; Ghiglione, E.; Pappalardo, I.; Zaccone, G.; Paglia, A.; Morelli, N. Concomitant myasthenia gravis, myositis, myocarditis and polyneuropathy, induced by immune-checkpoint inhibitors: A life-threatening continuum of neuromuscular and cardiac toxicity. eNeurologicalSci 2019, 14, 4–5. [Google Scholar] [CrossRef]
- Filley, A.C.; Henriquez, M.; Dey, M. Recurrent glioma clinical trial, CheckMate-143: The game is not over yet. Oncotarget 2017, 8, 91779–91794. [Google Scholar] [CrossRef] [Green Version]
- Omuro, A.; Vlahovic, G.; Lim, M.; Sahebjam, S.; Baehring, J.; Cloughesy, T.; Voloschin, A.; Ramkissoon, S.H.; Ligon, K.L.; Latek, R.; et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: Results from exploratory phase I cohorts of CheckMate 143. Neuro. Oncol. 2018, 20, 674–686. [Google Scholar] [CrossRef]
- Emens, L.A. Breast Cancer Immunotherapy: Facts and Hopes. Clin. Cancer Res. 2018, 24, 511–520. [Google Scholar] [CrossRef] [Green Version]
- Conry, R.M.; Westbrook, B.; McKee, S.; Norwood, T.G. Talimogene laherparepvec: First in class oncolytic virotherapy. Hum. Vaccin. Immunother. 2018, 14, 839–846. [Google Scholar] [CrossRef]
- Siurala, M.; Havunen, R.; Saha, D.; Lumen, D.; Airaksinen, A.J.; Tahtinen, S.; Cervera-Carrascon, V.; Bramante, S.; Parviainen, S.; Vaha-Koskela, M.; et al. Adenoviral Delivery of Tumor Necrosis Factor-alpha and Interleukin-2 Enables Successful Adoptive Cell Therapy of Immunosuppressive Melanoma. Mol. Ther. 2016, 24, 1435–1443. [Google Scholar] [CrossRef] [Green Version]
- Tahtinen, S.; Blattner, C.; Vaha-Koskela, M.; Saha, D.; Siurala, M.; Parviainen, S.; Utikal, J.; Kanerva, A.; Umansky, V.; Hemminki, A. T-Cell Therapy Enabling Adenoviruses Coding for IL2 and TNFalpha Induce Systemic Immunomodulation in Mice With Spontaneous Melanoma. J. Immunother. 2016, 39, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Martinet, O.; Divino, C.M.; Zang, Y.; Gan, Y.; Mandeli, J.; Thung, S.; Pan, P.Y.; Chen, S.H. T cell activation with systemic agonistic antibody versus local 4-1BB ligand gene delivery combined with interleukin-12 eradicate liver metastases of breast cancer. Gene Ther. 2002, 9, 786–792. [Google Scholar] [CrossRef] [Green Version]
- Kurokawa, C.; Iankov, I.D.; Anderson, S.K.; Aderca, I.; Leontovich, A.A.; Maurer, M.J.; Oberg, A.L.; Schroeder, M.A.; Giannini, C.; Greiner, S.M.; et al. Constitutive Interferon Pathway Activation in Tumors as an Efficacy Determinant Following Oncolytic Virotherapy. J. Natl. Cancer Inst. 2018, 110, 1123–1132. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Genomic Modification | Cancer Model | RoA | Dose (pfu) | Efficacy | Ref. |
|---|---|---|---|---|---|---|
| G47Δ-mIL12 | ΔICP6, Δ∆ICP34.5, ΔICP47, ◦LacZ, ◦mIL-12 | Intracranial 005 GSC (Glioblastoma) | I.T. | 5 × 105 | Inhibited intracranial tumor growth and extended survival. Promoted IL-12 expression, stimulated IFN-γ production, upregulated IP-10, and inhibited VEGF. Polarized TH1 response and inhibited T-regs function. | [19,30] |
| T-mfIL12 | ΔICP6, Δ∆ICP34.5, ΔICP47, ◦mIL-12 | Intracerebral Neuro2a (neuroblastma) | I.V. | 5 × 106 | Prolonged survival (Mock vs. T-mfIL12, p < 0.05), although not statistically significant versus T-01 treatments. | [31] |
| NV1042 | ΔICP0, ΔICP4, ΔICP34.5, ΔUL56, ΔICP47, Us11Δ, Us10Δ, UL56 (duplicated), ◦mIL-12 | Subcutaneous SCC VII (Squamous Cell Carcinoma) | I.T. | 1 × 107 | Reduced tumor volume and improved survival (3 doses of 2 × 107 pfu). 57% of mice from NV1042 group rejected subsequent SCC re-challenge in the contralateral flank compared with 14% in NV1023 or NV1034 group. | [32] |
| I.V. | 5 × 107 | NV1042 treatment resulted in 100% survival, in contrast to 70% of NV1023 and 0% of PBS. | [33] | |||
| M002 | ∆ΔICP34.5, ◦mIL-12 | Intracranial X21415 (Pediatric embryonal tumor); D456 (pediatric glioblastoma); GBM-12 and UAB106 (adult glioblastoma) | I.T. | 1 × 107 | M002 significantly prolonged survival in mice bearing all 4 types of tumor compared to saline. No difference in survival was observed compared with G207, excluding X21415 with high levels of nectin-1 | [34] |
| Intracranial SCK (brain metastasized breast cancer) | I.T. | 1.5 × 107 | Single injection of M002 extended the survival of treated animals more effectively than a non-cytokine control virus. | [35] | ||
| Xenograft SK-N-AS and SK-N-BE (2) (human neuroblastoma); subcutaneous Neuro-2a (murine neuroblastoma) | I.T. | 1 × 107 | Significant decrease in tumor growth were observed in both SK-N-AS and SK-N-BE (2) cell lines. Extended median survival compared to the parent R3659. | [36] | ||
| HuH6 (human hepatoblastoma; G401 (human malignant rhabdoid kidney tumor); SK-NEP-1 (renal Ewing sarcoma) | I.T. | 1 × 107 | M002 significantly reduced tumor volume and increased survival over those treated with vehicle alone in all three different xenograft models. | [37] | ||
| R-115 | Virulent with retargeted HER-2, ◦mIL-12 | pLV-HER2- nectin-puro | I.P. | 1 × 108 to 2 × 109 | Induced greater local and systemic anti-tumor immunity and durable response than unarmed R-LM113 in both early and late schedule. All mice that survived from primary tumor challenge were protected from the distant challenge tumor and subsequent re-challenge. Increased number of CD8+ and CD141+ cells, PD-L1+ tumor cells, and Treg with a decrease in the number of CD11b+ cells. Enhanced Th1 polarization and increased expression of IFN-γ, IL-2, Granzyme B, T-bet and TNFα and tumor infiltrating lymphocytes | [38] |
| Orthotopic mHGGpdgf- hHER2 (glioblastoma) | I.T. | Low dose: 2 × 106 High dose: 1 × 108 | 27% of mice treated with R-115 (n = 6, 4 low-dose arm and 2 high-dose arm) alive 100 days after the virus treatment versus all mice died within 48 days. Increased infiltrating CD4+ and CD8+, and expression of IFN-γ | [39] | ||
| vHSV-IL-12 | ΔICP6, Δ∆ICP34.5, ◦mIL-12 | Subcutaneous Neuro2a (neuroblastoma) | I.T. | 1 × 104 | Significantly reduced tumor growth versus vHSV-null and other cytokine armed groups. | [40] |
| T2850 | ∆IR 15,091bp, ◦mIL-12 | Subcutaneous A20 (Murine B Lymphoma), MC38 (colon adenocarcinoma), MFC (Murine Forestomach Carcinoma) | I.T. | 1 × 107 | Reduced tumor volume compared to IL-12 unarmed parental group. IFN-γ level was markedly increased in the tumor bed and sera of mice infected with both T2850 and T3855 by day 4. | [41] |
| T3855 | ∆IR 15,091bp, ◦mIL-12, ◦mPD-1 | Subcutaneous B16 (melanoma) | 5 × 106, 1 × 107, 3 × 107 | |||
| T3011 | ∆IR15,091bp, ◦hIL-12, ◦hPD-1 | Subcutaneous B16 (melanoma) | I.T. | 5 × 106, 1 × 107 or 3 × 107 | Reduced tumor volume as compared with control group. | [41] |
| Virus | Strain | Cancer Model | RoA | Dose | Efficacy | Ref |
|---|---|---|---|---|---|---|
| Adenovirus | Ad5-yCD/ mutTKSR39 rep-mIL12 | Subcutaneous TRAMP (C2 prostate adenocarcinoma) | I.T. | 5 × 108 pfu | Improved local and metastatic tumor control. Increased NK and CTL cytolytic activities. Significantly increased survival, levels of IL-12 and IFN-ƴ in serum and tumor. | [42,43] |
| Ad-TD-IL-12, Ad-TD-nsIL-12 | Subcutaneous HPD1NR (pancreatic cancer) | I.T. | 1 × 109 pfu | 100% tumor eradication and survival of both IL-12 modified Adenovirus treated animals. Ad-TD-IL-12, but not Ad-TD-nsIL-12 resulted in a significant increase in CD3+CD4−CD8+ populations in the spleen. Level of splenic IFN-γ, IP-10 and lymph node IFN-γ were lower in Ad-TD-nsIL-12 compared to Ad-TD-IL-12 treated hamster. | [44] | |
| Ad-ΔB7/IL12/GMCSF | Subcutaneous B16-F10 (melanoma) | I.T. | 5 × 107 pfu | Primary tumor growth was better controlled in Ad-ΔB7/IL12/GMCSF and Ad-ΔB7/IL12 compared to Ad-ΔB7GMCSF or PBS. Increased tumor infiltrating CD86+ APCs and enhanced CD4+ and CD8+ T cell-mediated Th1 antitumor immune response.Reduced VEGF expression in the tumor treated with oncolytic Ad co-expressing IL-12 and GM-CSF or IL-12 alone. IFN-γ, TNF-α and IL-6 were higher in Ad-ΔB7/IL12/GMCSF and Ad-ΔB7/IL12 compared to Ad-ΔB7GMCSF or PBS. | [45] | |
| RdB/IL-12/ IL-18 | Subcutaneous B16-F10 (melanoma) | I.T. | 1 × 108 pfu | 95% and 99% tumor growth inhibition was observed in treatment with RdB/IL-12 and RdB/IL-12/IL-18, respectively. Increased Th1/Th2 cytokine ratio and increased tumor infiltration of CD4+ T, CD8+ T and NK cells. Promoted differentiation of T cells expressing IL-12Rβ2 or IL-18Rα. | [46] | |
| RdB/IL12/ DCN | Orthotopic 4T1 (Triple negative breast cancer) | I.T. | 2 × 1010 VP | Both of the IL-12-expressing oncolytic Ads showed similar tumor growth inhibition up to day 9 after initial treatment. RdB/IL12/DCN increased upregulation of IFN-γ, TNF-α, infiltrating cytotoxic lymphocytes, downregulation of TGF-β expression and T-regs compared to RdB/IL12 and RdB/DCN. | [47] | |
| YKL-IL12/B7 | Subcutaneous B16-F10 (melanoma) | I.T. | 5 × 108 pfu | Tumor growth was suppressed in both YKL-IL12 and YKL-IL12/B7 treated mice vs PBS. YKL-IL12- or YKL-IL12/B7-treated mice produced a significantly greater level of IFN-γ, infiltrating APCs, CD4+, CD8+ compared with PBS. | [48] | |
| Ad-ΔB7/ IL-12/4-1BBL | Subcutaneous B16-F10 (melanoma) | I.T. | 5 × 109 VP | 100% of mice in the Ad-ΔB7/IL-12/4-1BBL group survived >30 days after initial viral injection compared with 20% of that in virus expressing either IL-12 or 4-1BBL. Mice treated with Ad-ΔB7/IL-12 or Ad-ΔB7/IL-12/4-1BBL had greater amount of tumor infiltrating CD4+ and CD8+ compared to Ad-ΔB7/4-1BBL and Ad-ΔB7. | [49] | |
| Measles virus | MeVac FmIL-12 | Subcutaneous MC38ce (colon carcinoma) | I.T. | 5 × 105–1× 106 ciu | Tumor remissions in 90% of animals. Driven polarization of Th1-associated immune response and increased tumor infiltrating CD8+ T cells. Increased IFN-γ and TNF-α, and polarization of Th1-associated immune response. Co-expression of IL-12 and IL-15 showed synergistic effect. | [20,50] |
| Maraba Virus | MG1-IL12- ICV | CT26 and B16F10 peritoneal carcinomatosis | I.P | Seeding dose 5 × 105, then 1 × 104 on day 3 | Reduced tumor burden and improved mouse survival. Activated and matured DCs to secrete IP-10, and activated and recruited NK cells. Increased production of IFN-γ | [51] |
| Newcastle disease virus | rClone30– IL-12 | Orthotopic H22 (hepatocarcinoma) | I.T. | 1 × 107 pfu | Reduced tumor volume and improved percentage of survival. Increased IFN-γ and IP-10. Co-expression of IL-12 and IL-2 showed synergistic effect. | [52] |
| Semliki Forest virus | rSFV/IL12 | Subcutaneous B16 (melanoma) | I.T. | 107 IU | Single injection with SFV-IL12 resulted in significant tumor regression. 2 days after injection, IFN-γ production increased with inhibition of tumor vascularization. Splenic IP-10 and MIG expression was increased. | [53] |
| SFV/IL12 | Subcutaneous P815 (mastocytoma) | I.T. | 106 IU | Significantly delayed P815 tumor growth. 40–53% of mice exhibited complete tumor regressions. Induced high levels of IFN-γ production in draining lymph nodes. | [54] | |
| SFV/IL12 | Subcutaneous MC38 (colon adenocarcinoma) | I.T. | 108 particles | Reduced tumor volume and improved percentage of survival. Increased tumor-specific CD8+ T lymphocytes. Enhanced the expression of CD11c, CD8α, CD40, and CD86 of tumor-infiltrating M-MDSCs in the presence of an intact endogenous IFN-I system. | [55] | |
| SFV-VLP- | Syngeneic RG2 (rat glioma) | I.T. | 5 × 107 (low-dose) or 5 × 108 (high-dose) | Reduction in tumor volume (70%—low dose; 87%—high dose) | [56] | |
| Vesicular stomatitis virus (VSV) | VSV-IL12 | Orthotopic SCC VII (squamous cell Carcinoma) | I.T. | MOI 0.01 | Significant reduction in tumor volume, and prolonged survival. | [57] |
| Sindbis virus | Sin/IL12 | Orthotopic ES-2 cells (ovarian clear cell Carcinoma) | I.P | 107 pfu | Reduced tumor growth and improved survival. Activated and matured DCs, activated and recruited NK cells. Increased production of IFN-γ. | [58] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, H.-M.; Guz-Montgomery, K.; Saha, D. Oncolytic Virus Encoding a Master Pro-Inflammatory Cytokine Interleukin 12 in Cancer Immunotherapy. Cells 2020, 9, 400. https://doi.org/10.3390/cells9020400
Nguyen H-M, Guz-Montgomery K, Saha D. Oncolytic Virus Encoding a Master Pro-Inflammatory Cytokine Interleukin 12 in Cancer Immunotherapy. Cells. 2020; 9(2):400. https://doi.org/10.3390/cells9020400
Chicago/Turabian StyleNguyen, Hong-My, Kirsten Guz-Montgomery, and Dipongkor Saha. 2020. "Oncolytic Virus Encoding a Master Pro-Inflammatory Cytokine Interleukin 12 in Cancer Immunotherapy" Cells 9, no. 2: 400. https://doi.org/10.3390/cells9020400
APA StyleNguyen, H. -M., Guz-Montgomery, K., & Saha, D. (2020). Oncolytic Virus Encoding a Master Pro-Inflammatory Cytokine Interleukin 12 in Cancer Immunotherapy. Cells, 9(2), 400. https://doi.org/10.3390/cells9020400