Interaction of the Anti-Proliferative GPER Inverse Agonist ERα17p with the Breast Cancer Cell Plasma Membrane: From Biophysics to Biology



 , ,
, ,  and
and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials and Cell Cultures
2.2. Preparation of Large Unilamellar Vesicles (LUV)
2.3. Far-UV Circular Dichroism (CD) Spectroscopy
2.4. Plasmon Waveguide Resonance (PWR)
2.5. Membrane Permeability Assay in Large Unilamellar Vesicles
2.6. Scanning (SEM) and Transmission (TEM) Electron Microscopy
2.7. Cytotoxicity Assays
3. Results and Discussion
3.1. ERα17p Interacts with Lipidic Bilayers Mimicking the Eukaryote Plasma Membrane and Alters Their Integrity
3.1.1. Far-UV Circular Dichroism (CD)
3.1.2. Plasmon Waveguide Resonance (PWR)
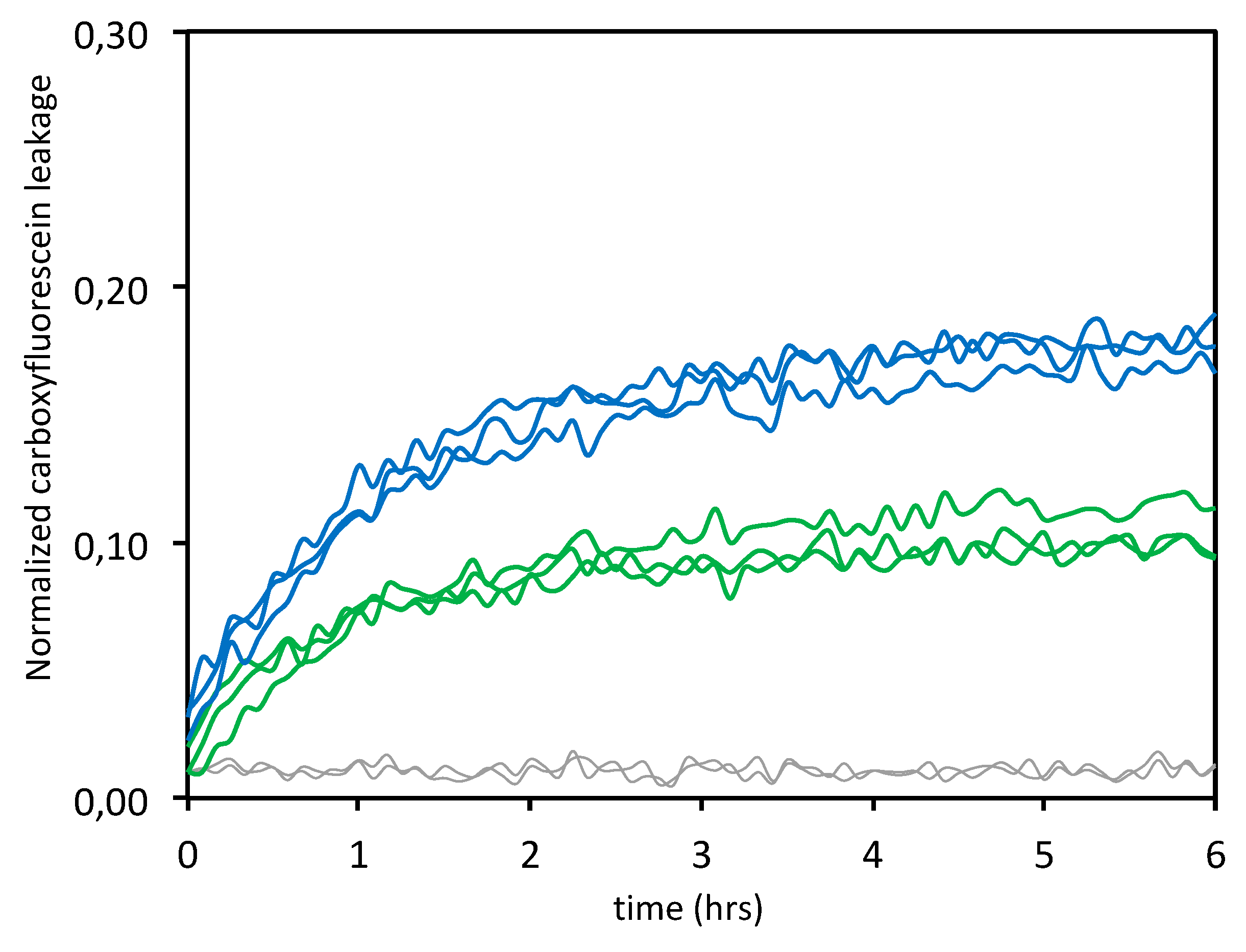
3.1.3. Leakage Experiments
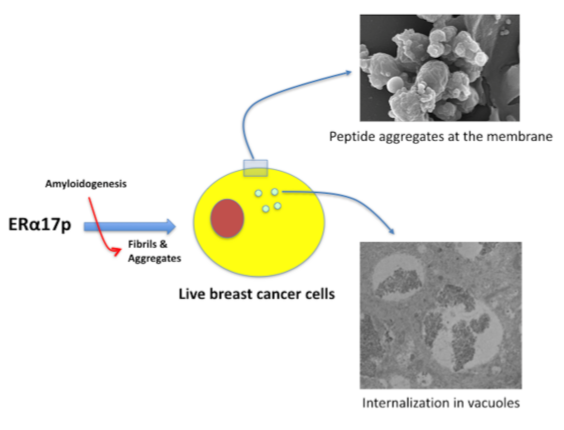
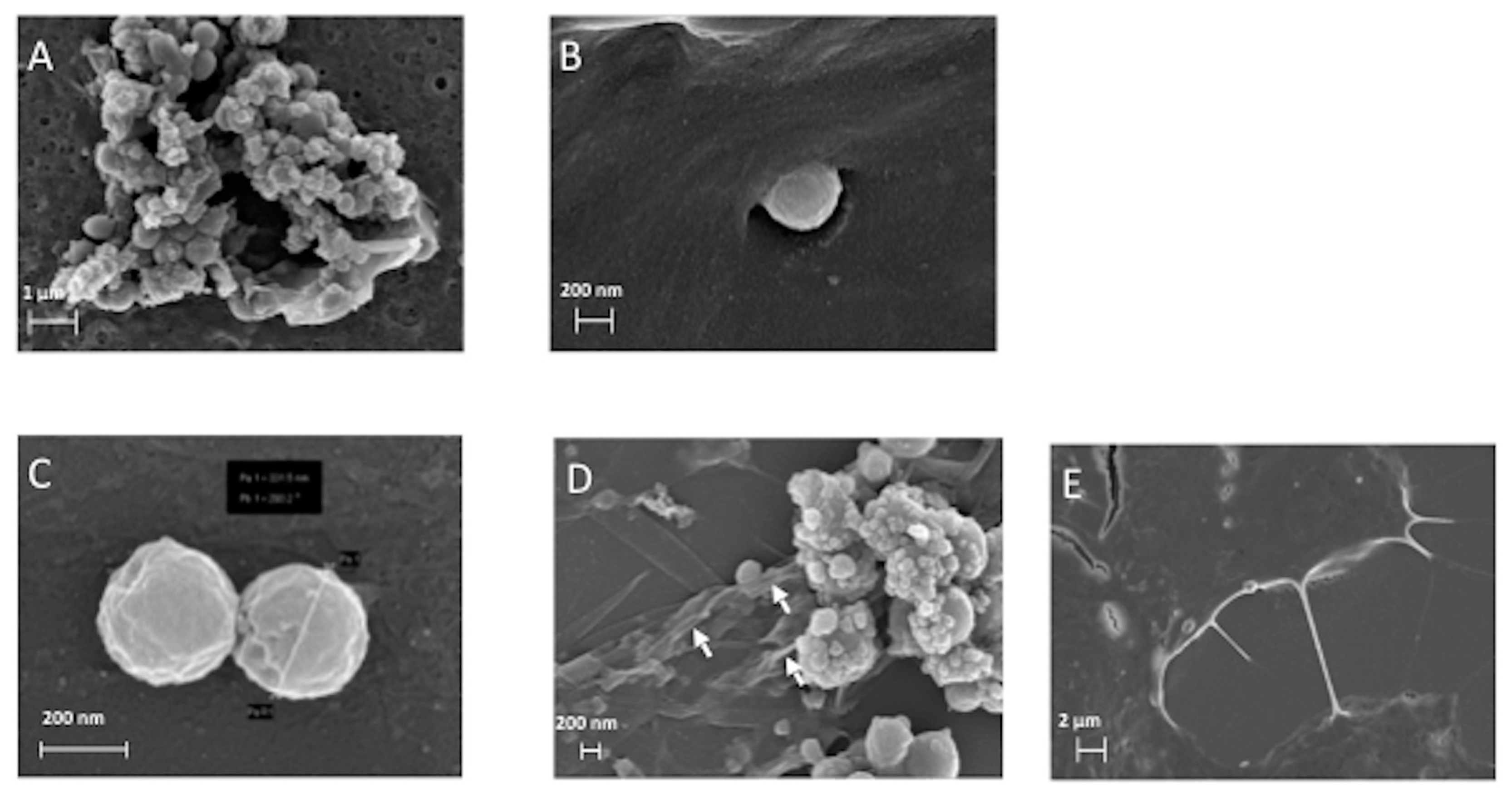
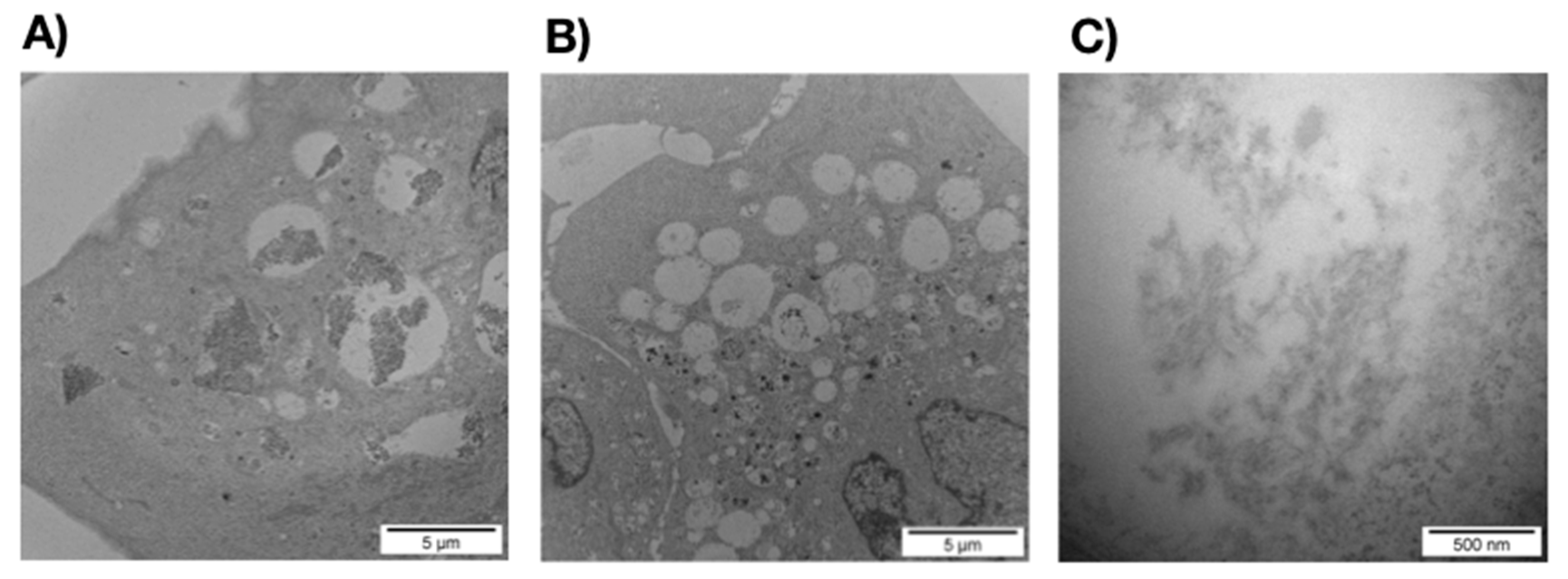
3.2. The ERα17p Forms Peptide Aggregates Interacting with Plasma Membrane
3.2.1. SEM and TEM Experiments
3.2.2. Cytotoxicity Assays of ERα17p Aggregates
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bouhoute, A.; Leclercq, G. Modulation of estradiol and DNA binding to estrogen receptor upon association with calmodulin. Biochem. Biophys. Res. Commun. 1995, 208, 748–755. [Google Scholar] [CrossRef]
- Gallo, D.; Haddad, I.; Duvillier, H.; Jacquemotte, F.; Laïos, I.; Laurent, G.; Jacquot, Y.; Vinh, J.; Leclercq, G. Trophic effect in MCF-7 cells of ERα17p, a peptide corresponding to a platform regulatory motif of the estrogen receptor α—Underlying mechanisms. J. Steroid. Biochem. Mol. Biol. 2008, 109, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Cubellis, M.V.; Caillez, F.; Blundell, T.L.; Lovell, S.C. Properties of polyproline II, a secondary structure element implicated in protein-protein interactions. Proteins 2005, 58, 880–892. [Google Scholar] [CrossRef] [PubMed]
- Jacquot, Y.; Gallo, D.; Leclercq, G. Estrogen receptor alpha—Identification by a modeling approach of a potential polyproline II recognizing domain within the AF-2 region of the receptor that would play a role of prime importance in its mechanism of action. J. Steroid Biochem. Mol. Biol. 2007, 104, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gallo, D.; Leclercq, G.; Jacquot, Y. The N-terminal part of the ligand-binding domain of the human estrogen receptor α: A new target for estrogen disruptors. In Medicinal Chemistry Research Progress, 1st ed.; Colombo, G.P., Ricci, S., Eds.; Nova: New York, NY, USA, 2009; pp. 207–224. [Google Scholar]
- Seielstad, D.A.; Carlson, K.E.; Kushner, P.J.; Greene, G.L.; Katzenellenbogen, J.A. Analysis of the structure core of the human estrogen receptor ligand binding domain by selective proteolysis/mass spectrometric analysis. Biochemistry 1995, 34, 12605–12615. [Google Scholar] [CrossRef] [PubMed]
- Ylikomi, T.; Bocquel, M.T.; Berry, M.; Gronemeyer, H.; Chambon, P. Cooperation of proto-signals for nuclear accumulation of estrogen and progesterone receptors. EMBO J. 1992, 11, 3681–3694. [Google Scholar] [CrossRef] [PubMed]
- Norris, J.D.; Fan, D.; Kerner, S.A.; McDonnell, D.P. Identification of a third autonomous activation domain with the human estrogen receptor. Mol. Endocrinol. 1997, 11, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.; Parrish, E.; Edmiston, S.N.; Tolbert, D.; Tse, C.K.; Geradts, J.; Livasy, C.A.; Singh, H.; Newman, B.; Millikan, R.C. The estrogen receptor-alpha A908G (K303R) mutation occurs at a low frquency in invasive breast tumors: Results from a population-based study. Breast Cancer Res. 2005, 7, R871–R880. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, K.; Jia, D.; Kapoor-Vazirani, P.; Powell, D.R.; Collins, R.E.; Sharma, D.; Peng, J.; Cheng, X.; Vertino, P.M. Regulation of estrogen receptor alpha by the SET7 lysine methyltransferase. Mol. Cell. 2008, 30, 336–347. [Google Scholar] [CrossRef] [Green Version]
- Gallo, D.; Jacquemotte, F.; Cleeren, A.; Laïos, I.; Hadiy, S.; Rowlands, M.G.; Caille, O.; Nonclercq, D.; Laurent, G.; Jacquot, Y.; et al. Calmodulin-independent, agonistic properties of a peptide containing the calmodulin binding site of estrogen receptor α. Mol. Cell. Endocrinol. 2007, 268, 37–49. [Google Scholar] [CrossRef] [Green Version]
- Gallo, D.; Jacquot, Y.; Cleeren, A.; Jacquemotte, F.; Laïos, I.; Laurent, G.; Leclercq, G. Molecular basis of agonistic activity of ERα17p, a synthetic peptide corresponding to a sequence located at the N-terminal part of the estrogen receptor α ligand binding domain. Lett. Drug Design Discov. 2007, 4, 346–355. [Google Scholar] [CrossRef]
- Notas, G.; Kampa, M.; Pelekanou, V.; Troullinaki, M.; Jacquot, Y.; Leclercq, G.; Castanas, E. Whole transcriptome analysis of the ERα synthetic fragment P295-T311 (ERα17p)-isoform (ERα, ERα36)-dependent and -independent actions in breast cancer cells. Mol. Oncol. 2013, 7, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Leiber, D.; Burlina, F.; Byrne, C.; Robin, P.; Piesse, C.; Gonzalez, L.; Leclercq, G.; Tanfin, Z.; Jacquot, Y. The sequence Pro295-Thr311 of the hinge region of oestrogen receptor α is involved in ERK1/2 activation via GPR30 in leiomyoma cells. Biochem. J. 2015, 472, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Pelekanou, V.; Kampa, M.; Gallo, D.; Notas, G.; Troullinaki, M.; Duvillier, H.; Jacquot, Y.; Stathopoulos, E.N.; Castanas, E.; Leclercq, G. The estrogen receptor alpha-derived peptide ERα17p (P205-T311) exerts pro-apoptotic actions in breast cancer cells in vitro and in vivo, independently from their ERα status. Mol. Oncol. 2011, 5, 36–47. [Google Scholar] [CrossRef]
- Kampa, M.; Pelekanou, V.; Gallo, D.; Notas, G.; Troullinaki, M.; Pediaditakis, I.; Charalampoulos, I.; Jacquot, Y.; Leclercq, G.; Castanas, E. ERα17p, an ERα P295-T311 fragment, modifies the migration of breast cancer cells, through actin cytoskeleton rearrangements. J. Cell. Biochem. 2011, 112, 3786–3796. [Google Scholar] [CrossRef]
- Lappano, R.; Mallet, C.; Rizzuti, B.; Grande, F.; Galli, G.R.; Byrne, C.; Broutin, I.; Boudieu, L.; Eschalier, A.; Jacquot, Y.; et al. The peptide ERα17p is a GPER inverse agonist that exerts antiproliferative effects in breast cancer cells. Cells 2019, 8, 590. [Google Scholar] [CrossRef] [Green Version]
- Yip, F.; Nemati, F.; El Botty, R.; Belnou, M.; Decaudin, D.; Mansuy, C.; Jacquot, Y. Improvement of the antiproliferative activity of the peptide ERα17p in MCF-7 breast cancer cells using nanodiamonds. Ann. Pharm. Fr. 2019, 77, 488–495. [Google Scholar] [CrossRef]
- Craig Jordan, V.; Lewis-Wambi, J.S.; Patel, R.R.; Kim, H.; Ariazi, E.A. New hypotheses and opportunities in endocrine therapy: Amplification of oestrogen-induced apoptosis. Breast 2009, 18, S10–S17. [Google Scholar] [CrossRef]
- Chimento, A.; Casaburi, I.; Rosano, C.; Avena, P.; De Luca, A.; Campana, C.; Martire, E.; Santolla, M.F.; Maggiolini, M.; Pezzi, V.; et al. Oleuropein and hydroxytyrosol activate GPER/GPR30-dependent pathways leading to apoptosis of ER-negative SKBR3 breast cancer cells. Mol. Nutr. Food Res. 2014, 58, 478–489. [Google Scholar] [CrossRef]
- Maggiolini, M.; Santolla, M.F.; Avino, S.; Aiello, F.; Rosano, C.; Garofalo, A.; Grande, F. Identification of two benzopyrroloxazines acting as selective GPER antagonists in breast cancer cells and cancer-associated fibroblasts. Future Med. Chem. 2015, 7, 437–448. [Google Scholar] [CrossRef]
- Rosano, C.; Ponassi, M.; Santolla, M.F.; Pisano, A.; Felli, L.; Vivacqua, A.; Maggiolini, M.; Lappano, R. Macromolecular modelling and docking simulations for the discovery of selective GPER ligands. AAPS J. 2016, 18, 41–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Méndez-Muna, D.; Bello, M.; Correa-Basurto, J. Understanding the molecular basis of agonist/antagonist mechanism of GPER1/GPR30 through structural and energetic analyses. J. Steroid Biochem. Mol. Biol. 2016, 158, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.; Carey, L.A. Understanding and treating triple-negative breast cancer. Oncology 2008, 22, 1233–1243. [Google Scholar] [PubMed]
- Byrne, C.; Khemtémourian, L.; Pelekanou, V.; Kampa, M.; Leclercq, G.; Sagan, S.; Castanas, E.; Burlina, F.; Jacquot, Y. ERα17p, a peptide reproducing the hinge region of the estrogen receptor α associates to biological membranes: A biophysical approach. Steroids 2012, 77, 979–987. [Google Scholar] [CrossRef]
- Ruggeri, F.S.; Byrne, C.; Khemtemourian, L.; Ducouret, G.; Dietler, G.; Jacquot, Y. Concentration-dependent and surface-assisted self-assembly properties of a bioactive estrogen receptor α-derived peptide. J. Pept. Sci. 2015, 21, 95–104. [Google Scholar] [CrossRef]
- Zachowski, A. Phospholipids in animal eukaryotic membranes: Transverse asymmetric and movement. Biochem. J. 1993, 294, 1–14. [Google Scholar] [CrossRef]
- Rustenbeck, I.; Matthies, A.; Lenzen, S. Lipid composition of glucose-stimulated pancreatic islets and insulin-secreting tumor cells. Lipids 1994, 29, 685–692. [Google Scholar] [CrossRef]
- Khemtémourian, L.; Engel, M.F.M.; Liskamp, R.M.J.; Höppener, J.W.M.; Killian, J.A. The N-terminal fragment of human islet amyloid polypeptide is non-fibrillogenic in the presence of membranes and does not cause leakage of bilayers of physiologically relevant lipid composition. Biochim. Biophys. Acta 2010, 1798, 1805–1811. [Google Scholar] [CrossRef]
- Engberg, O.; Yasuda, T.; Hautala, V.; Matsumori, N.; Nyholm, T.K.M.; Murata, M.; Slotte, J.P. Lipid interactions and organization in complex bilayer membranes. Biophys. J. 2016, 110, 1563–1573. [Google Scholar] [CrossRef] [Green Version]
- Rouser, G.; Siakotos, A.N.; Fleischer, S. Quantitative analysis of phospholipids by thin-layer chromatography and phosphorus analysis of spots. Lipids 1966, 1, 85–86. [Google Scholar] [CrossRef]
- Harte, E.; Maalouli, N.; Shalabney, A.; Texier, E.; Berthelot, K.; Lecomte, S.; Alves, I.D. Probing the kinetics of lipid membrane formation and the interaction of a nontoxic and a toxic amyloid with plasmon waveguide resonance. Chem. Comm. 2014, 50, 4168–4171. [Google Scholar] [CrossRef] [PubMed]
- Salamon, Z.; Macleod, H.A.; Tollin, G. Coupled plasmon-waveguide resonators: A new spectroscopic tool for probing proteolipid film structure and properties. Biophys. J. 1997, 73, 2791–2797. [Google Scholar] [CrossRef] [Green Version]
- Salamon, Z.; Lindblom, G.; Tollin, G. Plasmon-waveguide resonance and impedance spectroscopy studies of the interaction between penetratin and supported lipid bilayer membranes. Biophys. J. 2003, 84, 1796–1807. [Google Scholar] [CrossRef] [Green Version]
- Mueller, P.; Rudin, D.O.; Tien, H.T.; Wescott, W.C. Reconstitution of cell membrane structure in vitro and its transformation into an excitable system. Nature 1962, 194, 979–980. [Google Scholar] [CrossRef]
- Mulgrew-Nesbitt, A.; Diraviyam, K.; Wang, J.; Singh, S.; Lurray, P.; Li, Z.; Rogers, L.; Mirkovic, N.; Murray, D. The role of electrostatics in protein—Membrane interactions. Biochim. Biophys. Acta 2006, 1761, 812–826. [Google Scholar] [CrossRef]
- Emmelot, P. The organization of the plasma membrane of mammalian cells: Structure in relation to function. In Mammalian Cell Membranes: The Diversity of Membranes; Jamieson, G.A., Robinson, D.M., Eds.; Butterworth-Heinemann: London, UK, 1977; Volume 2, pp. 1–54. [Google Scholar]
- Blondelle, S.E.; Lohner, K.; Aguilar, M.I. Lipid-induced conformation and lipid-binding properties of cytosolic and antimicrobial peptides: Determination and biological specificity. Biochim. Biophys. Acta 1999, 1462, 89–108. [Google Scholar] [CrossRef] [Green Version]
- Woody, R.W. Circular dichroism of peptides. In The Peptides; Hruby, V.J., Ed.; Academic Press: New York, NY, USA, 1985; Volume 7, pp. 15–114. [Google Scholar]
- Woody, R.W. Houben-Weyl: Methods of Organic Chemistry; Goodman, M., Felix, A., Moroder, L., Toniolo, C., Eds.; Thieme: Stuttgart, Germany, 2003; Volume E22b, pp. 739–765. [Google Scholar]
- Juszczyk, P.; Kolodziejczyk, A.S.; Grzonka, Z. Circular dichroism and aggregation studies of amyloid β (11-28) fragment and its variants. Acta Biochim. Pol. 2005, 52, 425–431. [Google Scholar] [CrossRef]
- Reed, J.; Reed, T.A. A set of constructed type spectra for the practical estimation of peptide secondary structure from circular dichroism. Anal. Biochem. 1997, 254, 36–40. [Google Scholar] [CrossRef]
- Ringstad, L.; Schmidtchen, A.; Malmsten, M. Effect of peptide length on the interaction between consensus peptides and DOPC/DOPA bilayers. Langmuir 2006, 22, 5042–5050. [Google Scholar] [CrossRef]
- Ambroggio, E.E.; Separovic, F.; Bowie, J.H.; Fidelio, G.D.; Bagatolli, L.A. Direct visualization of membrane leakage induced by the antibiotic peptides: Maculatin, citropin, and aurein. Biophys. J. 2005, 89, 1874–1881. [Google Scholar] [CrossRef] [Green Version]
- Leite, N.B.; Aufderhorst-Roberts, A.; Palma, M.S.; Connell, S.D.; Ruggiero Neto, J.; Beales, P.A. PE and PS lipids synergistically enhance membrane poration by a peptide with anticancer properties. Biophys. J. 2015, 109, 936–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utsugi, T.; Schroit, A.J.; Connor, J.; Bucana, C.D.; Fidler, I.J. Elevated expression of phosphatidylserine in the outer membrane leaflet of human tumor cells and recognition by activated human blood monocytes. Cancer Res. 1991, 51, 3062–3066. [Google Scholar] [PubMed]
- Zhao, H.; Tuominen, E.K.; Kinnunen, P.K. Formation of amyloid fibers triggered by phosphatidylserine-containing membranes. Biochemistry 2004, 43, 10302–10307. [Google Scholar] [CrossRef] [PubMed]
- Reuter, M.; Schwieger, C.; Meister, A.; Karlsson, G.; Blume, A. Poly-L-lysines and poly-L-arginines induce leakage of negatively charged phospholipid vesicles and translocate through the lipid bilayer upon electrostatic binding to the membrane. Biophys. J. 2009, 144, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Sharma, B.; Kanwar, S.S. Phosphatidylserine: A cancer cell targeting biomarker. Semin. Cancer Biol. 2018, 52, 17–25. [Google Scholar] [CrossRef]
- Couceiro, J.R.; Gallardo, R.; De Smet, F.; De Baets, G.; Baatsen, P.; Annaert, W.; Roose, K.; Saelens, X.; Schymkovitz, J.; Rousseau, F. Sequence-dependent internalization of aggregating peptides. J. Biol. Chem. 2015, 290, 242–258. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, J.M. Resolving the kinetics of lipid, protein and peptide diffusion in membranes. Mol. Membr. Biol. 2012, 29, 118–143. [Google Scholar] [CrossRef] [Green Version]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [Green Version]
- Veloria, J.R.; Chen, L.; Li, L.; Breen, G.A.M.; Lee, J.; Goux, W.J. Novel cell-penetrating-amyloid peptide conjugates preferentially kill cancer cells. Medchemcomm 2018, 9, 121–130. [Google Scholar] [CrossRef]
- Strober, W. Trypan blue exclusion test of cell viability. Curr. Protoc. Immunol. 2005. [Google Scholar] [CrossRef]
- Flanagan, M.T.; Ainsworth, S. The binding of aromatic sulphonic acids to bovine serum albumin. Biochim. Biophys. Acta 1968, 168, 16–26. [Google Scholar] [CrossRef]
- Chamchoy, K.; Pakotiprapha, D.; Pumirat, P.; Leartsakulpanich, U.; Boonyuen, U. Application of WST-8 based colorimetric NAD(P)H detection for quantitative dehydrogenase assays. BMC Biochem 2019, 20, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaji-Hasegawa, A.; Tsujimoto, M. Asymmetric distribution of phospholipids in biomembranes. Biol. Pharm. Bull. 2006, 29, 1547–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasaki, T.; Ishibashi, J.; Tanaka, H.; Sato, M.; Asaoka, A.; Taylor, D.; Yamakawa, M. Selective cancer cell cytotoxicity of enantiomeric 9-mer peptides derived from beetle defensins depends on negatively charged phosphatidylserine on cell surface. Peptides 2009, 30, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Dong, S.; Zhang, L.; Zhao, Y.; Huang, L.; Gong, X.; Wang, H.; Shang, D. Cell surface binding, uptaking and anticancer activity of L-K6, a lysine/leucine-rich peptide, on human breast cancer MCF-7 cells. Sci. Rep. 2017, 7, 8293. [Google Scholar] [CrossRef] [PubMed]
- De, M.; Ghosh, S.; Sen, T.; Shadab, M.; Banerjee, I.; Basu, S.; Ali, N. A novel therapeutic strategy for cancer using phosphatidylserine targeting stearylamine-bearing cationic liposomes. Mol. Ther. Nucleic Acids 2018, 10, 9–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipid | Kd (μM) | Resonance Minimum Shifts (mDeg) | Rate Constants (sec−1) | ||
|---|---|---|---|---|---|
| p-pol | s-pol | p-pol | s-pol | ||
| POPC/POPS/Chol | 1.2 ± 0.3 | 5 ± 1 | 5 ± 1 | 0.0045 ± 0.002 | 0.0026 ± 0.0016 |
| Viability (1 h Incubation) | Viability (48 h Incubation) | |
|---|---|---|
| 1 μM ERα17p | 100 ± 1 | 104 ± 2 |
| 10 μM ERα17p | 98 ± 2 | 107 ± 3 |
| Control | 100 ± 1 | 100 ± 3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trichet, M.; Lappano, R.; Belnou, M.; Salazar Vazquez, L.S.; Alves, I.; Ravault, D.; Sagan, S.; Khemtemourian, L.; Maggiolini, M.; Jacquot, Y. Interaction of the Anti-Proliferative GPER Inverse Agonist ERα17p with the Breast Cancer Cell Plasma Membrane: From Biophysics to Biology. Cells 2020, 9, 447. https://doi.org/10.3390/cells9020447
Trichet M, Lappano R, Belnou M, Salazar Vazquez LS, Alves I, Ravault D, Sagan S, Khemtemourian L, Maggiolini M, Jacquot Y. Interaction of the Anti-Proliferative GPER Inverse Agonist ERα17p with the Breast Cancer Cell Plasma Membrane: From Biophysics to Biology. Cells. 2020; 9(2):447. https://doi.org/10.3390/cells9020447
Chicago/Turabian StyleTrichet, Michaël, Rosamaria Lappano, Mathilde Belnou, Lilian Shadai Salazar Vazquez, Isabel Alves, Delphine Ravault, Sandrine Sagan, Lucie Khemtemourian, Marcello Maggiolini, and Yves Jacquot. 2020. "Interaction of the Anti-Proliferative GPER Inverse Agonist ERα17p with the Breast Cancer Cell Plasma Membrane: From Biophysics to Biology" Cells 9, no. 2: 447. https://doi.org/10.3390/cells9020447
APA StyleTrichet, M., Lappano, R., Belnou, M., Salazar Vazquez, L. S., Alves, I., Ravault, D., Sagan, S., Khemtemourian, L., Maggiolini, M., & Jacquot, Y. (2020). Interaction of the Anti-Proliferative GPER Inverse Agonist ERα17p with the Breast Cancer Cell Plasma Membrane: From Biophysics to Biology. Cells, 9(2), 447. https://doi.org/10.3390/cells9020447