The Role of Gamma Delta T Cells in Autoimmune Rheumatic Diseases

Abstract
:1. Introduction
2. Rheumatoid Arthritis
2.1. Numerical Alterations of γδ T Cells in RA
2.2. γδ T Cells in Rheumatoid Synovium
2.3. TCR Gene Expression
2.4. Functions of γδ T Cells in RA
2.5. Responses to Putative Antigen
2.6. Relationship to Disease Activity and Severity
3. Rodent Models of Rheumatoid Arthritis
3.1. Rat Models
3.2. Murine Model
4. Juvenile Idiopathic Arthritis
4.1. Numerical Evaluation and Relationship to Disease Activity
4.2. Functional Characteristics
5. Murine Models Relevant to JIA
6. Ankylosing Spondylitis (AS)
Murine Model Relevant to Ankylosing Spondylitis
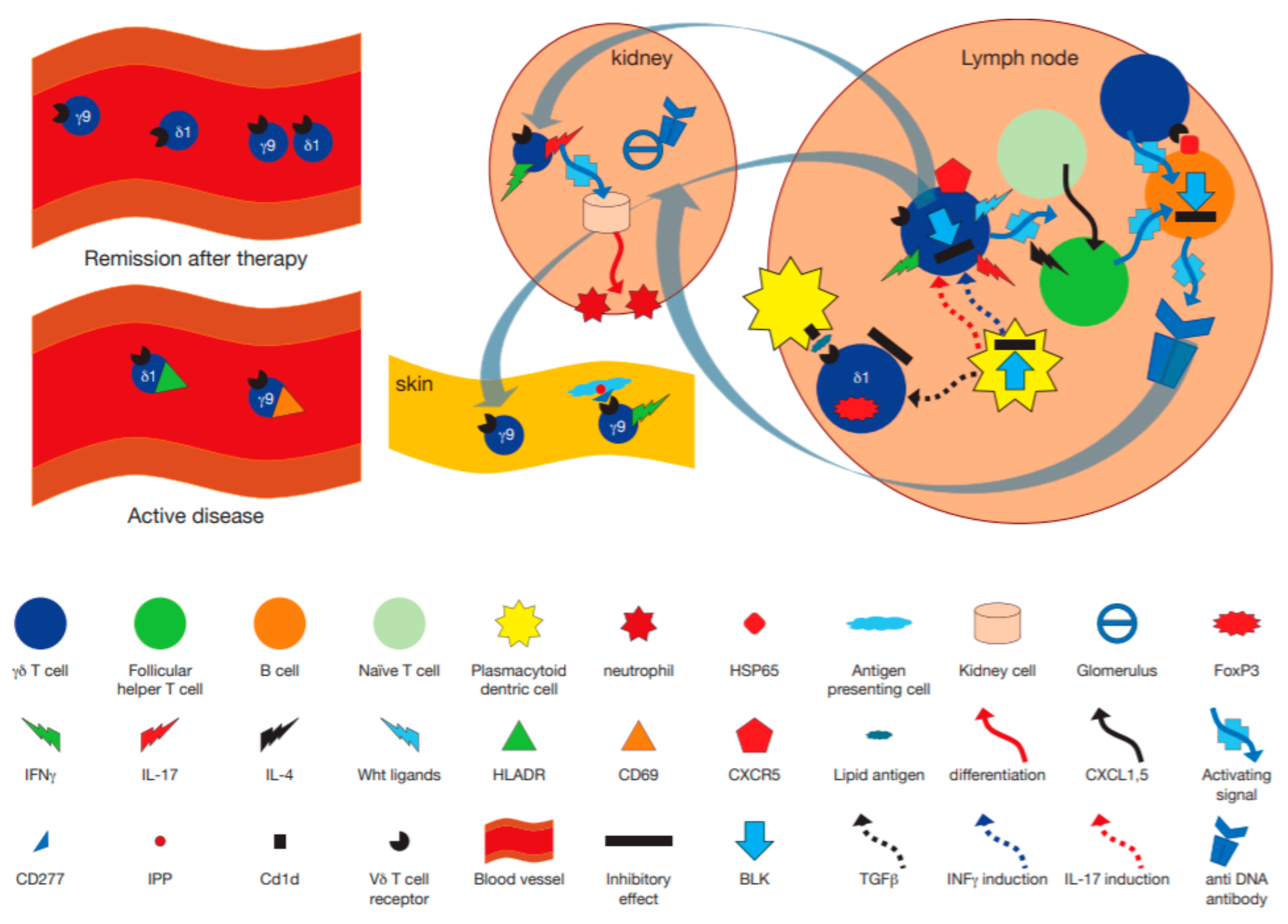
7. Systemic Lupus Erythematosus
7.1. In Vivo Levels of γδ T Cells and Their Correlation with Disease Activity
7.2. In Vitro Studies of γδ T Cells from SLE Patients
8. Murine Models
8.1. MRL/lpr Model
8.2. NZB/NZW Model
8.3. Pristane Induced Model
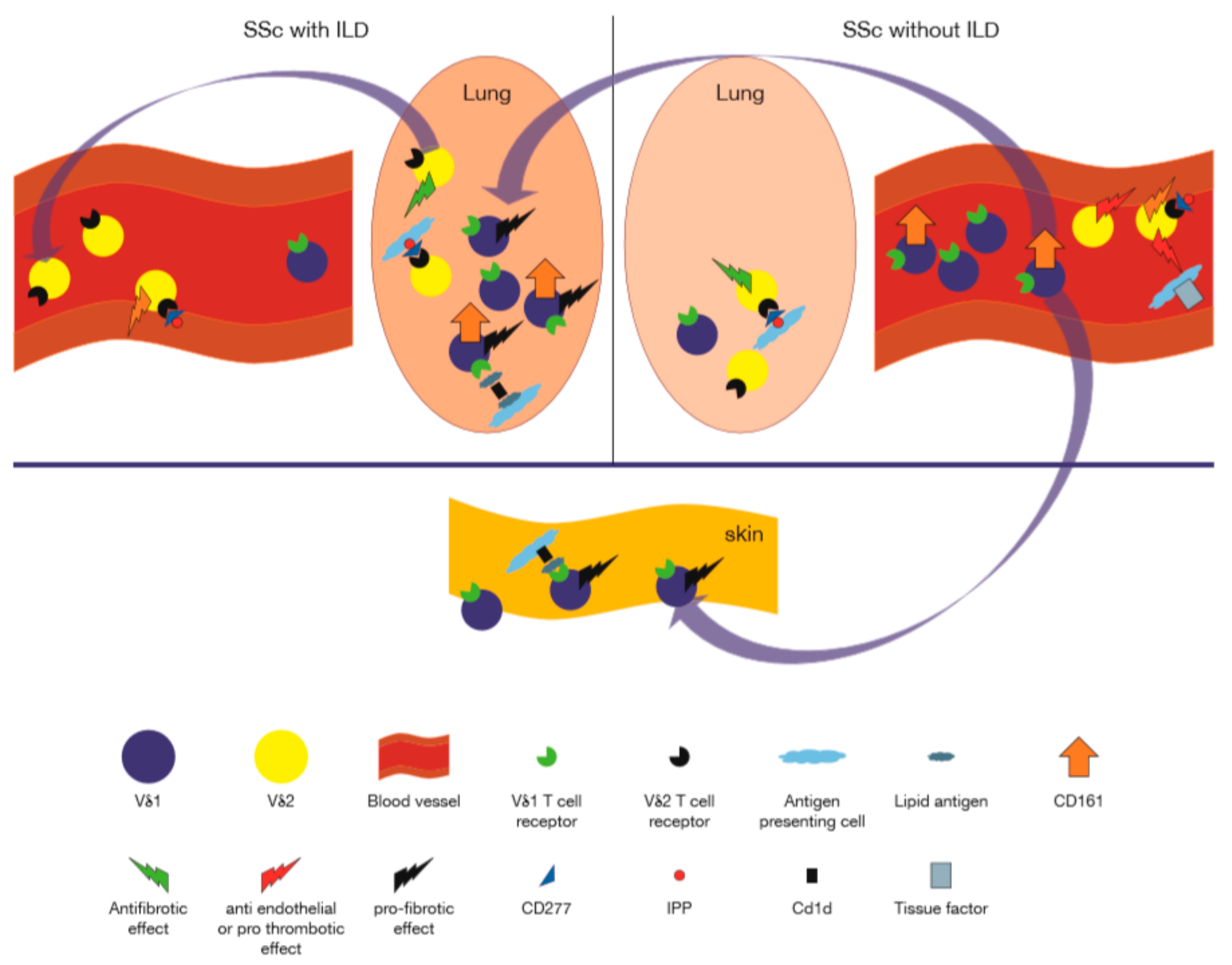
9. Systemic Sclerosis
9.1. γδ T Cells in PB and Tissue of SSc Patients
9.2. Functions and Subsets of γδ T Cells in SSc
10. Concluding Remarks
Funding
Conflicts of Interest
References
- Allison, J.P.; Ridge, L.; Lund, J.; Gross-Pelose, J.; Lanier, L.; McIntyre, B.W. The murine T cell antigen receptor and associated structures. Immunol. Rev. 1984, 81, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Acuto, O.; Hussey, R.E.; Fitzgerald, K.A.; Protentis, J.P.; Meuer, S.C.; Schlossman, S.F.; Reinherz, E.L. The human T cell receptor: Appearance in ontogeny and biochemical relationship of alpha and beta subunits on IL-2 dependent clones and T cell tumors. Cell 1983, 34, 717–726. [Google Scholar] [CrossRef]
- Saito, H.; Kranz, D.M.; Takagaki, Y.; Hayday, A.C.; Eisen, H.N.; Tonegawa, S. A third rearranged and expressed gene in a clone of cytotoxic T lymphocytes. Nature 1984, 312, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Bank, I.; DePinho, R.A.; Brenner, M.B.; Cassimeris, J.; Alt, F.W.; Chess, L. A functional T3 molecule associated with a novel heterodimer on the surface of immature human thymocytes. Nature 1986, 322, 179–181. [Google Scholar] [CrossRef]
- Brenner, M.B.; McLean, J.; Dialynas, D.P.; Strominger, J.L.; Smith, J.A.; Owen, F.L.; Seidman, J.G.; Ip, S.; Rosen, F.; Krangel, M.S. Identification of a putative second T-cell receptor. Nature 1986, 322, 145–149. [Google Scholar] [CrossRef]
- Chien, Y.H.; Iwashima, M.; Kaplan, K.B.; Elliott, J.F.; Davis, M.M. A new T-cell receptor gene located within the alpha locus and expressed early in T-cell differentiation. Nature 1987, 327, 677–682. [Google Scholar] [CrossRef]
- Vermijlen, D.; Gatti, D.; Kouzeli, A.; Rus, T.; Eberl, M. gammadelta T cell responses: How many ligands will it take till we know? Semin Cell Dev. Biol. 2018, 84, 75–86. [Google Scholar] [CrossRef]
- Yang, Y.; Li, L.; Yuan, L.; Zhou, X.; Duan, J.; Xiao, H.; Cai, N.; Han, S.; Ma, X.; Liu, W.; et al. A Structural Change in Butyrophilin upon Phosphoantigen Binding Underlies Phosphoantigen-Mediated Vγ9Vδ2 T Cell Activation. Immunity 2019, 50, 1043.e5–1053.e5. [Google Scholar] [CrossRef] [Green Version]
- Melandri, D.; Zlatareva, I.; Chaleil, R.A.G.; Dart, R.J.; Chancellor, A.; Nussbaumer, O.; Polyakova, O.; Roberts, N.A.; Wesch, D.; Kabelitz, D.; et al. The γδTCR combines innate immunity with adaptive immunity by utilizing spatially distinct regions for agonist selection and antigen responsiveness. Nat. Immunol. 2018, 19, 1352–1365. [Google Scholar] [CrossRef]
- Adams, E.J.; Gu, S.; Luoma, A.M. Human gamma delta T cells: Evolution and ligand recognition. Cell Immunol. 2015, 296, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Hayday, A.C. gammadelta T Cell Update: Adaptate Orchestrators of Immune Surveillance. J. Immunol. 2019, 203, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Vantourout, P.; Hayday, A. Six-of-the-best: Unique contributions of gammadelta T cells to immunology. Nat. Rev. Immunol. 2013, 13, 88–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papotto, P.H.; Reinhardt, A.; Prinz, I.; Silva-Santos, B. Innately versatile: gammadelta17 T cells in inflammatory and autoimmune diseases. J. Autoimmun. 2018, 87, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.H.; Meyer, C.; Bonneville, M. gammadelta T cells: First line of defense and beyond. Annu Rev. Immunol. 2014, 32, 121–155. [Google Scholar] [CrossRef]
- Bank, I.; Marcu-Malina, V. Quantitative peripheral blood perturbations of γδ T cells in human disease and their clinical implications. Clin. Rev. Allergy Immunol. 2014, 47, 311–333. [Google Scholar] [CrossRef]
- Silva-Santos, B.; Mensurado, S.; Coffelt, S.B. gammadelta T cells: Pleiotropic immune effectors with therapeutic potential in cancer. Nat. Rev. Cancer 2019, 19, 392–404. [Google Scholar] [CrossRef] [Green Version]
- Rudan, I.; Sidhu, S.; Papana, A.; Meng, S.J.; Yu, X.-W.; Wang, W.; Campbell-Page, R.M.; Demaio, A.R.; Nair, H.; Sridhar, D.; et al. Prevalence of rheumatoid arthritis in low- and middle-income countries: A systematic review and analysis. J. Glob. Health 2015, 5, 010409. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.F.; Yang, C.Y.; Chao, S.C.; Li, J.S.; Weng, T.H.; Lei, H.Y. Distribution of double-negative (CD4- CD8-, DN) T subsets in blood and synovial fluid from patients with rheumatoid arthritis. Clin. Rheumatol. 1999, 18, 227–231. [Google Scholar] [CrossRef]
- Gaur, P.; Misra, R.; Aggarwal, A. Natural killer cell and gamma delta T cell alterations in enthesitis related arthritis category of juvenile idiopathic arthritis. Clin. Immunol. 2015, 161, 163–169. [Google Scholar] [CrossRef]
- Hassan, J.; Feighery, C.; Bresnihan, B.; Whelan, A. Effect of gold therapy on CD5+ B-cells and TCR gamma delta+ T-cells in patients with rheumatoid arthritis. Rheumatol. Int. 1991, 11, 175–178. [Google Scholar] [CrossRef]
- Abuzakouk, M.; Feighery, C.; Kelleher, D.; O’Briain, D.S.; Jones, E.; Weir, D.; Casey, E.; O’Farrelly, C. Increased HLA-DR and CD44 antigen expression in the gut: Evidence of extraarticular immunological activity in rheumatoid arthritis. J. Rheumatol. 1999, 26, 1869–1876. [Google Scholar]
- Mitogawa, T.; Nishiya, K.; Ota, Z. Frequency of gamma delta T cells in peripheral blood, synovial fluid, synovial membrane and lungs from patients with rheumatoid arthritis. Acta. Med. Okayama 1992, 46, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Brennan, F.; Plater-Zyberk, C.; Maini, R.N.; Feldmann, M. Coordinate expansion of ‘fetal type’ lymphocytes (TCR gamma delta+T and CD5+B) in rheumatoid arthritis and primary Sjogren’s syndrome. Clin. Exp. Immunol. 1989, 77, 175–178. [Google Scholar] [PubMed]
- Hu, C.; Qian, L.; Miao, Y.; Huang, Q.; Miao, P.; Wang, P.; Yu, Q.; Nie, H.; Zhang, J.; He, D.; et al. Antigen-presenting effects of effector memory Vgamma9Vdelta2 T cells in rheumatoid arthritis. Cell Mol. Immunol. 2012, 9, 245–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guggino, G.; Orlando, V.; Saieva, L.; Ruscitti, P.; Cipriani, P.; La Manna, M.P.; Giacomelli, R.; Alessandro, R.; Triolo, G.; Ciccia, F.; et al. Downregulation of miRNA17-92 cluster marks Vgamma9Vdelta2 T cells from patients with rheumatoid arthritis. Arthritis. Res. Ther. 2018, 20, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, D.; Shen, M.; Gu, B.; Wang, X.; Wang, D.; Li, X.; Sun, L. (99) Tc-methylene diphosphonate improves rheumatoid arthritis disease activity by increasing the frequency of peripheral gammadelta T cells and CD4(+) CD25(+) Foxp3(+) Tregs. Int. J. Rheum. Dis. 2016, 19, 586–593. [Google Scholar] [CrossRef]
- Mo, W.X.; Yin, S.S.; Chen, H.; Zhou, C.; Zhou, J.X.; Zhao, L.D.; Fei, Y.Y.; Yang, H.X.; Guo, J.B.; Mao, Y.J.; et al. Chemotaxis of Vdelta2 T cells to the joints contributes to the pathogenesis of rheumatoid arthritis. Ann. Rheum. Dis. 2017, 76, 2075–2084. [Google Scholar] [CrossRef]
- Lamour, A.; Jouen-Beades, F.; Lees, O.; Gilbert, D.; Le Loet, X.; Tron, F. Analysis of T cell receptors in rheumatoid arthritis: The increased expression of HLA-DR antigen on circulating gamma delta+ T cells is correlated with disease activity. Clin. Exp. Immunol. 1992, 89, 217–222. [Google Scholar] [CrossRef]
- Schwaneck, E.C.; Renner, R.; Junker, L.; Einsele, H.; Gadeholt, O.; Geissinger, E.; Kleinert, S.; Gernert, M.; Tony, H.P.; Schmalzing, M. Prevalence and Characteristics of Persistent Clonal T Cell Large Granular Lymphocyte Expansions in Rheumatoid Arthritis: A Comprehensive Analysis of 529 Patients. Okayama Rheumatol. 2018, 70, 1914–1922. [Google Scholar] [CrossRef] [Green Version]
- Yabe, M.; Medeiros, L.J.; Wang, S.A.; Konoplev, S.; Ok, C.Y.; Loghavi, S.; Lu, G.; Flores, L.; Khoury, J.D.; Cason, R.C.; et al. Clinicopathologic, Immunophenotypic, Cytogenetic, and Molecular Features of gammadelta T-Cell Large Granular Lymphocytic Leukemia: An Analysis of 14 Patients Suggests Biologic Differences With alphabeta T-Cell Large Granular Lymphocytic Leukemia. [corrected]. Am. J. Clin. Pathol. 2015, 144, 607–619. [Google Scholar] [CrossRef]
- Bourgault-Rouxel, A.S.; Loughran, T.P., Jr.; Zambello, R.; Epling-Burnette, P.K.; Semenzato, G.; Donadieu, J.; Amiot, L.; Fest, T.; Lamy, T. Clinical spectrum of gammadelta+ T cell LGL leukemia: Analysis of 20 cases. Leuk. Res. 2008, 32, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.R.; Haynes, B.F. Increase in TCR gamma delta T lymphocytes in synovia from rheumatoid arthritis patients with active synovitis. J. Clin. Immunol. 1992, 12, 130–138. [Google Scholar] [CrossRef] [PubMed]
- el-Gabalawy, H.S.; Keillor, J. Immunohistologic study of T-cell receptor delta-chain expression in rheumatoid synovial membranes. Semin. Arthritis Rheumatol. 1992, 21, 239–245. [Google Scholar] [CrossRef]
- Bodman-Smith, M.D.; Anand, A.; Durand, V.; Youinou, P.Y.; Lydyard, P.M. Decreased expression of FcgammaRIII (CD16) by gammadelta T cells in patients with rheumatoid arthritis. Immunology 2000, 99, 498–503. [Google Scholar] [CrossRef]
- Andreu, J.L.; Trujillo, A.; Alonso, J.M.; Mulero, J.; Martinez, C. Selective expansion of T cells bearing the gamma/delta receptor and expressing an unusual repertoire in the synovial membrane of patients with rheumatoid arthritis. Arthritis Rheumatol. 1991, 34, 808–814. [Google Scholar] [CrossRef]
- Soderstrom, K.; Bucht, A.; Halapi, E.; Lundqvist, C.; Gronberg, A.; Nilsson, E.; Orsini, D.L.; van de Wal, Y.; Koning, F.; Hammarstrom, M.L.; et al. High expression of V gamma 8 is a shared feature of human gamma delta T cells in the epithelium of the gut and in the inflamed synovial tissue. J. Immunol. 1994, 152, 6017–6027. [Google Scholar]
- Kageyama, Y.; Koide, Y.; Miyamoto, S.; Inoue, T.; Yoshida, T.O. The biased V gamma gene usage in the synovial fluid of patients with rheumatoid arthritis. Eur. J. Immunol. 1994, 24, 1122–1129. [Google Scholar] [CrossRef] [Green Version]
- Olive, C.; Gatenby, P.A.; Serjeantson, S.W. Variable gene usage of T cell receptor gamma- and delta-chain transcripts expressed in synovia and peripheral blood of patients with rheumatoid arthritis. Clin. Exp. Immunol. 1992, 87, 172–177. [Google Scholar] [CrossRef]
- Doherty, P.J.; Inman, R.D.; Laxer, R.M.; Silverman, E.D.; Yang, S.X.; Suurmann, I.; Pan, S. Analysis of T cell receptor gamma transcripts in right and left knee synovial fluids of patients with rheumatoid arthritis. J. Rheumatol. 1996, 23, 1143–1150. [Google Scholar]
- Olive, C.; Gatenby, P.A.; Serjeantson, S.W. Evidence for oligoclonality of T cell receptor delta chain transcripts expressed in rheumatoid arthritis patients. Eur. J. Immunol. 1992, 22, 2587–2593. [Google Scholar] [CrossRef]
- Le Nours, J.; Gherardin, N.A.; Ramarathinam, S.H.; Awad, W.; Wiede, F.; Gully, B.S.; Khandokar, Y.; Praveena, T.; Wubben, J.M.; Sandow, J.J.; et al. A class of gammadelta T cell receptors recognize the underside of the antigen-presenting molecule MR1. Science 2019, 366, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Chomarat, P.; Kjeldsen-Kragh, J.; Quayle, A.J.; Natvig, J.B.; Miossec, P. Different cytokine production profiles of gamma delta T cell clones: Relation to inflammatory arthritis. Eur. J. Immunol. 1994, 24, 2087–2091. [Google Scholar] [CrossRef] [PubMed]
- Bank, I.; Tanay, A.; Migdal, A.; Book, M.; Livneh, A. V gamma 9-V delta 2+ gamma delta T cells from a patient with Felty syndrome that exhibit aberrant response to triggering of the CD3 molecule can regulate immunoglobulin secretion by B cells. Clin. Immunol. Immunopathol. 1995, 74, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Pollinger, B.; Junt, T.; Metzler, B.; Walker, U.A.; Tyndall, A.; Allard, C.; Bay, S.; Keller, R.; Raulf, F.; Di Padova, F.; et al. Th17 cells, not IL-17+ gammadelta T cells, drive arthritic bone destruction in mice and humans. J. Immunol. 2011, 186, 2602–2612. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Sano, S.; Nieves, E.; De Libero, G.; Rosa, D.; Modlin, R.L.; Brenner, M.B.; Bloom, B.R.; Morita, C.T. Nonpeptide ligands for human gamma delta T cells. Proc. Natl. Acad. Sci. USA 1994, 91, 8175–8179. [Google Scholar] [CrossRef] [Green Version]
- Laurent, A.J.; Bindslev, N.; Johansson, B.; Berg, L. Synergistic effects of ethanol and isopentenyl pyrophosphate on expansion of gammadelta T cells in synovial fluid from patients with arthritis. PLoS ONE 2014, 9, e103683. [Google Scholar] [CrossRef]
- Li, N.L.; Zhang, D.Q.; Zhou, K.Y.; Cartman, A.; Leroux, J.Y.; Poole, A.R.; Zhang, Y.P. Isolation and characteristics of autoreactive T cells specific to aggrecan G1 domain from rheumatoid arthritis patients. Cell Res. 2000, 10, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Ohta, M.; Sato, N. The cytotoxic analysis of T cell receptor V delta 1+ T cell lines derived from the synovial fluid of rheumatoid arthritis patients. Clin. Exp. Immunol. 1994, 97, 193–199. [Google Scholar] [CrossRef]
- Kogure, A.; Miyata, M.; Nishimaki, T.; Kasukawa, R. Proliferative response of synovial fluid mononuclear cells of patients with rheumatoid arthritis to mycobacterial 65 kDa heat shock protein and its association with HLA-DR+.gamma delta+ T cells. J. Rheumatol. 1994, 21, 1403–1408. [Google Scholar]
- Holoshitz, J.; Vila, L.M.; Keroack, B.J.; McKinley, D.R.; Bayne, N.K. Dual antigenic recognition by cloned human gamma delta T cells. J. Clin. Invest. 1992, 89, 308–314. [Google Scholar] [CrossRef]
- Bank, I.; Cohen, L.; Mouallem, M.; Farfel, Z.; Grossman, E.; Ben-Nun, A. gammadelta T cell subsets in patients with arthritis and chronic neutropenia. Ann. Rheum. Dis. 2002, 61, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Pelegri, C.; Kuhnlein, P.; Buchner, E.; Schmidt, C.B.; Franch, A.; Castell, M.; Hunig, T.; Emmrich, F.; Kinne, R.W. Depletion of gamma/delta T cells does not prevent or ameliorate, but rather aggravates, rat adjuvant arthritis. Arthritis Rheumatol. 1996, 39, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, S.; Schlipkoter, E.; Kinne, R.; Hunig, T.; Emmrich, F. Suppression and prevention of adjuvant arthritis in rats by a monoclonal antibody to the alpha/beta T cell receptor. Eur. J. Immunol. 1990, 20, 2805–2808. [Google Scholar] [CrossRef] [PubMed]
- Jansson, A.M.; Lorentzen, J.C.; Bucht, A. CD8+ cells suppress oil-induced arthritis. Clin. Exp. Immunol. 2000, 120, 532–536. [Google Scholar] [CrossRef]
- Carlson, B.C.; Jansson, A.M.; Larsson, A.; Bucht, A.; Lorentzen, J.C. The endogenous adjuvant squalene can induce a chronic T-cell-mediated arthritis in rats. Am. J. Pathol. 2000, 156, 2057–2065. [Google Scholar] [CrossRef] [Green Version]
- Corthay, A.; Johansson, A.; Vestberg, M.; Holmdahl, R. Collagen-induced arthritis development requires alpha beta T cells but not gamma delta T cells: Studies with T cell-deficient (TCR mutant) mice. Int. Immunol. 1999, 11, 1065–1073. [Google Scholar] [CrossRef]
- Arai, K.; Yamamura, S.; Hanyu, T.; Takahashi, H.E.; Umezu, H.; Watanabe, H.; Abo, T. Extrathymic differentiation of resident T cells in the joints of mice with collagen-induced arthritis. J. Immunol. 1996, 157, 5170–5177. [Google Scholar]
- Roark, C.L.; French, J.D.; Taylor, M.A.; Bendele, A.M.; Born, W.K.; O’Brien, R.L. Exacerbation of collagen-induced arthritis by oligoclonal, IL-17-producing gamma delta T cells. J. Immunol. 2007, 179, 5576–5583. [Google Scholar] [CrossRef] [Green Version]
- Cornelissen, F.; Mus, A.M.; Asmawidjaja, P.S.; van Hamburg, J.P.; Tocker, J.; Lubberts, E. Interleukin-23 is critical for full-blown expression of a non-autoimmune destructive arthritis and regulates interleukin-17A and RORgammat in gammadelta T cells. Arthritis. Res. Ther. 2009, 11, R194. [Google Scholar] [CrossRef] [Green Version]
- Xue, X.; Soroosh, P.; De Leon-Tabaldo, A.; Luna-Roman, R.; Sablad, M.; Rozenkrants, N.; Yu, J.; Castro, G.; Banie, H.; Fung-Leung, W.P.; et al. Pharmacologic modulation of RORgammat translates to efficacy in preclinical and translational models of psoriasis and inflammatory arthritis. Sci. Rep. 2016, 6, 37977. [Google Scholar] [CrossRef] [Green Version]
- Blazek, K.; Eames, H.L.; Weiss, M.; Byrne, A.J.; Perocheau, D.; Pease, J.E.; Doyle, S.; McCann, F.; Williams, R.O.; Udalova, I.A. IFN-lambda resolves inflammation via suppression of neutrophil infiltration and IL-1beta production. J. Exp. Med. 2015, 212, 845–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harnett, M.M.; Harnett, W.; Pineda, M.A. The parasitic worm product ES-62 up-regulates IL-22 production by gammadelta T cells in the murine model of Collagen-Induced Arthritis. Inflamm. Cell Signal. 2014, 1. [Google Scholar] [CrossRef]
- Pineda, M.A.; McGrath, M.A.; Smith, P.C.; Al-Riyami, L.; Rzepecka, J.; Gracie, J.A.; Harnett, W.; Harnett, M.M. The parasitic helminth product ES-62 suppresses pathogenesis in collagen-induced arthritis by targeting the interleukin-17-producing cellular network at multiple sites. Arthritis Rheumatol. 2012, 64, 3168–3178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, J.; Kamanaka, M.; Hao, J.; Hao, Z.; Jiang, X.; Craft, J.E.; Flavell, R.A.; Wu, Z.; Hong, Z.; Zhao, L.; et al. IL-10 signaling in CD4+ T cells is critical for the pathogenesis of collagen-induced arthritis. Arthritis. Res. Ther. 2011, 13, R212. [Google Scholar] [CrossRef] [Green Version]
- Ito, Y.; Usui, T.; Kobayashi, S.; Iguchi-Hashimoto, M.; Ito, H.; Yoshitomi, H.; Nakamura, T.; Shimizu, M.; Kawabata, D.; Yukawa, N.; et al. Gamma/delta T cells are the predominant source of interleukin-17 in affected joints in collagen-induced arthritis, but not in rheumatoid arthritis. Arthritis Rheumatol. 2009, 60, 2294–2303. [Google Scholar] [CrossRef]
- Evans-Marin, H.; Rogier, R.; Koralov, S.B.; Manasson, J.; Roeleveld, D.; van der Kraan, P.M.; Scher, J.U.; Koenders, M.I.; Abdollahi-Roodsaz, S. Microbiota-Dependent Involvement of Th17 Cells in Murine Models of Inflammatory Arthritis. Arthritis. Rheumatol. 2018, 70, 1971–1983. [Google Scholar] [CrossRef]
- Bouchareychas, L.; Grossinger, E.M.; Kang, M.; Adamopoulos, I.E. gammadeltaTCR regulates production of interleukin-27 by neutrophils and attenuates inflammatory arthritis. Sci. Rep. 2018, 8, 7590. [Google Scholar] [CrossRef] [Green Version]
- Akitsu, A.; Ishigame, H.; Kakuta, S.; Chung, S.H.; Ikeda, S.; Shimizu, K.; Kubo, S.; Liu, Y.; Umemura, M.; Matsuzaki, G.; et al. IL-1 receptor antagonist-deficient mice develop autoimmune arthritis due to intrinsic activation of IL-17-producing CCR2(+)Vgamma6(+)gammadelta T cells. Nat. Commun. 2015, 6, 7464. [Google Scholar] [CrossRef] [Green Version]
- Noto Llana, M.; Sarnacki, S.H.; Morales, A.L.; Aya Castaneda, M.D.R.; Giacomodonato, M.N.; Blanco, G.; Cerquetti, M.C. Activation of iNKT Cells Prevents Salmonella-Enterocolitis and Salmonella-Induced Reactive Arthritis by Downregulating IL-17-Producing gammadeltaT Cells. Front. Cell Infect. Microbiol. 2017, 7, 398. [Google Scholar] [CrossRef]
- Khmaladze, I.; Kelkka, T.; Guerard, S.; Wing, K.; Pizzolla, A.; Saxena, A.; Lundqvist, K.; Holmdahl, M.; Nandakumar, K.S.; Holmdahl, R. Mannan induces ROS-regulated, IL-17A-dependent psoriasis arthritis-like disease in mice. Proc. Natl. Acad. Sci. USA 2014, 111, E3669–E3678. [Google Scholar] [CrossRef] [Green Version]
- Massa, M.; de Benedetti, F.; Robbioni, P.; Ramenghi, B.; Albani, S.; Martini, A. Association of methotrexate treatment with a decrease of double negative (CD4-CD8-) and gamma/delta T cell levels in patients with juvenile rheumatoid arthritis. J. Rheumatol. 1993, 20, 1944–1948. [Google Scholar] [PubMed]
- Rosser, E.C.; Lom, H.; Bending, D.; Duurland, C.L.; Bajaj-Elliott, M.; Wedderburn, L.R. Innate Lymphoid Cells and T Cells Contribute to the Interleukin-17A Signature Detected in the Synovial Fluid of Patients With Juvenile Idiopathic Arthritis. Arthritis. Rheumatol. 2019, 71, 460–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macaubas, C.; Nguyen, K.; Deshpande, C.; Phillips, C.; Peck, A.; Lee, T.; Park, J.L.; Sandborg, C.; Mellins, E.D. Distribution of circulating cells in systemic juvenile idiopathic arthritis across disease activity states. Clin. Immunol. 2010, 134, 206–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Licciardi, F.; Ceci, M.; Toppino, C.; Turco, M.; Martino, S.; Ricotti, E.; Ferro, F.; Montin, D. Low synovial double negative T and gammadelta T cells predict longer free-disease survival in oligoarticular JIA. Cytometry B Clin. Cytom. 2018, 94, 423–427. [Google Scholar] [CrossRef]
- Kjeldsen-Kragh, J.; Quayle, A.J.; Vinje, O.; Natvig, J.B.; Forre, O. A high proportion of the V delta 1+ synovial fluid gamma delta T cells in juvenile rheumatoid arthritis patients express the very early activation marker CD69, but carry the high molecular weight isoform of the leucocyte common antigen (CD45RA). Clin. Exp. Immunol. 1993, 91, 202–206. [Google Scholar] [CrossRef]
- Berkun, Y.; Bendersky, A.; Gerstein, M.; Goldstein, I.; Padeh, S.; Bank, I. GammadeltaT cells in juvenile idiopathic arthritis: Higher percentages of synovial Vdelta1+ and Vgamma9+ T cell subsets are associated with milder disease. J. Rheumatol. 2011, 38, 1123–1129. [Google Scholar] [CrossRef]
- Kessel, C.; Lippitz, K.; Weinhage, T.; Hinze, C.; Wittkowski, H.; Holzinger, D.; Fall, N.; Grom, A.A.; Gruen, N.; Foell, D. Proinflammatory Cytokine Environments Can Drive Interleukin-17 Overexpression by gamma/delta T Cells in Systemic Juvenile Idiopathic Arthritis. Arthritis. Rheumatol. 2017, 69, 1480–1494. [Google Scholar] [CrossRef]
- Bendersky, A.; Marcu-Malina, V.; Berkun, Y.; Gerstein, M.; Nagar, M.; Goldstein, I.; Padeh, S.; Bank, I. Cellular interactions of synovial fluid gammadelta T cells in juvenile idiopathic arthritis. J. Immunol. 2012, 188, 4349–4359. [Google Scholar] [CrossRef] [Green Version]
- Avau, A.; Mitera, T.; Put, S.; Put, K.; Brisse, E.; Filtjens, J.; Uyttenhove, C.; Van Snick, J.; Liston, A.; Leclercq, G.; et al. Systemic juvenile idiopathic arthritis-like syndrome in mice following stimulation of the immune system with Freund’s complete adjuvant: Regulation by interferon-gamma. Arthritis. Rheumatol. 2014, 66, 1340–1351. [Google Scholar] [CrossRef]
- Kenna, T.J.; Davidson, S.I.; Duan, R.; Bradbury, L.A.; McFarlane, J.; Smith, M.; Weedon, H.; Street, S.; Thomas, R.; Thomas, G.P.; et al. Enrichment of circulating interleukin-17-secreting interleukin-23 receptor-positive gamma/delta T cells in patients with active ankylosing spondylitis. Arthritis Rheum. 2012, 64, 1420–1429. [Google Scholar] [CrossRef] [Green Version]
- Venken, K.; Jacques, P.; Mortier, C.; Labadia, M.E.; Decruy, T.; Coudenys, J.; Hoyt, K.; Wayne, A.L.; Hughes, R.; Turner, M.; et al. RORgammat inhibition selectively targets IL-17 producing iNKT and gammadelta-T cells enriched in Spondyloarthritis patients. Nat. Commun. 2019, 10, 9. [Google Scholar] [CrossRef] [Green Version]
- Al-Mossawi, M.H.; Chen, L.; Fang, H.; Ridley, A.; de Wit, J.; Yager, N.; Hammitzsch, A.; Pulyakhina, I.; Fairfax, B.P.; Simone, D.; et al. Unique transcriptome signatures and GM-CSF expression in lymphocytes from patients with spondyloarthritis. Nat. Commun. 2017, 8, 1510. [Google Scholar] [CrossRef]
- Toussirot, E.; Laheurte, C.; Gaugler, B.; Gabriel, D.; Saas, P. Increased IL-22- and IL-17A-Producing Mucosal-Associated Invariant T Cells in the Peripheral Blood of Patients With Ankylosing Spondylitis. Front. Immunol. 2018, 9, 1610. [Google Scholar] [CrossRef] [PubMed]
- Gracey, E.; Qaiyum, Z.; Kuruvilla, J.; Inman, R.D. Gamma Delta T Cell Subset V Gamma 2+ Expansion Associated with Longterm Infliximab Treatment in a Patient with Ankylosing Spondylitis. J. Rheumatol. 2016, 43, 2079–2082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermann, E.; Ackermann, B.; Duchmann, R.; Meyer zum Buschenfelde, K.H. Synovial fluid MHC-unrestricted gamma delta-T lymphocytes contribute to antibacterial and anti-self cytotoxicity in the spondylarthropathies. Clin. Exp. Rheumatol. 1995, 13, 187–191. [Google Scholar] [PubMed]
- Reinhardt, A.; Yevsa, T.; Worbs, T.; Lienenklaus, S.; Sandrock, I.; Oberdorfer, L.; Korn, T.; Weiss, S.; Forster, R.; Prinz, I. Interleukin-23-Dependent gamma/delta T Cells Produce Interleukin-17 and Accumulate in the Enthesis, Aortic Valve, and Ciliary Body in Mice. Arthritis Rheumatol. 2016, 68, 2476–2486. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Kang, N.; Zhou, J.; Guo, Y.; Zhang, X.; Cui, L.; Ba, D.; He, W. Downregulation of CD94/NKG2A inhibitory receptor on decreased gammadelta T cells in patients with systemic lupus erythematosus. Scand. J. Immunol. 2012, 76, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Yuan, Y.; Zhao, L.; Ye, Z.; Xu, J.; Li, M.; Jiang, Z.; Jiang, Y. Association of gammadelta T Cell Compartment Size to Disease Activity and Response to Therapy in SLE. PLoS ONE 2016, 11, e0157772. [Google Scholar] [CrossRef]
- Li, X.; Kang, N.; Zhang, X.; Dong, X.; Wei, W.; Cui, L.; Ba, D.; He, W. Generation of human regulatory gammadelta T cells by TCRgammadelta stimulation in the presence of TGF-beta and their involvement in the pathogenesis of systemic lupus erythematosus. J. Immunol. 2011, 186, 6693–6700. [Google Scholar] [CrossRef] [Green Version]
- Robak, E.; Niewiadomska, H.; Robak, T.; Bartkowiak, J.; Blonski, J.Z.; Wozniacka, A.; Pomorski, L.; Sysa-Jedrezejowska, A. Lymphocyctes Tgammadelta in clinically normal skin and peripheral blood of patients with systemic lupus erythematosus and their correlation with disease activity. Mediators. Inflamm. 2001, 10, 179–189. [Google Scholar] [CrossRef]
- Holcombe, R.F.; Baethge, B.A.; Wolf, R.E.; Betzing, K.W.; Stewart, R.M. Natural killer cells and gamma delta T cells in scleroderma: Relationship to disease duration and anti-Scl-70 antibodies. Ann. Rheum. Dis. 1995, 54, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Zordan, T.; Tsokos, G.C.; Datta, S.K. Pathogenic anti-DNA autoantibody-inducing T helper cell lines from patients with active lupus nephritis: Isolation of CD4-8- T helper cell lines that express the gamma delta T-cell antigen receptor. Proc. Natl. Acad. Sci. USA 1990, 87, 7020–7024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuelson, E.M.; Laird, R.M.; Papillion, A.M.; Tatum, A.H.; Princiotta, M.F.; Hayes, S.M. Reduced B lymphoid kinase (Blk) expression enhances proinflammatory cytokine production and induces nephrosis in C57BL/6-lpr/lpr mice. PLoS ONE 2014, 9, e92054. [Google Scholar] [CrossRef] [PubMed]
- Page, N.; Schall, N.; Strub, J.M.; Quinternet, M.; Chaloin, O.; Decossas, M.; Cung, M.T.; Van Dorsselaer, A.; Briand, J.P.; Muller, S. The spliceosomal phosphopeptide P140 controls the lupus disease by interacting with the HSC70 protein and via a mechanism mediated by gammadelta T cells. PLoS ONE 2009, 4, e5273. [Google Scholar] [CrossRef]
- Giacomelli, R.; Matucci-Cerinic, M.; Cipriani, P.; Ghersetich, I.; Lattanzio, R.; Pavan, A.; Pignone, A.; Cagnoni, M.L.; Lotti, T.; Tonietti, G. Circulating Vdelta1+ T cells are activated and accumulate in the skin of systemic sclerosis patients. Arthritis Rheumatol. 1998, 41, 327–334. [Google Scholar] [CrossRef]
- Wood, G.M.; Dawisha, S.; Gourley, M. Characteristics of HPRT-mutant T cell lines in a lupus patient treated with cyclophosphamide. Arthritis Rheumatol. 1994, 37, 1548–1552. [Google Scholar] [CrossRef]
- Peng, S.L.; Madaio, M.P.; Hayday, A.C.; Craft, J. Propagation and regulation of systemic autoimmunity by gammadelta T cells. J. Immunol. 1996, 157, 5689–5698. [Google Scholar]
- Qiu, F.; Li, T.; Zhang, K.; Wan, J.; Qi, X. CD4(+)B220(+)TCRgammadelta(+) T cells produce IL-17 in lupus-prone MRL/lpr mice. Int. Immunopharmacol. 2016, 38, 31–39. [Google Scholar] [CrossRef]
- Jacinto, J.; Kim, P.J.; Singh, R.R. Disparate effects of depletion of CD1d-reactive T cells during early versus late stages of disease in a genetically susceptible model of lupus. Lupus 2012, 21, 485–490. [Google Scholar] [CrossRef] [Green Version]
- Rezende, R.M.; Lanser, A.J.; Rubino, S.; Kuhn, C.; Skillin, N.; Moreira, T.G.; Liu, S.; Gabriely, G.; David, B.A.; Menezes, G.B.; et al. gammadelta T cells control humoral immune response by inducing T follicular helper cell differentiation. Nat. Commun. 2018, 9, 3151. [Google Scholar] [CrossRef] [Green Version]
- Riedel, J.H.; Paust, H.J.; Krohn, S.; Turner, J.E.; Kluger, M.A.; Steinmetz, O.M.; Krebs, C.F.; Stahl, R.A.; Panzer, U. IL-17F Promotes Tissue Injury in Autoimmune Kidney Diseases. J. Am. Soc. Nephrol. 2016, 27, 3666–3677. [Google Scholar] [CrossRef] [PubMed]
- Riccieri, V.; Parisi, G.; Spadaro, A.; Scrivo, R.; Barone, F.; Moretti, T.; Bernardini, G.; Strom, R.; Taccari, E.; Valesini, G. Reduced circulating natural killer T cells and gamma/delta T cells in patients with systemic sclerosis. J. Rheumatol. 2005, 32, 283–286. [Google Scholar] [PubMed]
- Bendersky, A.; Markovits, N.; Bank, I. Vgamma9+ gammadelta T cells in systemic sclerosis patients are numerically and functionally preserved and induce fibroblast apoptosis. Immunobiology 2010, 215, 380–394. [Google Scholar] [CrossRef]
- Segawa, S.; Goto, D.; Horikoshi, M.; Kondo, Y.; Umeda, N.; Hagiwara, S.; Yokosawa, M.; Hirota, T.; Miki, H.; Tsuboi, H.; et al. Involvement of CD161+ Vdelta1+ gammadelta T cells in systemic sclerosis: Association with interstitial pneumonia. Rheumatology 2014, 53, 2259–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda-Hayakawa, I.; Hasegawa, M.; Hamaguchi, Y.; Takehara, K.; Fujimoto, M. Circulating gamma/delta T cells in systemic sclerosis exhibit activated phenotype and enhance gene expression of proalpha2(I) collagen of fibroblasts. J. Dermatol. Sci. 2013, 69, 54–60. [Google Scholar] [CrossRef]
- Henriques, A.; Silva, C.; Santiago, M.; Henriques, M.J.; Martinho, A.; Trindade, H.; da Silva, J.A.; Silva-Santos, B.; Paiva, A. Subset-specific alterations in frequencies and functional signatures of gammadelta T cells in systemic sclerosis patients. Inflamm. Res. 2016, 65, 985–994. [Google Scholar] [CrossRef]
- Kahaleh, M.B.; Fan, P.S.; Otsuka, T. Gammadelta receptor bearing T cells in scleroderma: Enhanced interaction with vascular endothelial cells in vitro. Clin. Immunol. 1999, 91, 188–195. [Google Scholar] [CrossRef]
- Yurovsky, V.V.; Sutton, P.A.; Schulze, D.H.; Wigley, F.M.; Wise, R.A.; Howard, R.F.; White, B. Expansion of selected V delta 1+ gamma delta T cells in systemic sclerosis patients. J. Immunol. 1994, 153, 881–891. [Google Scholar]
- Migalovich Sheikhet, H.; Villacorta Hidalgo, J.; Fisch, P.; Balbir-Gurman, A.; Braun-Moscovici, Y.; Bank, I. Dysregulated CD25 and Cytokine Expression by gammadelta T Cells of Systemic Sclerosis Patients Stimulated With Cardiolipin and Zoledronate. Front. Immunol. 2018, 9, 753. [Google Scholar] [CrossRef]
- Marcu-Malina, V.; Balbir-Gurman, A.; Dardik, R.; Braun-Moscovici, Y.; Segel, M.J.; Bank, I. A Novel Prothrombotic Pathway in Systemic Sclerosis Patients: Possible Role of Bisphosphonate-Activated gammadelta T Cells. Front. Immunol. 2014, 5, 414. [Google Scholar] [CrossRef] [Green Version]
- Markovits, N.; Bendersky, A.; Loebstein, R.; Brusel, M.; Kessler, E.; Bank, I. Anti-fibrotic characteristics of Vgamma9+ gammadelta T cells in systemic sclerosis. Clin. Exp. Rheumatol. 2016, 34 (Suppl. 100), 23–29. [Google Scholar] [PubMed]
- Carbone, L.D.; Warrington, K.J.; Barrow, K.D.; Pugazhenthi, M.; Watsky, M.A.; Somes, G.; Ingels, J.; Postlethwaite, A.E. Pamidronate infusion in patients with systemic sclerosis results in changes in blood mononuclear cell cytokine profiles. Clin. Exp. Immunol. 2006, 146, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Bank, I. The Role of gammadelta T Cells in Fibrotic Diseases. Rambam Maimonides Med. J. 2016, 7. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Disease (Tissue) | Total γδ T Cells (Relative to Normal) | Vδ1+ T Cells (Relative to Normal) | Vγ9Vδ2 T Cells (Relative to Normal) | References |
|---|---|---|---|---|
| RA (PB) | Equal or decreased | Ratio relative to Vδ2 is increased. Sometimes includes oligoclonal expansions | Equal or decreased (in established long term disease), increased VγδVδ2 TEMRA, decreased naïve Vγ9Vδ2 T cells. Sometimes include oligoclonal expansions. Increases noted after anti TNFα and gold salt therapy. Negative association with disease activity | |
| RA (synovium) | Polyclonal repertoire sometimes containing oligoclonal expansions common to different joints. HLADR expression increased, CD16 decreased. | Increased relative to Vδ2. Often using Vγ8 or Vγ3 along with Vδ1 in the TCR | Relatively expanded compared to the PB, may use Jδ2. | [32] |
| JIA (PB) | May be Increased in oligoarticular and quiescent systemic JIA otherwise equal. Increase of IL-17 producers in SJIA | Increase of Vδ1+CD69+ T cells | Increase of Vδ2+CD69+ T cells | [71] |
| JIA (synovium) | Higher than PB in oligoarticular JIA. Otherwise equal to percentage in PB | Higher CD69+ than in PB, usually CD45RA+, higher in ANA+ patients, inversely associated with age at onset, and with recurrence of synovitis | Higher CD69+ than PB. Usually CD45RO+ inversely associated with age at onset, positively with recovery | [74] |
| AS (pB) | Total decreased, but enriched for IL23R+ γδ T cells secreting IL-17 and in IL-17 and GM-CSF double producing γδ T cells | Elevated in AS patients receiving anti TNFα, secrete IFNγ. | [80,82,83,84] | |
| AS (enthesium/synovium) | RORγt+ iNKT and γδ-hi T cells increased, producing IL-17 | Enriched for IL-23+ RORγt+ iNKT and γδ-hi | [80,82,83,84] | |
| SLE (pB) | Decreased, but increase of γδ T cells expressing CD69 and HLADR, and decrease of TNFα and IL-17 secreting cells. Inverse correlation with disease activity. γδ lines help anti DNA production by B cells | decreased, but increased in inactive SLE | decreased | [18,87,88] |
| SLE (skin) | increased | increased | [90] | |
| SSc [pB) | Decreased especially in early term disease (less than 3 years), diffuse disease and in SCL70+ patient. Otherwise equal. | Increased Vδ1+ and CD161+Vδ1+ especially in patients without ILD. Increase of Vδ1+CD49d+, and HLADR+ cells. May be profibrotic in vitro, may respond to cardiolipin via CD1d | Unchanged, decreased, or increased in some patients with ILD, increased granzyme expression, cytotoxic to endothelial cells. Induce fibroblast apoptosis. May be anti fibrotic. | [91] |
| SSC (skin) | Increased, restricted clonality | [88,90,92,93,94,95] |
| Disease Model | Role of γδ T Cells | References |
|---|---|---|
| Rat adjuvant arthritis | No role in disease induction. Possible role in effector phase of disease. | [52,53,54,55] |
| Murine Collagen induced arthritis | Vγ4/Vδ4+ cells producing IL-17 are pathogenic. IL-17 production can be suppressed by inhibitor of RORγt and by IL-28A. ES-62, a phosphorylcholine containing glycoprotein and IL-10 reduce migration of IL-17 producing γδ T cells to the inflamed joint, which are maintained by IL-23, and are not associated with bone destruction. | [58] |
| Murine BSA induced arthritis | (RORγ)t+ IL-17 producing γδ T cells dependent upon IL-23 accumulated in arthritic joints. | [59] |
| Murine non antigen dependent arthritis | IL-1R and IL-23R expressing Vγ6+ γδ IL 17 cells are the main producers of IL-17 in joints of Il1rn -/- mice spontaneously developing arthritis. γδ T cells are responsible for arthritis in B10.RIII mice induced by gene transfer of IL-23. Arthritis induced by intraperitoneal injection of mannan is dependent upon IL-17 secreting γδ T cells. | [67,68,70] |
| Murine IFNγ-knockout (KO) | IL-17 secreting γδ T cells were shown to participate in arthritis and the systemic response to complete Freund adjuvant injection developing in these mice. | [79] |
| Murine IL-23 gene introduction | increased number of γδ T cells are found in Achilles tendon enthesis, aortic root, and adjacent to the ciliary body and secreted IL-17. | [86] |
| Murine MRL/lpr model of SLE | γδ T cells are protective from development of glomerulonephritis in the presence of αβ T cells, but mediate a less severe form of disease in their absence, mediated by cytokines and help for B cells. With age, some γδ T cells acquire a CD4+B220+ phenotype, and produce IL-17. In BLK+/-.lpr mice expressing low levels of Bruton lymphocyte kinase gene IL-17 and IFNγ producing γδ T cells are increased enhanced and mediate glomerular damage. γδ T cells induce phosphopeptide P140 mediated apoptosis of lymphocytes, which is associated with amelioration of disease in MRL/lpr mice. | [93,94,97] |
| lupus-prone NZB/NZW mice | CD1d restricted γδ T cells may be protective in young, and pathogenic in old mice. | [99] |
| Pristane induced model of SLE | γδ T cells in the kidney expressed IL-17F and A and attracted neutrophils to the kidney. TCRδ-/- mice developed milder glomerulonephritis, due to decreased T follicular helper cell differentiation dependent upon γδ T cell secretion of Wnt ligands. | [101] |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bank, I. The Role of Gamma Delta T Cells in Autoimmune Rheumatic Diseases. Cells 2020, 9, 462. https://doi.org/10.3390/cells9020462
Bank I. The Role of Gamma Delta T Cells in Autoimmune Rheumatic Diseases. Cells. 2020; 9(2):462. https://doi.org/10.3390/cells9020462
Chicago/Turabian StyleBank, Ilan. 2020. "The Role of Gamma Delta T Cells in Autoimmune Rheumatic Diseases" Cells 9, no. 2: 462. https://doi.org/10.3390/cells9020462
APA StyleBank, I. (2020). The Role of Gamma Delta T Cells in Autoimmune Rheumatic Diseases. Cells, 9(2), 462. https://doi.org/10.3390/cells9020462