Computational Identification and Analysis of Ubiquinone-Binding Proteins

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Benchmark Datasets
2.2. Feature Construction
2.2.1. Amino Acid Composition (AAC)
2.2.2. Dipeptide Composition (DC)
2.2.3. Position-Specific Scoring Matrix (PSSM)
2.3. Feature Selection Strategy
2.4. Binary Prediction Model
2.5. Parameter Tuning
2.6. Performance Evaluation
3. Results and Discussion
3.1. Comparison of Different Classifiers
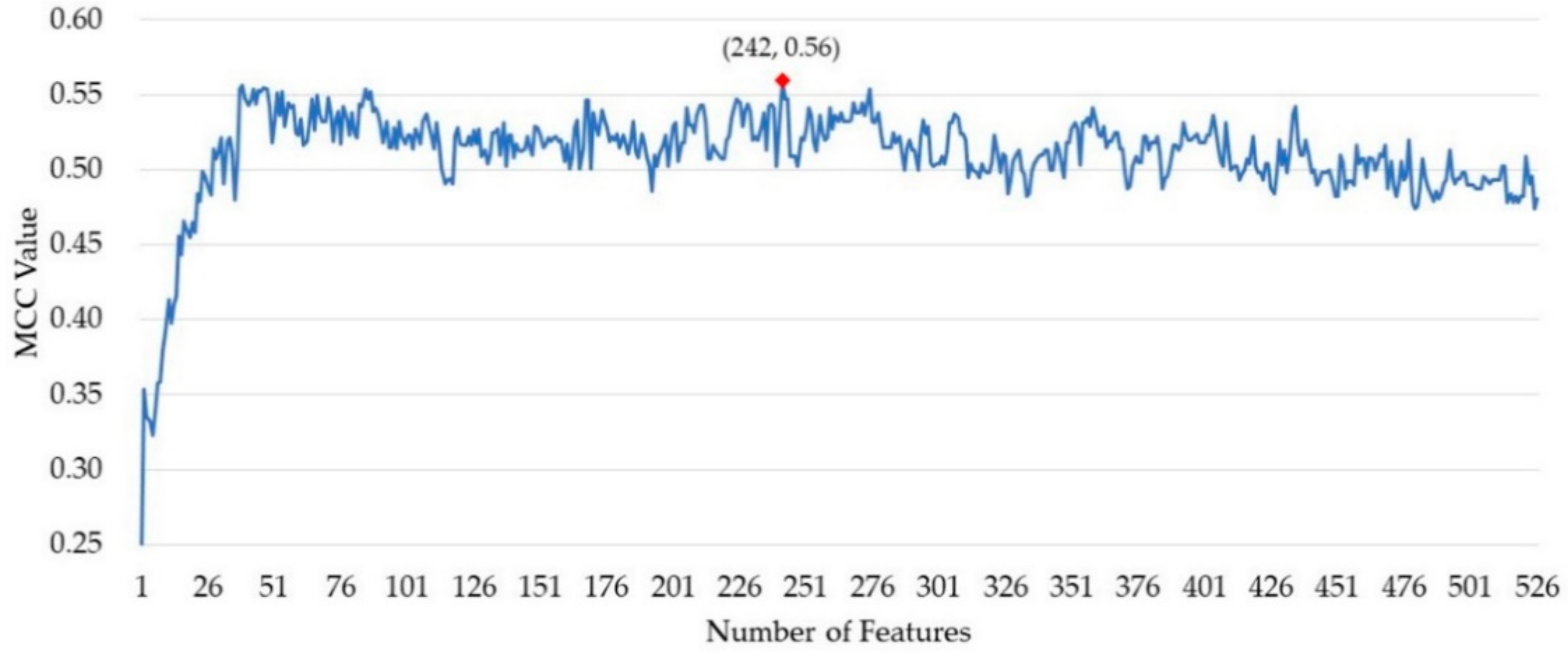
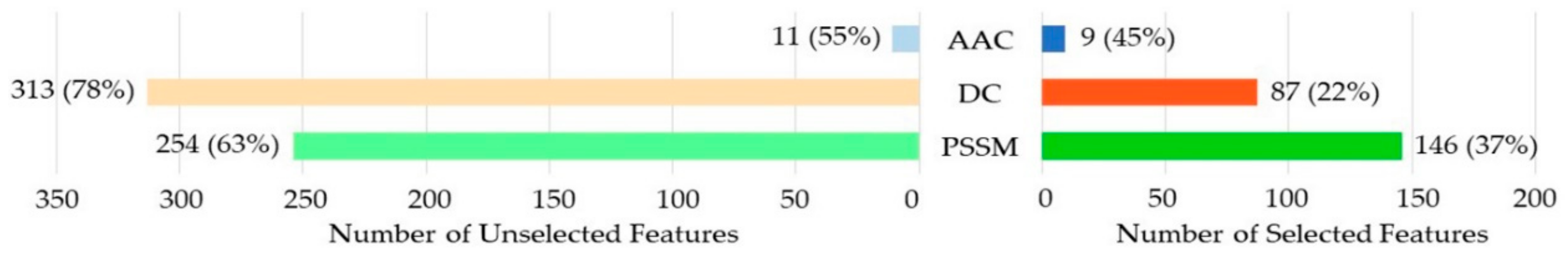
3.2. The Feature Selection Result
3.3. The Result of Parameter Tuning
3.4. Case Studies
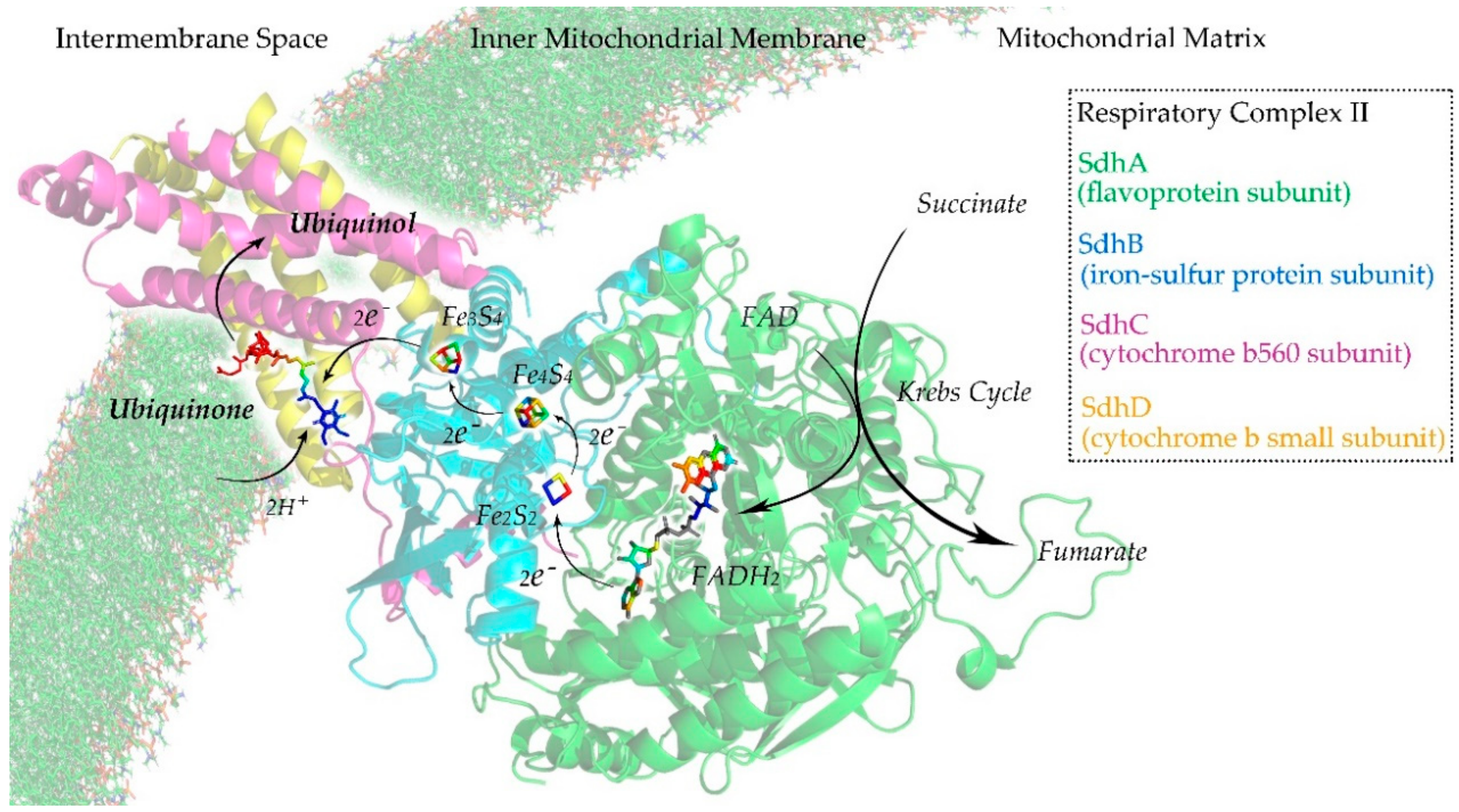
3.5. Ubiquinone-Binding Domain Analysis
3.6. Distribution of UBPs
3.6.1. Most UBPs are Membrane Proteins
3.6.2. Superfamilies of UBPs
3.7. Gene Ontology Enrichment Analysis
3.8. KEGG Pathway Enrichment Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ernster, L.; Dallner, G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim. Biophys. Acta 1995, 1271, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hekimi, S. Understanding Ubiquinone. Trends Cell Biol. 2016, 26, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Crane, F.L. Biochemical functions of coenzyme Q10. J. Am. Coll. Nutr. 2001, 20, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Jafari, M.; Mousavi, S.M.; Asgharzadeh, A.; Yazdani, N. Coenzyme Q10 in the treatment of heart failure: A systematic review of systematic reviews. Indian Heart J. 2018, 70, S111–S117. [Google Scholar] [CrossRef] [PubMed]
- Sobirin, M.A.; Herry, Y.; Sofia, S.N.; Uddin, I.; Rifqi, S.; Tsutsui, H. Effects of coenzyme Q10 supplementation on diastolic function in patients with heart failure with preserved ejection fraction. Drug Discov. Ther. 2019, 13, 38–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Shi, Z.; Liu, Q.; Quan, H.; Cheng, X. Effects of coenzyme Q10 intervention on diabetic kidney disease: A systematic review and meta-analysis. Medicine 2019, 98, e15850. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, J.; Han, E.; Wang, Y.; Gao, J. Efficacy of coenzyme Q10 in patients with chronic kidney disease: Protocol for a systematic review. BMJ Open 2019, 9, e029053. [Google Scholar] [CrossRef]
- Tafazoli, A. Coenzyme Q10 in breast cancer care. Future Oncol. 2017, 13, 1035–1041. [Google Scholar] [CrossRef]
- Vetvicka, V.; Vetvickova, J. Combination Therapy with Glucan and Coenzyme Q10 in Murine Experimental Autoimmune Disease and Cancer. Anticancer Res. 2018, 38, 3291–3297. [Google Scholar] [CrossRef] [Green Version]
- Tuz, K.; Li, C.; Fang, X.; Raba, D.A.; Liang, P.D.; Minh, D.D.L.; Juarez, O. Identification of the Catalytic Ubiquinone-binding Site of Vibrio cholerae Sodium-dependent NADH Dehydrogenase A NOVEL UBIQUINONE-BINDING MOTIF. J. Biol. Chem. 2017, 292, 3039–3048. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, B.J.; Daly, T.M.; Morrisey, J.M.; Mather, M.W.; Vaidya, A.B.; Bergman, L.W. Characterization of a Plasmodium falciparum Orthologue of the Yeast Ubiquinone-Binding Protein, Coq10p. PLoS ONE 2016, 11, e0152197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fathima, A.J.; Murugaboopathi, G.; Selvam, P. Pharmacophore Mapping of Ligand Based Virtual Screening, Molecular Docking and Molecular Dynamic Simulation Studies for Finding Potent NS2B/NS3 Protease Inhibitors as Potential Anti-dengue Drug Compounds. Curr. Bioinform. 2018, 13, 606–616. [Google Scholar] [CrossRef]
- Basith, S.; Manavalan, B.; Shin, T.H.; Lee, G. iGHBP: Computational identification of growth hormone binding proteins from sequences using extremely randomised tree. Comput. Struct. Biotec. J. 2018, 16, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.C.T.; Ahmad, S.D.R. Enabling full-length evolutionary profiles based deep convolutional neural network for predicting DNA-binding proteins from sequence. Proteins 2019, 88, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.Y.; Rijnbeek, P.; Yan, J.C.; Shen, H.B. Prediction of RNA-protein sequence and structure binding preferences using deep convolutional and recurrent neural networks. BMC Genomics 2018, 19, 511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- UniProt, C. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Zou, Q.; Lin, G.; Jiang, X.; Liu, X.; Zeng, X. Sequence clustering in bioinformatics: An empirical study. Brief. Bioinform. 2018, 21(1), 1–10. [Google Scholar] [CrossRef]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef]
- Zhang, J.; Chai, H.T.; Guo, S.; Guo, H.P.; Li, Y.L. High-Throughput Identification of Mammalian Secreted Proteins Using Species-Specific Scheme and Application to Human Proteome. Molecules 2018, 23, 1448. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chai, H.; Gao, B.; Yang, G.; Ma, Z. HEMEsPred: Structure-Based Ligand-Specific Heme Binding Residues Prediction by Using Fast-Adaptive Ensemble Learning Scheme. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 15, 147–156. [Google Scholar] [CrossRef]
- Khan, M.; Hayat, M.; Khan, S.A.; Iqbal, N. Unb-DPC: Identify mycobacterial membrane protein types by incorporating un-biased dipeptide composition into Chou’s general PseAAC. J. Theor. Biol. 2017, 415, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chai, H.T.; Yang, G.F.; Ma, Z.Q. Prediction of bioluminescent proteins by using sequence-derived features and lineage-specific scheme. BMC Bioinform. 2017, 18, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, J.C.; Lin, X.; Chen, X.W. On position-specific scoring matrix for protein function prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2011, 8, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.; Honigschmid, P.; Frishman, D. Residue co-evolution helps predict interaction sites in alpha-helical membrane proteins. J. Struct. Biol. 2019, 206, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Zangooei, M.H.; Jalili, S. Protein secondary structure prediction using DWKF based on SVR-NSGAII. Neurocomputing 2012, 94, 87–101. [Google Scholar] [CrossRef]
- Qu, K.Y.; Wei, L.Y.; Zou, Q. A Review of DNA-binding Proteins Prediction Methods. Curr. Bioinform. 2019, 14, 246–254. [Google Scholar] [CrossRef]
- Wei, L.; Tang, J.; Zou, Q. Local-DPP: An improved DNA-binding protein prediction method by exploring local evolutionary information. Inform. Sci. 2017, 384, 135–144. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Ru, X.; Li, L.; Zou, Q. Incorporating Distance-Based Top-n-gram and Random Forest To Identify Electron Transport Proteins. J. Proteome Res. 2019, 18, 2931–2939. [Google Scholar] [CrossRef]
- Lv, Z.; Jin, S.; Ding, H.; Zou, Q. A Random Forest Sub-Golgi Protein Classifier Optimized via Dipeptide and Amino Acid Composition Features. Front. Bioeng. Biotechnol. 2019, 7, 215. [Google Scholar] [CrossRef] [Green Version]
- Breiman, L. Random Forests. Machine Learning 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Zhu, R.Q.; Zeng, D.L.; Kosorok, M.R. Reinforcement Learning Trees. J. Am. Stat. Assoc. 2015, 110, 1770–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Setiono, R. Incremental feature selection. Appl. Intell. 1998, 9, 217–230. [Google Scholar] [CrossRef]
- Chen, T.Q.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Kdd’16: Proceedings of the 22nd Acm Sigkdd International Conference on Knowledge Discovery and Data Mining; ACM: San Francisco, CA, USA, 2016; pp. 785–794. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Yin, Y.; Quan, X.; Zhang, H. Gene Expression Value Prediction Based on XGBoost Algorithm. Front. Genet. 2019, 10, 1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, L.; Wang, J.; Zhao, L.; Wang, C.; Zhan, H. A Novel Protein Subcellular Localization Method With CNN-XGBoost Model for Alzheimer’s Disease. Front. Genet. 2018, 9, 751. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gribskov, M. IRESpy: An XGBoost model for prediction of internal ribosome entry sites. BMC Bioinform. 2019, 20, 409. [Google Scholar] [CrossRef]
- Coello, C.C.; Lechuga, M.S. MOPSO: A proposal for multiple objective particle swarm optimization. In Proceedings of the 2002 Congress on Evolutionary Computation. CEC’02 (Cat. No. 02TH8600), Honolulu, HI, USA, 12–17 May 2002; Volume 2, pp. 1051–1056. [Google Scholar] [CrossRef]
- Eberhart, R.; Kennedy, J. Particle swarm optimization. Proc. IEEE Int. Conf. Neural Netw. 1995, 4, 1942–1948. [Google Scholar] [CrossRef]
- Huang, L.S.; Sun, G.; Cobessi, D.; Wang, A.C.; Shen, J.T.; Tung, E.Y.; Anderson, V.E.; Berry, E.A. 3-Nitropropionic acid is a suicide inhibitor of mitochondrial respiration that, upon oxidation by Complex II, forms a covalent adduct with a catalytic base arginine in the active site of the enzyme. J. Biol Chem 2006, 281, 5965–5972. [Google Scholar] [CrossRef] [Green Version]
- Horsefield, R.; Yankovskaya, V.; Sexton, G.; Whittingham, W.; Shiomi, K.; Omura, S.; Byrne, B.; Cecchini, G.; Iwata, S. Structural and computational analysis of the quinone-binding site of complex II (succinate-ubiquinone oxidoreductase): A mechanism of electron transfer and proton conduction during ubiquinone reduction. J. Biol Chem 2006, 281, 7309–7316. [Google Scholar] [CrossRef] [Green Version]
- Ishii, N.; Fujii, M.; Hartman, P.S.; Tsuda, M.; Yasuda, K.; Senoo-Matsuda, N.; Yanase, S.; Ayusawa, D.; Suzuki, K. A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and ageing in nematodes. Nature 1998, 394, 694–697. [Google Scholar] [CrossRef]
- Ishii, T.; Yasuda, K.; Akatsuka, A.; Hino, O.; Hartman, P.S.; Ishii, N. A mutation in the SDHC gene of complex II increases oxidative stress, resulting in apoptosis and tumorigenesis. Cancer Res. 2005, 65, 203–209. [Google Scholar] [PubMed]
- Krebs, H.A. The citric acid cycle and the Szent-Gyorgyi cycle in pigeon breast muscle. Biochem. J. 1940, 775–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oyedotun, K.S.; Lemire, B.D. The quaternary structure of the Saccharomyces cerevisiae succinate dehydrogenase. Homology modeling, cofactor docking, and molecular dynamics simulation studies. J. Biol. Chem. 2004, 279, 9424–9431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, F.; Huo, X.; Zhai, Y.; Wang, A.; Xu, J.; Su, D.; Bartlam, M.; Rao, Z. Crystal structure of mitochondrial respiratory membrane protein complex II. Cell 2005, 121, 1043–1057. [Google Scholar] [CrossRef] [Green Version]
- Yankovskaya, V.; Horsefield, R.; Tornroth, S.; Luna-Chavez, C.; Miyoshi, H.; Leger, C.; Byrne, B.; Cecchini, G.; Iwata, S. Architecture of succinate dehydrogenase and reactive oxygen species generation. Science 2003, 299, 700–704. [Google Scholar] [CrossRef] [Green Version]
- Schneider, H.; Lemasters, J.J.; Hackenbrock, C.R. Lateral diffusion of ubiquinone during electron transfer in phospholipid- and ubiquinone-enriched mitochondrial membranes. J. Biol. Chem. 1982, 257, 10789–10793. [Google Scholar]
- Aberg, F.; Appelkvist, E.L.; Dallner, G.; Ernster, L. Distribution and redox state of ubiquinones in rat and human tissues. Arch. Biochem. Biophys. 1992, 295, 230–234. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Bashton, M.; Chothia, C. The geometry of domain combination in proteins. J. Mol. Biol 2002, 315, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, C.R.D.; Kroger, A.; Auer, M.; Michel, H. Structure of fumarate reductase from Wolinella succinogenes at 2.2 angstrom resolution. Nature 1999, 402, 377–385. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Description | Default | Threshold | Tuned |
|---|---|---|---|---|
| Booster Parameters | ||||
| learning_rate | Step size shrinkage | 0.10 | [0,0.5] | 0.08 |
| n_estimators | Number of trees | 100 | [100,2,000] | 162 |
| max_depth | The maximum depth of a tree | 3 | [1,10] | 8 |
| subsample | Percentage of samples used per tree | 1.00 | [0,1] | 0.75 |
| colsample_bytree | Percentage of features used per tree | 1.00 | [0,1] | 0.12 |
| Learning Task Parameters | ||||
| gamma | Controls a given node will split or not | 0 | [0,1] | 0.83 |
| reg_alpha | L1 regularization term on weight | 0 | [0,1] | 0.08 |
| reg_lambda | L2 regularization term on weights | 1.00 | [0,2] | |
| Classifier | Sen 1 | Spe 2 | Pre 3 | ACC 4 | F1 5 | MCC 6 |
|---|---|---|---|---|---|---|
| NB | 0.536 | 0.767 | 0.696 | 0.650 | 0.604 | 0.311 |
| MLP | 0.594 | 0.738 | 0.744 | 0.675 | 0.629 | 0.377 |
| SVM | 0.688 | 0.705 | 0.698 | 0.695 | 0.692 | 0.393 |
| AdaBoost | 0.704 | 0.734 | 0.723 | 0.719 | 0.712 | 0.438 |
| RF | 0.651 | 0.814 7 | 0.781 | 0.734 | 0.708 | 0.474 |
| XGBoost | 0.754 | 0.759 | 0.756 | 0.755 | 0.753 | 0.511 |
| Models | Sen | Spe | Pre | ACC | F1 | MCC |
|---|---|---|---|---|---|---|
| Cross-Validation | ||||||
| Default parameters | 0.759 | 0.786 | 0.779 | 0.772 | 0.768 | 0.545 |
| Tuned parameters | 0.746 * | 0.807 | 0.796 | 0.776 | 0.768 | 0.577 |
| Independent Validation | ||||||
| Default parameters | 0.649 | 0.760 | 0.727 | 0.705 | 0.686 | 0.411 |
| Tuned parameters | 0.703 | 0.811 | 0.788 | 0.757 | 0.743 | 0.517 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, C.; Jiang, W.; Wang, H.; Jiang, J.; Ma, Z.; Wang, H. Computational Identification and Analysis of Ubiquinone-Binding Proteins. Cells 2020, 9, 520. https://doi.org/10.3390/cells9020520
Lu C, Jiang W, Wang H, Jiang J, Ma Z, Wang H. Computational Identification and Analysis of Ubiquinone-Binding Proteins. Cells. 2020; 9(2):520. https://doi.org/10.3390/cells9020520
Chicago/Turabian StyleLu, Chang, Wenjie Jiang, Hang Wang, Jinxiu Jiang, Zhiqiang Ma, and Han Wang. 2020. "Computational Identification and Analysis of Ubiquinone-Binding Proteins" Cells 9, no. 2: 520. https://doi.org/10.3390/cells9020520
APA StyleLu, C., Jiang, W., Wang, H., Jiang, J., Ma, Z., & Wang, H. (2020). Computational Identification and Analysis of Ubiquinone-Binding Proteins. Cells, 9(2), 520. https://doi.org/10.3390/cells9020520