From Synthesis to Utilization: The Ins and Outs of Mitochondrial Heme

 ,
,  ,
, 
Abstract
:1. Introduction
2. Heme Biosynthesis
2.1. ALA Production
2.2. CPgenIII Formation
2.3. Coproporphyrinogen Oxidase (CPOX) and Protoporphyrinogen Oxidase (PPOX)
2.4. Ferrochelatase
2.5. Anemias and Porphyrias
3. From Heme b to Hemes c, o and a
3.1. Mitochondrial Heme b Pathways
3.2. Heme c Pathway
3.3. Heme a Pathway
3.3.1. Heme o Synthase
3.3.2. Heme a Synthase
3.3.3. Other Proteins Related to Heme a Biogenesis
3.4. Heme c and Heme a Pathway-Related Diseases
4. Extra-Mitochondrial Heme Trafficking
4.1. Exit of Mitochondrial Heme
4.2. Import of Exogenous Heme
4.3. Exogenous vs. Endogenous Heme
4.4. Heme Trafficking Factors
5. Multi-Model Comparison of Eukaryotic Heme Homeostasis
6. New Methods to Probe Heme Trafficking
7. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CO | Carbon monoxide |
| IMS | Mitochondrial intermembrane space |
| TCA | Tricarboxic acid |
| IMM | Inner mitochondrial membrane |
| 5-ALA | 5-aminolevulunic acid |
| ALAS | Aminolevulunic acid synthase |
| PBGS | Porphobilinogen synthase |
| PBG | Porphobilinogen |
| HMB | Hydroxymethylbilane |
| HMBS | Hydroxymethylbilane synthase |
| UPgen III | Uroporphyrinogen III |
| UROS | Uroporphyrinogen synthase |
| CPgen III | Coproporphyrinogen III |
| UROD | Uroporphyrinogen decarboxylase |
| PPgen IX | Protoporphyrinogen IX |
| CPOX | Coproporphyrinogen oxidase |
| Fe-PPIX | Iron-protoporphyrin IX |
| PPIX | Protoporphyrin IX |
| PPOX | Protoporphyrinogen oxidase |
| FECH | Ferrochelatase |
| PLP | Pyridoxal 5’-phosphate |
| OMM | Outer mitochondrial membrane |
| SLC | Solute carrier |
| α-KG | A-ketoglutarate |
| MTS | Mitochondria-targeting sequence |
| HRM | Heme regulatory motif |
| IRP | Iron regulatory protein |
| ATP | Adenosine triphosphate |
| ADP | Adenosine diphosphate |
| AAA+ | ATP hydrolase associate with various cellular activities |
| ANT | Adenine nucleotide translocator |
| KDH | A-ketoglutarate dehydrogenase |
| CcO | Cytochrome c oxidase |
| ETC | Electron transport chain |
| SDH | Succinate dehydrogenase |
| CCHL | Cytochrome c heme lyase |
| HCCS | Holocytochrome c synthase |
| MLS | Microphthalmia with linear skin defects |
| TMD | Transmembrane domain |
| FDX | Ferredoxin |
| MAMs | Mitochondria-associated membranes |
| ERMES | Endoplasmic reticulum-mitochondria encounter structure |
| MDV | Mitochondrial-derived vesicle |
| HRG | Heme responsive gene |
| ZnMP | Zinc mesoporphyrin |
| CHO | Chinese hamster ovary |
| LDL | Low-density lipoprotein |
| HDL | High-density lipoprotein |
| HO | Heme oxygenase |
| FeLV | Feline leukemia virus |
| ABC | ATP-binding cassette |
| BRCP | Breast cancer resistance protein |
| HBP | Heme-binding protein |
| FABP | Fatty acid-binding protein |
| GAPDH | Glyceraldehyde phosphate dehydrogenase |
| PGRMC1/2 | Progesterone receptor membrane component 1/2 |
| EGFP | Enhanced green fluorescent protein |
| ECFP | Enhanced cyan fluorescent protein |
| EYFP | Enhanced yellow fluorescent protein |
| HS | Heme sensor |
| FRET | Forster resonance energy transfer |
References
- Severance, S.; Hamza, I. Trafficking of heme and porphyrins in metazoa. Chem. Rev. 2009, 109, 4596–4616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, D.A.; Martinez-Guzman, O.; Reddi, A.R. Heme gazing: Illuminating eukaryotic heme trafficking, dynamics, and signaling with fluorescent heme sensors. Biochemistry 2017, 56, 1815–1823. [Google Scholar] [CrossRef] [PubMed]
- Reddi, A.R.; Hamza, I. Heme mobilization in animals: A metallolipid’s journey. Acc. Chem. Res. 2016, 49, 1104–1110. [Google Scholar] [CrossRef] [Green Version]
- Ponka, P. Cell biology of heme. Am. J. Med. Sci. 1999, 318, 241–256. [Google Scholar] [CrossRef]
- Tsiftsoglou, A.S.; Tsamadou, A.I.; Papadopoulou, L.C. Heme as key regulator of major mammalian cellular functions: Molecular, cellular, and pharmacological aspects. Pharmacol. Ther. 2006, 111, 327–345. [Google Scholar] [CrossRef]
- Kim, H.J.; Khalimonchuk, O.; Smith, P.M.; Winge, D.R. Structure, function, and assembly of heme centers in mitochondrial respiratory complexes. Biochim. Biophys. Acta 2012, 1823, 1604–1616. [Google Scholar] [CrossRef] [Green Version]
- Mense, S.M.; Zhang, L. Heme: A versatile signaling molecule controlling the activities of diverse regulators ranging from transcription factors to map kinases. Cell Res. 2006, 16, 681–692. [Google Scholar] [CrossRef] [Green Version]
- Faller, M.; Matsunaga, M.; Yin, S.; Loo, J.A.; Guo, F. Heme is involved in microrna processing. Nat. Struct. Mol. Biol. 2007, 14, 23–29. [Google Scholar] [CrossRef]
- Quick-Cleveland, J.; Jacob, J.P.; Weitz, S.H.; Shoffner, G.; Senturia, R.; Guo, F. The dgcr8 rna-binding heme domain recognizes primary micrornas by clamping the hairpin. Cell Rep. 2014, 7, 1994–2005. [Google Scholar] [CrossRef] [Green Version]
- Igarashi, K.; Sun, J. The heme-bach1 pathway in the regulation of oxidative stress response and erythroid differentiation. Antioxid Redox Signal 2006, 8, 107–118. [Google Scholar] [CrossRef]
- Raghuram, S.; Stayrook, K.R.; Huang, P.; Rogers, P.M.; Nosie, A.K.; McClure, D.B.; Burris, L.L.; Khorasanizadeh, S.; Burris, T.P.; Rastinejad, F. Identification of heme as the ligand for the orphan nuclear receptors rev-erbalpha and rev-erbbeta. Nat. Struct. Mol. Biol. 2007, 14, 1207–1213. [Google Scholar] [CrossRef] [Green Version]
- Hamza, I.; Dailey, H.A. One ring to rule them all: Trafficking of heme and heme synthesis intermediates in the metazoans. Biochim. Biophys. Acta 2012, 1823, 1617–1632. [Google Scholar] [CrossRef] [Green Version]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Mercurio, S.; Tolosano, E. Heme in pathophysiology: A matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharmacol. 2014, 5, 61. [Google Scholar] [CrossRef] [Green Version]
- Keel, S.B.; Doty, R.T.; Yang, Z.; Quigley, J.G.; Chen, J.; Knoblaugh, S.; Kingsley, P.D.; De Domenico, I.; Vaughn, M.B.; Kaplan, J.; et al. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science 2008, 319, 825–828. [Google Scholar] [CrossRef] [Green Version]
- Haldar, M.; Kohyama, M.; So, A.Y.; Kc, W.; Wu, X.; Briseno, C.G.; Satpathy, A.T.; Kretzer, N.M.; Arase, H.; Rajasekaran, N.S.; et al. Heme-mediated spi-c induction promotes monocyte differentiation into iron-recycling macrophages. Cell 2014, 156, 1223–1234. [Google Scholar] [CrossRef] [Green Version]
- Dutra, F.F.; Bozza, M.T. Heme on innate immunity and inflammation. Front. Pharmacol. 2014, 5, 115. [Google Scholar] [CrossRef] [Green Version]
- Desmard, M.; Boczkowski, J.; Poderoso, J.; Motterlini, R. Mitochondrial and cellular heme-dependent proteins as targets for the bioactive function of the heme oxygenase/carbon monoxide system. Antioxid Redox Signal 2007, 9, 2139–2155. [Google Scholar] [CrossRef]
- Kim, H.P.; Ryter, S.W.; Choi, A.M. Co as a cellular signaling molecule. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 411–449. [Google Scholar] [CrossRef]
- Shen, J.; Sheng, X.; Chang, Z.; Wu, Q.; Wang, S.; Xuan, Z.; Li, D.; Wu, Y.; Shang, Y.; Kong, X.; et al. Iron metabolism regulates p53 signaling through direct heme-p53 interaction and modulation of p53 localization, stability, and function. Cell Rep. 2014, 7, 180–193. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Bandyopadhyay, U. Free heme toxicity and its detoxification systems in human. Toxicol. Lett. 2005, 157, 175–188. [Google Scholar] [CrossRef]
- Sassa, S. Why heme needs to be degraded to iron, biliverdin ixalpha, and carbon monoxide? Antioxid Redox Signal 2004, 6, 819–824. [Google Scholar]
- Wu, M.L.; Ho, Y.C.; Lin, C.Y.; Yet, S.F. Heme oxygenase-1 in inflammation and cardiovascular disease. Am. J. Cardiovasc. Dis. 2011, 1, 150–158. [Google Scholar]
- Schipper, H.M.; Song, W.; Zukor, H.; Hascalovici, J.R.; Zeligman, D. Heme oxygenase-1 and neurodegeneration: Expanding frontiers of engagement. J. Neurochem. 2009, 110, 469–485. [Google Scholar] [CrossRef]
- Atamna, H.; Killilea, D.W.; Killilea, A.N.; Ames, B.N. Heme deficiency may be a factor in the mitochondrial and neuronal decay of aging. Proc. Natl. Acad. Sci. USA 2002, 99, 14807–14812. [Google Scholar] [CrossRef] [Green Version]
- Atamna, H.; Frey, W.H., 2nd. A role for heme in alzheimer’s disease: Heme binds amyloid beta and has altered metabolism. Proc. Natl. Acad. Sci. USA 2004, 101, 11153–11158. [Google Scholar] [CrossRef] [Green Version]
- Dailey, H.A.; Dailey, T.A.; Gerdes, S.; Jahn, D.; Jahn, M.; O’Brian, M.R.; Warren, M.J. Prokaryotic heme biosynthesis: Multiple pathways to a common essential product. Microbiol. Mol. Biol. Rev. 2017, 81, e00048-16. [Google Scholar] [CrossRef] [Green Version]
- Labbe-Bois, R.A.L.P. Tetrapyrrole and heme biosynthesis in the yeast sacchromyces cerevisiae. In Biosynthesis of Heme and Cholorophylls; Dailey, H.A., Ed.; Green Pub. Associates and Wiley-Interscience: New York, NY, USA, 1990; pp. 235–285. [Google Scholar]
- Ajioka, R.S.; Phillips, J.D.; Kushner, J.P. Biosynthesis of heme in mammals. Biochim. Biophys. Acta 2006, 1763, 723–736. [Google Scholar] [CrossRef] [Green Version]
- May, B.K.; Dogra, S.C.; Sadlon, T.J.; Bhasker, C.R.; Cox, T.C.; Bottomley, S.S. Molecular regulation of heme biosynthesis in higher vertebrates. Prog. Nucleic Acid Res. Mol. Biol. 1995, 51, 1–51. [Google Scholar]
- Sun, F.; Cheng, Y.; Chen, C. Regulation of heme biosynthesis and transport in metazoa. Sci. China Life Sci. 2015, 58, 757–764. [Google Scholar] [CrossRef] [Green Version]
- Hunter, G.A.; Ferreira, G.C. 5-aminolevulinate synthase: Catalysis of the first step of heme biosynthesis. Cell Mol. Biol. (Noisy-le-grand) 2009, 55, 102–110. [Google Scholar]
- Hunter, G.A.; Ferreira, G.C. Molecular enzymology of 5-aminolevulinate synthase, the gatekeeper of heme biosynthesis. Biochim. Biophys. Acta 2011, 1814, 1467–1473. [Google Scholar] [CrossRef] [Green Version]
- Guernsey, D.L.; Jiang, H.; Campagna, D.R.; Evans, S.C.; Ferguson, M.; Kellogg, M.D.; Lachance, M.; Matsuoka, M.; Nightingale, M.; Rideout, A.; et al. Mutations in mitochondrial carrier family gene slc25a38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat. Genet. 2009, 41, 651–653. [Google Scholar] [CrossRef]
- Lunetti, P.; Damiano, F.; De Benedetto, G.; Siculella, L.; Pennetta, A.; Muto, L.; Paradies, E.; Marobbio, C.M.; Dolce, V.; Capobianco, L. Characterization of human and yeast mitochondrial glycine carriers with implications for heme biosynthesis and anemia. J. Biol. Chem. 2016, 291, 19746–19759. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Murray, J.P.; Prykhozhij, S.V.; Dufay, J.N.; Steele, S.L.; Gaston, D.; Nasrallah, G.K.; Coombs, A.J.; Liwski, R.S.; Fernandez, C.V.; Berman, J.N.; et al. Glycine and folate ameliorate models of congenital sideroblastic anemia. PLoS Genet. 2016, 12, e1005783. [Google Scholar] [CrossRef] [Green Version]
- Shemin, D.; Kumin, S. The mechanism of porphyrin formation; the formation of a succinyl intermediate from succinate. J. Biol. Chem. 1952, 198, 827–837. [Google Scholar]
- Labbe, R.F.; Kurumada, T.; Onisawa, J. The role of succinyl-coa synthetase in the control of heme biosynthesis. Biochim. Biophys. Acta 1965, 111, 403–415. [Google Scholar] [CrossRef]
- Furuyama, K.; Sassa, S. Interaction between succinyl coa synthetase and the heme-biosynthetic enzyme alas-e is disrupted in sideroblastic anemia. J. Clin. Investig. 2000, 105, 757–764. [Google Scholar] [CrossRef] [Green Version]
- Burch, J.S.; Marcero, J.R.; Maschek, J.A.; Cox, J.E.; Jackson, L.K.; Medlock, A.E.; Phillips, J.D.; Dailey, H.A., Jr. Glutamine via alpha-ketoglutarate dehydrogenase provides succinyl-coa for heme synthesis during erythropoiesis. Blood 2018, 132, 987–998. [Google Scholar] [CrossRef] [Green Version]
- Astner, I.; Schulze, J.O.; Van den Heuvel, J.; Jahn, D.; Schubert, W.D.; Heinz, D.W. Crystal structure of 5-aminolevulinate synthase, the first enzyme of heme biosynthesis, and its link to xlsa in humans. EMBO J. 2005, 24, 3166–3177. [Google Scholar] [CrossRef] [Green Version]
- Brown, B.L.; Kardon, J.R.; Sauer, R.T.; Baker, T.A. Structure of the mitochondrial aminolevulinic acid synthase, a key heme biosynthetic enzyme. Structure 2018, 26, 580–589 e584. [Google Scholar] [CrossRef] [Green Version]
- Fratz, E.J.; Clayton, J.; Hunter, G.A.; Ducamp, S.; Breydo, L.; Uversky, V.N.; Deybach, J.C.; Gouya, L.; Puy, H.; Ferreira, G.C. Human erythroid 5-aminolevulinate synthase mutations associated with x-linked protoporphyria disrupt the conformational equilibrium and enhance product release. Biochemistry 2015, 54, 5617–5631. [Google Scholar] [CrossRef]
- Duncan, R.; Faggart, M.A.; Cornell, N.W. Phylogenetic analysis of the 5-aminolevulinate synthase gene. Biol. Bull. 1997, 193, 247–248. [Google Scholar] [CrossRef]
- Lathrop, J.T.; Timko, M.P. Regulation by heme of mitochondrial protein transport through a conserved amino acid motif. Science 1993, 259, 522–525. [Google Scholar] [CrossRef]
- Dailey, T.A.; Woodruff, J.H.; Dailey, H.A. Examination of mitochondrial protein targeting of haem synthetic enzymes: In vivo identification of three functional haem-responsive motifs in 5-aminolaevulinate synthase. Biochem. J. 2005, 386, 381–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, M.; Gora, M.; Rytka, J. Identification of rate-limiting steps in yeast heme biosynthesis. Biochem. Biophys. Res. Commun. 2003, 310, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Furuyama, K.; Kaneko, K.; Vargas, P.D. Heme as a magnificent molecule with multiple missions: Heme determines its own fate and governs cellular homeostasis. Tohoku J. Exp. Med. 2007, 213, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishop, D.F.; Henderson, A.S.; Astrin, K.H. Human delta-aminolevulinate synthase: Assignment of the housekeeping gene to 3p21 and the erythroid-specific gene to the x chromosome. Genomics 1990, 7, 207–214. [Google Scholar] [CrossRef]
- Yamamoto, M.; Hayashi, N.; Kikuchi, G. Evidence for the transcriptional inhibition by heme of the synthesis of delta-aminolevulinate synthase in rat liver. Biochem. Biophys. Res. Commun. 1982, 105, 985–990. [Google Scholar] [CrossRef]
- Sassa, S.; Granick, S. Induction of -aminolevulinic acid synthetase in chick embryo liver cells in cluture. Proc. Natl. Acad. Sci. USA 1970, 67, 517–522. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Hayashi, N.; Kikuchi, G. Translational inhibition by heme of the synthesis of hepatic delta-aminolevulinate synthase in a cell-free system. Biochem. Biophys. Res. Commun. 1983, 115, 225–231. [Google Scholar] [CrossRef]
- Hamilton, J.W.; Bement, W.J.; Sinclair, P.R.; Sinclair, J.F.; Alcedo, J.A.; Wetterhahn, K.E. Heme regulates hepatic 5-aminolevulinate synthase mrna expression by decreasing mrna half-life and not by altering its rate of transcription. Arch. Biochem. Biophys. 1991, 289, 387–392. [Google Scholar] [CrossRef]
- Tian, Q.; Li, T.; Hou, W.; Zheng, J.; Schrum, L.W.; Bonkovsky, H.L. Lon peptidase 1 (lonp1)-dependent breakdown of mitochondrial 5-aminolevulinic acid synthase protein by heme in human liver cells. J. Biol. Chem. 2011, 286, 26424–26430. [Google Scholar] [CrossRef] [Green Version]
- Munakata, H.; Sun, J.Y.; Yoshida, K.; Nakatani, T.; Honda, E.; Hayakawa, S.; Furuyama, K.; Hayashi, N. Role of the heme regulatory motif in the heme-mediated inhibition of mitochondrial import of 5-aminolevulinate synthase. J. Biochem. 2004, 136, 233–238. [Google Scholar] [CrossRef]
- Aich, A.; Freundlich, M.; Vekilov, P.G. The free heme concentration in healthy human erythrocytes. Blood Cells Mol. Dis. 2015, 55, 402–409. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, T.; O’Geen, H.; Keles, S.; Blahnik, K.; Linnemann, A.K.; Kang, Y.A.; Choi, K.; Farnham, P.J.; Bresnick, E.H. Discovering hematopoietic mechanisms through genome-wide analysis of gata factor chromatin occupancy. Mol. Cell 2009, 36, 667–681. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, K.; Furuyama, K.; Fujiwara, T.; Kobayashi, R.; Ishida, H.; Harigae, H.; Shibahara, S. Identification of a novel erythroid-specific enhancer for the alas2 gene and its loss-of-function mutation which is associated with congenital sideroblastic anemia. Haematologica 2014, 99, 252–261. [Google Scholar] [CrossRef] [Green Version]
- Doyle, F.; Tenenbaum, S.A. Trans-regulation of rna-binding protein motifs by microrna. Front. Genet. 2014, 5, 79. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, M.; Galy, B.; Schwanhaeusser, B.; Blake, J.; Bahr-Ivacevic, T.; Benes, V.; Selbach, M.; Muckenthaler, M.U.; Hentze, M.W. Iron regulatory protein-1 and -2: Transcriptome-wide definition of binding mrnas and shaping of the cellular proteome by iron regulatory proteins. Blood 2011, 118, e168–e179. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.; Anderson, S.A.; Gwynn, B.; Deck, K.M.; Chen, M.J.; Langer, N.B.; Shaw, G.C.; Huston, N.C.; Boyer, L.F.; Datta, S.; et al. Iron regulatory protein-1 protects against mitoferrin-1-deficient porphyria. J. Biol. Chem. 2014, 289, 7835–7843. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, H.; Kato, M.; Hori, H.; Ishimori, K.; Kirisako, T.; Tokunaga, F.; Iwai, K. Involvement of heme regulatory motif in heme-mediated ubiquitination and degradation of irp2. Mol. Cell 2005, 19, 171–181. [Google Scholar] [CrossRef]
- Yien, Y.Y.; Ducamp, S.; Van der Vorm, L.N.; Kardon, J.R.; Manceau, H.; Kannengiesser, C.; Bergonia, H.A.; Kafina, M.D.; Karim, Z.; Gouya, L.; et al. Mutation in human clpx elevates levels of delta-aminolevulinate synthase and protoporphyrin ix to promote erythropoietic protoporphyria. Proc. Natl. Acad. Sci. USA 2017, 114, e8045–e8052. [Google Scholar] [CrossRef] [Green Version]
- Abu-Farha, M.; Niles, J.; Willmore, W.G. Erythroid-specific 5-aminolevulinate synthase protein is stabilized by low oxygen and proteasomal inhibition. Biochem. Cell Biol. 2005, 83, 620–630. [Google Scholar] [CrossRef]
- Nilsson, R.; Schultz, I.J.; Pierce, E.L.; Soltis, K.A.; Naranuntarat, A.; Ward, D.M.; Baughman, J.M.; Paradkar, P.N.; Kingsley, P.D.; Culotta, V.C.; et al. Discovery of genes essential for heme biosynthesis through large-scale gene expression analysis. Cell Metab. 2009, 10, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Schubert, H.L.; Erskine, P.T.; Cooper, J.B. 5-aminolaevulinic acid dehydratase, porphobilinogen deaminase and uroporphyrinogen iii synthase. In Tetrapyrroles: Birth, Life, and Death; Warren, M.J., Smith, A.G., Eds.; Landes Bioscience: Austin, TX, USA, 2009; pp. 43–73. [Google Scholar]
- Krishnamurthy, P.C.; Du, G.; Fukuda, Y.; Sun, D.; Sampath, J.; Mercer, K.E.; Wang, J.; Sosa-Pineda, B.; Murti, K.G.; Schuetz, J.D. Identification of a mammalian mitochondrial porphyrin transporter. Nature 2006, 443, 586–589. [Google Scholar] [CrossRef]
- Helias, V.; Saison, C.; Ballif, B.A.; Peyrard, T.; Takahashi, J.; Takahashi, H.; Tanaka, M.; Deybach, J.C.; Puy, H.; Le Gall, M.; et al. Abcb6 is dispensable for erythropoiesis and specifies the new blood group system langereis. Nat. Genet. 2012, 44, 170–173. [Google Scholar] [CrossRef] [Green Version]
- Ulrich, D.L.; Lynch, J.; Wang, Y.; Fukuda, Y.; Nachagari, D.; Du, G.; Sun, D.; Fan, Y.; Tsurkan, L.; Potter, P.M.; et al. Atp-dependent mitochondrial porphyrin importer abcb6 protects against phenylhydrazine toxicity. J. Biol. Chem. 2012, 287, 12679–12690. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, Y.; Cheong, P.L.; Lynch, J.; Brighton, C.; Frase, S.; Kargas, V.; Rampersaud, E.; Wang, Y.; Sankaran, V.G.; Yu, B.; et al. The severity of hereditary porphyria is modulated by the porphyrin exporter and lan antigen abcb6. Nat. Commun. 2016, 7, 12353. [Google Scholar] [CrossRef]
- Rhee, H.W.; Zou, P.; Udeshi, N.D.; Martell, J.D.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 2013, 339, 1328–1331. [Google Scholar] [CrossRef] [Green Version]
- Azuma, M.; Kabe, Y.; Kuramori, C.; Kondo, M.; Yamaguchi, Y.; Handa, H. Adenine nucleotide translocator transports haem precursors into mitochondria. PLoS ONE 2008, 3, e3070. [Google Scholar] [CrossRef] [Green Version]
- Yien, Y.Y.; Robledo, R.F.; Schultz, I.J.; Takahashi-Makise, N.; Gwynn, B.; Bauer, D.E.; Dass, A.; Yi, G.; Li, L.; Hildick-Smith, G.J.; et al. Tmem14c is required for erythroid mitochondrial heme metabolism. J. Clin. Investig. 2014, 124, 4294–4304. [Google Scholar] [CrossRef] [Green Version]
- Shaw, G.C.; Cope, J.J.; Li, L.; Corson, K.; Hersey, C.; Ackermann, G.E.; Gwynn, B.; Lambert, A.J.; Wingert, R.A.; Traver, D.; et al. Mitoferrin is essential for erythroid iron assimilation. Nature 2006, 440, 96–100. [Google Scholar] [CrossRef]
- Muhlenhoff, U.; Hoffmann, B.; Richter, N.; Rietzschel, N.; Spantgar, F.; Stehling, O.; Uzarska, M.A.; Lill, R. Compartmentalization of iron between mitochondria and the cytosol and its regulation. Eur. J. Cell Biol. 2015, 94, 292–308. [Google Scholar] [CrossRef]
- Lane, D.J.; Merlot, A.M.; Huang, M.L.; Bae, D.H.; Jansson, P.J.; Sahni, S.; Kalinowski, D.S.; Richardson, D.R. Cellular iron uptake, trafficking and metabolism: Key molecules and mechanisms and their roles in disease. Biochim. Biophys. Acta 2015, 1853, 1130–1144. [Google Scholar] [CrossRef] [Green Version]
- Korolnek, T.; Hamza, I. Like iron in the blood of the people: The requirement for heme trafficking in iron metabolism. Front. Pharmacol. 2014, 5, 126. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Paw, B.H. Cellular and mitochondrial iron homeostasis in vertebrates. Biochim. Biophys. Acta 2012, 1823, 1459–1467. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.K.; Dailey, H.A.; Rose, J.P.; Burden, A.; Sellers, V.M.; Wang, B.C. The 2.0 a structure of human ferrochelatase, the terminal enzyme of heme biosynthesis. Nat. Struct. Biol. 2001, 8, 156–160. [Google Scholar] [CrossRef]
- Dailey, H.A.; Finnegan, M.G.; Johnson, M.K. Human ferrochelatase is an iron-sulfur protein. Biochemistry 1994, 33, 403–407. [Google Scholar] [CrossRef]
- Crouse, B.R.; Sellers, V.M.; Finnegan, M.G.; Dailey, H.A.; Johnson, M.K. Site-directed mutagenesis and spectroscopic characterization of human ferrochelatase: Identification of residues coordinating the [2fe-2s] cluster. Biochemistry 1996, 35, 16222–16229. [Google Scholar] [CrossRef]
- Shah, D.I.; Takahashi-Makise, N.; Cooney, J.D.; Li, L.; Schultz, I.J.; Pierce, E.L.; Narla, A.; Seguin, A.; Hattangadi, S.M.; Medlock, A.E.; et al. Mitochondrial atpif1 regulates haem synthesis in developing erythroblasts. Nature 2012, 491, 608–612. [Google Scholar] [CrossRef] [Green Version]
- Medlock, A.E.; Shiferaw, M.T.; Marcero, J.R.; Vashisht, A.A.; Wohlschlegel, J.A.; Phillips, J.D.; Dailey, H.A. Identification of the mitochondrial heme metabolism complex. PLoS ONE 2015, 10, e0135896. [Google Scholar] [CrossRef] [Green Version]
- Piel, R.B., 3rd; Shiferaw, M.T.; Vashisht, A.A.; Marcero, J.R.; Praissman, J.L.; Phillips, J.D.; Wohlschlegel, J.A.; Medlock, A.E. A novel role for progesterone receptor membrane component 1 (pgrmc1): A partner and regulator of ferrochelatase. Biochemistry 2016, 55, 5204–5217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Dailey, H.A.; Paw, B.H. Ferrochelatase forms an oligomeric complex with mitoferrin-1 and abcb10 for erythroid heme biosynthesis. Blood 2010, 116, 628–630. [Google Scholar] [CrossRef] [PubMed]
- Taketani, S.; Kakimoto, K.; Ueta, H.; Masaki, R.; Furukawa, T. Involvement of abc7 in the biosynthesis of heme in erythroid cells: Interaction of abc7 with ferrochelatase. Blood 2003, 101, 3274–3280. [Google Scholar] [CrossRef] [Green Version]
- Maio, N.; Kim, K.S.; Holmes-Hampton, G.; Singh, A.; Rouault, T.A. Dimeric ferrochelatase bridges abcb7 and abcb10 homodimers in an architecturally defined molecular complex required for heme biosynthesis. Haematologica 2019, 104, 1756–1767. [Google Scholar] [CrossRef]
- Thompson, A.M.; Reddi, A.R.; Shi, X.; Goldbeck, R.A.; Moenne-Loccoz, P.; Gibney, B.R.; Holman, T.R. Measurement of the heme affinity for yeast dap1p, and its importance in cellular function. Biochemistry 2007, 46, 14629–14637. [Google Scholar] [CrossRef] [Green Version]
- Craven, R.J.; Mallory, J.C.; Hand, R.A. Regulation of iron homeostasis mediated by the heme-binding protein dap1 (damage resistance protein 1) via the p450 protein erg11/cyp51. J. Biol. Chem. 2007, 282, 36543–36551. [Google Scholar] [CrossRef] [Green Version]
- Hughes, A.L.; Powell, D.W.; Bard, M.; Eckstein, J.; Barbuch, R.; Link, A.J.; Espenshade, P.J. Dap1/pgrmc1 binds and regulates cytochrome p450 enzymes. Cell Metab. 2007, 5, 143–149. [Google Scholar] [CrossRef] [Green Version]
- Galmozzi, A.; Kok, B.P.; Kim, A.S.; Montenegro-Burke, J.R.; Lee, J.Y.; Spreafico, R.; Mosure, S.; Albert, V.; Cintron-Colon, R.; Godio, C.; et al. Pgrmc2 is an intracellular haem chaperone critical for adipocyte function. Nature 2019, 576, 138–142. [Google Scholar] [CrossRef]
- Sweeny, E.A.; Singh, A.B.; Chakravarti, R.; Martinez-Guzman, O.; Saini, A.; Haque, M.M.; Garee, G.; Dans, P.D.; Hannibal, L.; Reddi, A.R.; et al. Glyceraldehyde-3-phosphate dehydrogenase is a chaperone that allocates labile heme in cells. J. Biol. Chem. 2018, 293, 14557–14568. [Google Scholar] [CrossRef] [Green Version]
- Hanna, D.A.; Harvey, R.M.; Martinez-Guzman, O.; Yuan, X.; Chandrasekharan, B.; Raju, G.; Outten, F.W.; Hamza, I.; Reddi, A.R. Heme dynamics and trafficking factors revealed by genetically encoded fluorescent heme sensors. Proc. Natl. Acad. Sci. USA 2016, 113, 7539–7544. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.D. Heme biosynthesis and the porphyrias. Mol. Genet. Metab. 2019, 128, 164–177. [Google Scholar] [CrossRef]
- Karim, Z.; Lyoumi, S.; Nicolas, G.; Deybach, J.C.; Gouya, L.; Puy, H. Porphyrias: A 2015 update. Clin. Res. Hepatol. Gastroenterol. 2015, 39, 412–425. [Google Scholar] [CrossRef]
- Schmitt, C.; Lenglet, H.; Yu, A.; Delaby, C.; Benecke, A.; Lefebvre, T.; Letteron, P.; Paradis, V.; Wahlin, S.; Sandberg, S.; et al. Recurrent attacks of acute hepatic porphyria: Major role of the chronic inflammatory response in the liver. J. Intern. Med. 2018, 284, 78–91. [Google Scholar] [CrossRef] [Green Version]
- Fraser, D.J.; Podvinec, M.; Kaufmann, M.R.; Meyer, U.A. Drugs mediate the transcriptional activation of the 5-aminolevulinic acid synthase (alas1) gene via the chicken xenobiotic-sensing nuclear receptor (cxr). J. Biol. Chem. 2002, 277, 34717–34726. [Google Scholar] [CrossRef] [Green Version]
- Rifkind, A.B.; Gillette, P.N.; Song, C.S.; Kappas, A. Induction of hepatic delta-amino-levulinic acid synthetase by oral contraceptive steroids. J. Clin. Endocrinol. Metab. 1970, 30, 330–335. [Google Scholar] [CrossRef]
- Handschin, C.; Lin, J.; Rhee, J.; Peyer, A.K.; Chin, S.; Wu, P.H.; Meyer, U.A.; Spiegelman, B.M. Nutritional regulation of hepatic heme biosynthesis and porphyria through pgc-1alpha. Cell 2005, 122, 505–515. [Google Scholar] [CrossRef] [Green Version]
- Whatley, S.D.; Ducamp, S.; Gouya, L.; Grandchamp, B.; Beaumont, C.; Badminton, M.N.; Elder, G.H.; Holme, S.A.; Anstey, A.V.; Parker, M.; et al. C-terminal deletions in the alas2 gene lead to gain of function and cause x-linked dominant protoporphyria without anemia or iron overload. Am. J. Hum. Genet. 2008, 83, 408–414. [Google Scholar] [CrossRef] [Green Version]
- Hoggins, M.; Dailey, H.A.; Hunter, C.N.; Reid, J.D. Direct measurement of metal ion chelation in the active site of human ferrochelatase. Biochemistry 2007, 46, 8121–8127. [Google Scholar] [CrossRef] [Green Version]
- Jeney, V.; Balla, J.; Yachie, A.; Varga, Z.; Vercellotti, G.M.; Eaton, J.W.; Balla, G. Pro-oxidant and cytotoxic effects of circulating heme. Blood 2002, 100, 879–887. [Google Scholar] [CrossRef] [Green Version]
- Mogi, T. Biosynthesis and role of heme o and heme a. In The Iron and Cobalt Pigments: Biosynthesis, Structure, and Degradation; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Academic Press: Amsterdam, The Netherlands, 2003; pp. 157–181. [Google Scholar]
- Barros, M.H.; Carlson, C.G.; Glerum, D.M.; Tzagoloff, A. Involvement of mitochondrial ferredoxin and cox15p in hydroxylation of heme o. FEBS Lett. 2001, 492, 133–138. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.R.; Allan, B.M.; Do, P.; Hegg, E.L. Identification of novel hemes generated by heme a synthase: Evidence for two successive monooxygenase reactions. Biochemistry 2002, 41, 10906–10913. [Google Scholar] [CrossRef]
- Brown, K.R.; Brown, B.M.; Hoagland, E.; Mayne, C.L.; Hegg, E.L. Heme a synthase does not incorporate molecular oxygen into the formyl group of heme a. Biochemistry 2004, 43, 8616–8624. [Google Scholar] [CrossRef]
- Hederstedt, L. Heme a biosynthesis. Biochim. Biophys. Acta 2012, 1817, 920–927. [Google Scholar] [CrossRef] [Green Version]
- Puustinen, A.; Wikstrom, M. The heme groups of cytochrome o from escherichia coli. Proc. Natl. Acad. Sci. USA 1991, 88, 6122–6126. [Google Scholar] [CrossRef] [Green Version]
- Mogi, T.; Saiki, K.; Anraku, Y. Biosynthesis and functional role of haem o and haem a. Mol. Microbiol. 1994, 14, 391–398. [Google Scholar] [CrossRef]
- Cassanova, N.; O’Brien, K.M.; Stahl, B.T.; McClure, T.; Poyton, R.O. Yeast flavohemoglobin, a nitric oxide oxidoreductase, is located in both the cytosol and the mitochondrial matrix: Effects of respiration, anoxia, and the mitochondrial genome on its intracellular level and distribution. J. Biol. Chem. 2005, 280, 7645–7653. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.H.; Crofts, A.R.; Gennis, R.B. Assignment of the histidine axial ligands to the cytochrome bh and cytochrome bl components of the bc1 complex from rhodobacter sphaeroides by site-directed mutagenesis. Biochemistry 1991, 30, 6747–6754. [Google Scholar] [CrossRef]
- Maklashina, E.; Rajagukguk, S.; McIntire, W.S.; Cecchini, G. Mutation of the heme axial ligand of escherichia coli succinate-quinone reductase: Implications for heme ligation in mitochondrial complex ii from yeast. Biochim. Biophys. Acta 2010, 1797, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Yankovskaya, V.; Horsefield, R.; Tornroth, S.; Luna-Chavez, C.; Miyoshi, H.; Leger, C.; Byrne, B.; Cecchini, G.; Iwata, S. Architecture of succinate dehydrogenase and reactive oxygen species generation. Science 2003, 299, 700–704. [Google Scholar] [CrossRef] [Green Version]
- Soto, I.C.; Fontanesi, F.; Myers, R.S.; Hamel, P.; Barrientos, A. A heme-sensing mechanism in the translational regulation of mitochondrial cytochrome c oxidase biogenesis. Cell Metab. 2012, 16, 801–813. [Google Scholar] [CrossRef] [Green Version]
- Djavadi-Ohaniance, L.; Rudin, Y.; Schatz, G. Identification of enzymically inactive apocytochrome c peroxidase in anaerobically grown saccharomyces cerevisiae. J. Biol. Chem. 1978, 253, 4402–4407. [Google Scholar] [PubMed]
- Guiard, B. Structure, expression and regulation of a nuclear gene encoding a mitochondrial protein: The yeast l(+)-lactate cytochrome c oxidoreductase (cytochrome b2). EMBO J. 1985, 4, 3265–3272. [Google Scholar] [CrossRef] [PubMed]
- Hildenbeutel, M.; Hegg, E.L.; Stephan, K.; Gruschke, S.; Meunier, B.; Ott, M. Assembly factors monitor sequential hemylation of cytochrome b to regulate mitochondrial translation. J. Cell Biol. 2014, 205, 511–524. [Google Scholar] [CrossRef] [Green Version]
- Kranz, R.G.; Richard-Fogal, C.; Taylor, J.S.; Frawley, E.R. Cytochrome c biogenesis: Mechanisms for covalent modifications and trafficking of heme and for heme-iron redox control. Microbiol. Mol. Biol. Rev. 2009, 73, 510–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babbitt, S.E.; San Francisco, B.; Mendez, D.L.; Lukat-Rodgers, G.S.; Rodgers, K.R.; Bretsnyder, E.C.; Kranz, R.G. Mechanisms of mitochondrial holocytochrome c synthase and the key roles played by cysteines and histidine of the heme attachment site, cys-xx-cys-his. J. Biol. Chem. 2014, 289, 28795–28807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, D.G.; Gabilly, S.T.; Dujardin, G.; Merchant, S.; Hamel, P.P. Overlapping specificities of the mitochondrial cytochrome c and c1 heme lyases. J. Biol. Chem. 2003, 278, 49732–49742. [Google Scholar] [CrossRef] [Green Version]
- Babbitt, S.E.; Sutherland, M.C.; San Francisco, B.; Mendez, D.L.; Kranz, R.G. Mitochondrial cytochrome c biogenesis: No longer an enigma. Trends Biochem. Sci. 2015, 40, 446–455. [Google Scholar] [CrossRef] [Green Version]
- Bernard, D.G.; Quevillon-Cheruel, S.; Merchant, S.; Guiard, B.; Hamel, P.P. Cyc2p, a membrane-bound flavoprotein involved in the maturation of mitochondrial c-type cytochromes. J. Biol. Chem. 2005, 280, 39852–39859. [Google Scholar] [CrossRef] [Green Version]
- Corvest, V.; Murrey, D.A.; Hirasawa, M.; Knaff, D.B.; Guiard, B.; Hamel, P.P. The flavoprotein cyc2p, a mitochondrial cytochrome c assembly factor, is a nad(p)h-dependent haem reductase. Mol. Microbiol. 2012, 83, 968–980. [Google Scholar] [CrossRef]
- Sun, Y.; Benabbas, A.; Zeng, W.; Kleingardner, J.G.; Bren, K.L.; Champion, P.M. Investigations of heme distortion, low-frequency vibrational excitations, and electron transfer in cytochrome c. Proc. Natl. Acad. Sci. USA 2014, 111, 6570–6575. [Google Scholar] [CrossRef] [Green Version]
- Khalimonchuk, O.; Rodel, G. Biogenesis of cytochrome c oxidase. Mitochondrion 2005, 5, 363–388. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, S.; Shinzawa-Itoh, K.; Nakashima, R.; Yaono, R.; Yamashita, E.; Inoue, N.; Yao, M.; Fei, M.J.; Libeu, C.P.; Mizushima, T.; et al. Redox-coupled crystal structural changes in bovine heart cytochrome c oxidase. Science 1998, 280, 1723–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalimonchuk, O.; Bestwick, M.; Meunier, B.; Watts, T.C.; Winge, D.R. Formation of the redox cofactor centers during cox1 maturation in yeast cytochrome oxidase. Mol. Cell Biol. 2010, 30, 1004–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalimonchuk, O.; Bestwick, M.; Meunier, B.; Watts, T.C.; Winge, D.R. Correction for khalimonchuk et al., “formation of the redox cofactor centers during cox1 maturation in yeast cytochrome oxidase”. Mol. Cell Biol. 2017, 37, 1004–1017. [Google Scholar] [CrossRef] [Green Version]
- Mick, D.U.; Fox, T.D.; Rehling, P. Inventory control: Cytochrome c oxidase assembly regulates mitochondrial translation. Nat. Rev. Mol. Cell Biol. 2011, 12, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Soto, I.C.; Fontanesi, F.; Liu, J.; Barrientos, A. Biogenesis and assembly of eukaryotic cytochrome c oxidase catalytic core. Biochim. Biophys. Acta 2012, 1817, 883–897. [Google Scholar] [CrossRef] [Green Version]
- Dennerlein, S.; Rehling, P. Human mitochondrial cox1 assembly into cytochrome c oxidase at a glance. J. Cell Sci. 2015, 128, 833–837. [Google Scholar] [CrossRef] [Green Version]
- Timon-Gomez, A.; Nyvltova, E.; Abriata, L.A.; Vila, A.J.; Hosler, J.; Barrientos, A. Mitochondrial cytochrome c oxidase biogenesis: Recent developments. Semin. Cell Dev. Biol. 2017, 76, 163–178. [Google Scholar] [CrossRef]
- Bestwick, M.; Khalimonchuk, O.; Pierrel, F.; Winge, D.R. The role of coa2 in hemylation of yeast cox1 revealed by its genetic interaction with cox10. Mol. Cell Biol. 2010, 30, 172–185. [Google Scholar] [CrossRef] [Green Version]
- Khalimonchuk, O.; Kim, H.; Watts, T.; Perez-Martinez, X.; Winge, D.R. Oligomerization of heme o synthase in cytochrome oxidase biogenesis is mediated by cytochrome oxidase assembly factor coa2. J. Biol. Chem. 2012, 287, 26715–26726. [Google Scholar] [CrossRef] [Green Version]
- Cheng, W.; Li, W. Structural insights into ubiquinone biosynthesis in membranes. Science 2014, 343, 878–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Wang, Y.; Hegg, E.L. Regulation of the heme a biosynthetic pathway: Differential regulation of heme a synthase and heme o synthase in saccharomyces cerevisiae. J. Biol. Chem. 2009, 284, 839–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reedy, C.J.; Gibney, B.R. Heme protein assemblies. Chem. Rev. 2004, 104, 617–649. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Reddi, A.R.; Wang, Z.; Khodaverdian, B.; Hegg, E.L.; Gibney, B.R. Evaluating the roles of the heme a side chains in cytochrome c oxidase using designed heme proteins. Biochemistry 2006, 45, 12530–12538. [Google Scholar] [CrossRef]
- Niwa, S.; Takeda, K.; Kosugi, M.; Tsutsumi, E.; Mogi, T.; Miki, K. Crystal structure of heme a synthase from bacillus subtilis. Proc. Natl. Acad. Sci. USA 2018, 115, 11953–11957. [Google Scholar] [CrossRef] [Green Version]
- Swenson, S.; Cannon, A.; Harris, N.J.; Taylor, N.G.; Fox, J.L.; Khalimonchuk, O. Analysis of oligomerization properties of heme a synthase provides insights into its function in eukaryotes. J. Biol. Chem. 2016, 291, 10411–10425. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Hach, A. Molecular mechanism of heme signaling in yeast: The transcriptional activator hap1 serves as the key mediator. Cell Mol. Life Sci. 1999, 56, 415–426. [Google Scholar] [CrossRef]
- Barros, M.H.; Nobrega, F.G.; Tzagoloff, A. Mitochondrial ferredoxin is required for heme a synthesis in saccharomyces cerevisiae. J. Biol. Chem. 2002, 277, 9997–10002. [Google Scholar] [CrossRef] [Green Version]
- Lill, R.; Muhlenhoff, U. Iron-sulfur protein biogenesis in eukaryotes: Components and mechanisms. Annu. Rev. Cell Dev. Biol. 2006, 22, 457–486. [Google Scholar] [CrossRef] [Green Version]
- Stehling, O.; Lill, R. The role of mitochondria in cellular iron-sulfur protein biogenesis: Mechanisms, connected processes, and diseases. Cold Spring Harb. Perspect Biol. 2013, 5, a011312. [Google Scholar] [CrossRef] [Green Version]
- Bureik, M.; Schiffler, B.; Hiraoka, Y.; Vogel, F.; Bernhardt, R. Functional expression of human mitochondrial cyp11b2 in fission yeast and identification of a new internal electron transfer protein, etp1. Biochemistry 2002, 41, 2311–2321. [Google Scholar] [CrossRef] [PubMed]
- Sheftel, A.D.; Stehling, O.; Pierik, A.J.; Elsasser, H.P.; Muhlenhoff, U.; Webert, H.; Hobler, A.; Hannemann, F.; Bernhardt, R.; Lill, R. Humans possess two mitochondrial ferredoxins, fdx1 and fdx2, with distinct roles in steroidogenesis, heme, and fe/s cluster biosynthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 11775–11780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McEwen, J.E.; Hong, K.H.; Park, S.; Preciado, G.T. Sequence and chromosomal localization of two pet genes required for cytochrome c oxidase assembly in saccharomyces cerevisiae. Curr. Genet. 1993, 23, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Barros, M.H.; Tzagoloff, A. Regulation of the heme a biosynthetic pathway in saccharomyces cerevisiae. FEBS Lett. 2002, 516, 119–123. [Google Scholar] [CrossRef] [Green Version]
- Taylor, N.G.; Swenson, S.; Harris, N.J.; Germany, E.M.; Fox, J.L.; Khalimonchuk, O. The assembly factor pet117 couples heme a synthase activity to cytochrome oxidase assembly. J. Biol. Chem. 2017, 292, 1815–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bareth, B.; Dennerlein, S.; Mick, D.U.; Nikolov, M.; Urlaub, H.; Rehling, P. The heme a synthase cox15 associates with cytochrome c oxidase assembly intermediates during cox1 maturation. Mol. Cell Biol. 2013, 33, 4128–4137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bundschuh, F.A.; Hannappel, A.; Anderka, O.; Ludwig, B. Surf1, associated with leigh syndrome in humans, is a heme-binding protein in bacterial oxidase biogenesis. J. Biol. Chem. 2009, 284, 25735–25741. [Google Scholar] [CrossRef] [Green Version]
- Hannappel, A.; Bundschuh, F.A.; Ludwig, B. Role of surf1 in heme recruitment for bacterial cox biogenesis. Biochim. Biophys. Acta 2012, 1817, 928–937. [Google Scholar] [CrossRef] [Green Version]
- Bestwick, M.; Jeong, M.Y.; Khalimonchuk, O.; Kim, H.; Winge, D.R. Analysis of leigh syndrome mutations in the yeast surf1 homolog reveals a new member of the cytochrome oxidase assembly factor family. Mol. Cell Biol. 2010, 30, 4480–4491. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Yao, J.; Johns, T.; Fu, K.; De Bie, I.; Macmillan, C.; Cuthbert, A.P.; Newbold, R.F.; Wang, J.; Chevrette, M.; et al. Surf1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in leigh syndrome. Nat. Genet. 1998, 20, 337–343. [Google Scholar] [CrossRef]
- Pierrel, F.; Khalimonchuk, O.; Cobine, P.A.; Bestwick, M.; Winge, D.R. Coa2 is an assembly factor for yeast cytochrome c oxidase biogenesis that facilitates the maturation of cox1. Mol. Cell Biol. 2008, 28, 4927–4939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalimonchuk, O.; Jeong, M.Y.; Watts, T.; Ferris, E.; Winge, D.R. Selective oma1 protease-mediated proteolysis of cox1 subunit of cytochrome oxidase in assembly mutants. J. Biol. Chem. 2012, 287, 7289–7300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babbitt, S.E.; San Francisco, B.; Bretsnyder, E.C.; Kranz, R.G. Conserved residues of the human mitochondrial holocytochrome c synthase mediate interactions with heme. Biochemistry 2014, 53, 5261–5271. [Google Scholar] [CrossRef] [PubMed]
- Indrieri, A.; Conte, I.; Chesi, G.; Romano, A.; Quartararo, J.; Tate, R.; Ghezzi, D.; Zeviani, M.; Goffrini, P.; Ferrero, I.; et al. The impairment of hccs leads to mls syndrome by activating a non-canonical cell death pathway in the brain and eyes. EMBO Mol. Med. 2013, 5, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Valnot, I.; Von Kleist-Retzow, J.C.; Barrientos, A.; Gorbatyuk, M.; Taanman, J.W.; Mehaye, B.; Rustin, P.; Tzagoloff, A.; Munnich, A.; Rotig, A. A mutation in the human heme a:Farnesyltransferase gene (cox10 ) causes cytochrome c oxidase deficiency. Hum. Mol. Genet. 2000, 9, 1245–1249. [Google Scholar] [CrossRef] [Green Version]
- Antonicka, H.; Leary, S.C.; Guercin, G.H.; Agar, J.N.; Horvath, R.; Kennaway, N.G.; Harding, C.O.; Jaksch, M.; Shoubridge, E.A. Mutations in cox10 result in a defect in mitochondrial heme a biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated cox deficiency. Hum. Mol. Genet. 2003, 12, 2693–2702. [Google Scholar] [CrossRef] [Green Version]
- Antonicka, H.; Mattman, A.; Carlson, C.G.; Glerum, D.M.; Hoffbuhr, K.C.; Leary, S.C.; Kennaway, N.G.; Shoubridge, E.A. Mutations in cox15 produce a defect in the mitochondrial heme biosynthetic pathway, causing early-onset fatal hypertrophic cardiomyopathy. Am. J. Hum. Genet. 2003, 72, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Alfadhel, M.; Lillquist, Y.P.; Waters, P.J.; Sinclair, G.; Struys, E.; McFadden, D.; Hendson, G.; Hyams, L.; Shoffner, J.; Vallance, H.D. Infantile cardioencephalopathy due to a cox15 gene defect: Report and review. Am. J. Med. Genet. A 2011, 155A, 840–844. [Google Scholar] [CrossRef]
- Reiter, L.T.; Murakami, T.; Koeuth, T.; Gibbs, R.A.; Lupski, J.R. The human cox10 gene is disrupted during homologous recombination between the 24 kb proximal and distal cmt1a-reps. Hum. Mol. Genet. 1997, 6, 1595–1603. [Google Scholar] [CrossRef] [Green Version]
- Oquendo, C.E.; Antonicka, H.; Shoubridge, E.A.; Reardon, W.; Brown, G.K. Functional and genetic studies demonstrate that mutation in the cox15 gene can cause leigh syndrome. J. Med. Genet. 2004, 41, 540–544. [Google Scholar] [CrossRef] [Green Version]
- Bugiani, M.; Tiranti, V.; Farina, L.; Uziel, G.; Zeviani, M. Novel mutations in cox15 in a long surviving leigh syndrome patient with cytochrome c oxidase deficiency. J. Med. Genet. 2005, 42, e28. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.L.; Lightowlers, R.N.; Turnbull, D.M. Molecular analysis of cytochrome c oxidase deficiency in leigh’s syndrome. Ann. Neurol. 1997, 41, 268–270. [Google Scholar] [CrossRef] [PubMed]
- Tiranti, V.; Hoertnagel, K.; Carrozzo, R.; Galimberti, C.; Munaro, M.; Granatiero, M.; Zelante, L.; Gasparini, P.; Marzella, R.; Rocchi, M.; et al. Mutations of surf-1 in leigh disease associated with cytochrome c oxidase deficiency. Am. J. Hum. Genet. 1998, 63, 1609–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teraoka, M.; Yokoyama, Y.; Ninomiya, S.; Inoue, C.; Yamashita, S.; Seino, Y. Two novel mutations of surf1 in leigh syndrome with cytochrome c oxidase deficiency. Hum. Genet. 1999, 105, 560–563. [Google Scholar] [CrossRef] [PubMed]
- Poyau, A.; Buchet, K.; Bouzidi, M.F.; Zabot, M.T.; Echenne, B.; Yao, J.; Shoubridge, E.A.; Godinot, C. Missense mutations in surf1 associated with deficient cytochrome c oxidase assembly in leigh syndrome patients. Hum. Genet. 2000, 106, 194–205. [Google Scholar] [PubMed]
- Piekutowska-Abramczuk, D.; Magner, M.; Popowska, E.; Pronicki, M.; Karczmarewicz, E.; Sykut-Cegielska, J.; Kmiec, T.; Jurkiewicz, E.; Szymanska-Debinska, T.; Bielecka, L.; et al. Surf1 missense mutations promote a mild leigh phenotype. Clin. Genet. 2009, 76, 195–204. [Google Scholar] [CrossRef]
- Coenen, M.J.; Van den Heuvel, L.P.; Ugalde, C.; Ten Brinke, M.; Nijtmans, L.G.; Trijbels, F.J.; Beblo, S.; Maier, E.M.; Muntau, A.C.; Smeitink, J.A. Cytochrome c oxidase biogenesis in a patient with a mutation in cox10 gene. Ann. Neurol. 2004, 56, 560–564. [Google Scholar] [CrossRef]
- Shoubridge, E.A. Cytochrome c oxidase deficiency. Am. J. Med. Genet. 2001, 106, 46–52. [Google Scholar] [CrossRef]
- Szklarczyk, R.; Wanschers, B.F.; Cuypers, T.D.; Esseling, J.J.; Riemersma, M.; Van den Brand, M.A.; Gloerich, J.; Lasonder, E.; Van den Heuvel, L.P.; Nijtmans, L.G.; et al. Iterative orthology prediction uncovers new mitochondrial proteins and identifies c12orf62 as the human ortholog of cox14, a protein involved in the assembly of cytochrome c oxidase. Genome. Biol. 2012, 13, R12. [Google Scholar] [CrossRef]
- Renkema, G.H.; Visser, G.; Baertling, F.; Wintjes, L.T.; Wolters, V.M.; Van Montfrans, J.; De Kort, G.A.P.; Nikkels, P.G.J.; Van Hasselt, P.M.; Van der Crabben, S.N.; et al. Mutated pet117 causes complex iv deficiency and is associated with neurodevelopmental regression and medulla oblongata lesions. Hum. Genet. 2017, 136, 759–769. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Rietzschel, N.; Kwon, H.; Walter Nuno, A.B.; Hanna, D.A.; Phillips, J.D.; Raven, E.L.; Reddi, A.R.; Hamza, I. Regulation of intracellular heme trafficking revealed by subcellular reporters. Proc. Natl. Acad. Sci. USA 2016, 113, e5144–e5152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiabrando, D.; Marro, S.; Mercurio, S.; Giorgi, C.; Petrillo, S.; Vinchi, F.; Fiorito, V.; Fagoonee, S.; Camporeale, A.; Turco, E.; et al. The mitochondrial heme exporter flvcr1b mediates erythroid differentiation. J. Clin. Investig. 2012, 122, 4569–4579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quigley, J.G.; Yang, Z.; Worthington, M.T.; Phillips, J.D.; Sabo, K.M.; Sabath, D.E.; Berg, C.L.; Sassa, S.; Wood, B.L.; Abkowitz, J.L. Identification of a human heme exporter that is essential for erythropoiesis. Cell 2004, 118, 757–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.A.; Quigley, J.G. Heme and flvcr-related transporter families slc48 and slc49. Mol. Aspects Med. 2013, 34, 669–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, K.; Thompson, A.M.; Goldbeck, R.A.; Shi, X.; Whitman, S.; Oh, E.; Zhiwu, Z.; Vulpe, C.; Holman, T.R. Spectroscopic and biochemical characterization of heme binding to yeast dap1p and mouse pgrmc1p. Biochemistry 2005, 44, 16729–16736. [Google Scholar] [CrossRef] [Green Version]
- Kaluka, D.; Batabyal, D.; Chiang, B.Y.; Poulos, T.L.; Yeh, S.R. Spectroscopic and mutagenesis studies of human pgrmc1. Biochemistry 2015, 54, 1638–1647. [Google Scholar] [CrossRef]
- Min, L.; Strushkevich, N.V.; Harnastai, I.N.; Iwamoto, H.; Gilep, A.A.; Takemori, H.; Usanov, S.A.; Nonaka, Y.; Hori, H.; Vinson, G.P.; et al. Molecular identification of adrenal inner zone antigen as a heme-binding protein. FEBS J. 2005, 272, 5832–5843. [Google Scholar] [CrossRef]
- Peluso, J.J.; Liu, X.; Gawkowska, A.; Lodde, V.; Wu, C.A. Progesterone inhibits apoptosis in part by pgrmc1-regulated gene expression. Mol. Cell Endocrinol. 2010, 320, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Kim, S.Y.; Choi, H.S.; An, S.; Ryu, C.J. Epitope mapping of anti-pgrmc1 antibodies reveals the non-conventional membrane topology of pgrmc1 on the cell surface. Sci. Rep. 2019, 9, 653. [Google Scholar] [CrossRef]
- Vance, J.E. Mam (mitochondria-associated membranes) in mammalian cells: Lipids and beyond. Biochim. Biophys. Acta 2014, 1841, 595–609. [Google Scholar] [CrossRef]
- Poston, C.N.; Duong, E.; Cao, Y.; Bazemore-Walker, C.R. Proteomic analysis of lipid raft-enriched membranes isolated from internal organelles. Biochem. Biophys. Res. Commun. 2011, 415, 355–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, I.T.; Adelmant, G.; Lim, Y.; Marto, J.A.; Cho, G.; Golden, J.A. Ascorbate peroxidase proximity labeling coupled with biochemical fractionation identifies promoters of endoplasmic reticulum-mitochondrial contacts. J. Biol. Chem. 2017, 292, 16382–16392. [Google Scholar] [CrossRef] [Green Version]
- Schumann, U.; Subramani, S. Special delivery from mitochondria to peroxisomes. Trends Cell Biol. 2008, 18, 253–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, A.U.; Carta, L.K.; Lesuisse, E.; Hamza, I. Lack of heme synthesis in a free-living eukaryote. Proc. Natl. Acad. Sci. USA 2005, 102, 4270–4275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajagopal, A.; Rao, A.U.; Amigo, J.; Tian, M.; Upadhyay, S.K.; Hall, C.; Uhm, S.; Mathew, M.K.; Fleming, M.D.; Paw, B.H.; et al. Haem homeostasis is regulated by the conserved and concerted functions of hrg-1 proteins. Nature 2008, 453, 1127–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Callaghan, K.M.; Ayllon, V.; O’Keeffe, J.; Wang, Y.; Cox, O.T.; Loughran, G.; Forgac, M.; O’Connor, R. Heme-binding protein hrg-1 is induced by insulin-like growth factor i and associates with the vacuolar h+-atpase to control endosomal ph and receptor trafficking. J. Biol. Chem. 2010, 285, 381–391. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Protchenko, O.; Philpott, C.C.; Hamza, I. Topologically conserved residues direct heme transport in hrg-1-related proteins. J. Biol. Chem. 2012, 287, 4914–4924. [Google Scholar] [CrossRef] [Green Version]
- White, C.; Yuan, X.; Schmidt, P.J.; Bresciani, E.; Samuel, T.K.; Campagna, D.; Hall, C.; Bishop, K.; Calicchio, M.L.; Lapierre, A.; et al. Hrg1 is essential for heme transport from the phagolysosome of macrophages during erythrophagocytosis. Cell Metab. 2013, 17, 261–270. [Google Scholar] [CrossRef] [Green Version]
- Duffy, S.P.; Shing, J.; Saraon, P.; Berger, L.C.; Eiden, M.V.; Wilde, A.; Tailor, C.S. The fowler syndrome-associated protein flvcr2 is an importer of heme. Mol. Cell Biol. 2010, 30, 5318–5324. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, T.; Yanagisawa, Y.; Nagai, T. Analysis of the affinity of each haptoglobin polymer for hemoglobin by two-dimensional affinity electrophoresis. Clin. Chim. Acta 1997, 258, 137–144. [Google Scholar] [CrossRef]
- Graversen, J.H.; Madsen, M.; Moestrup, S.K. Cd163: A signal receptor scavenging haptoglobin-hemoglobin complexes from plasma. Int. J. Biochem. Cell Biol. 2002, 34, 309–314. [Google Scholar] [CrossRef]
- Delaby, C.; Pilard, N.; Puy, H.; Canonne-Hergaux, F. Sequential regulation of ferroportin expression after erythrophagocytosis in murine macrophages: Early mrna induction by haem, followed by iron-dependent protein expression. Biochem. J. 2008, 411, 123–131. [Google Scholar] [CrossRef] [Green Version]
- Hrkal, Z.; Vodrázka, Z.; Kalousek, I. Transfer of heme from ferrihemoglobin and ferrihemoglobin isolated chains to hemopexin. Eur. J. Biochem. 1974, 43, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Vinchi, F.; Gastaldi, S.; Silengo, L.; Altruda, F.; Tolosano, E. Hemopexin prevents endothelial damage and liver congestion in a mouse model of heme overload. Am. J. Pathol. 2008, 173, 289–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hvidberg, V.; Maniecki, M.B.; Jacobsen, C.; Hojrup, P.; Moller, H.J.; Moestrup, S.K. Identification of the receptor scavenging hemopexin-heme complexes. Blood 2005, 106, 2572–2579. [Google Scholar] [CrossRef]
- Camus, S.M.; De Moraes, J.A.; Bonnin, P.; Abbyad, P.; Le Jeune, S.; Lionnet, F.; Loufrani, L.; Grimaud, L.; Lambry, J.C.; Charue, D.; et al. Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood 2015, 125, 3805–3814. [Google Scholar] [CrossRef] [Green Version]
- Donadee, C.; Raat, N.J.; Kanias, T.; Tejero, J.; Lee, J.S.; Kelley, E.E.; Zhao, X.; Liu, C.; Reynolds, H.; Azarov, I.; et al. Nitric oxide scavenging by red blood cell microparticles and cell-free hemoglobin as a mechanism for the red cell storage lesion. Circulation 2011, 124, 465–476. [Google Scholar] [CrossRef]
- Kim-Shapiro, D.B.; Lee, J.; Gladwin, M.T. Storage lesion: Role of red blood cell breakdown. Transfusion 2011, 51, 844–851. [Google Scholar] [CrossRef] [Green Version]
- Gaggar, A.; Patel, R.P. There is blood in the water: Hemolysis, hemoglobin, and heme in acute lung injury. Am. J. Physiol. Lung. Cell Mol. Physiol. 2016, 311, L714–L718. [Google Scholar] [CrossRef]
- Kim, Y.; Abplanalp, W.A.; Jung, A.D.; Schuster, R.M.; Lentsch, A.B.; Gulbins, E.; Caldwell, C.C.; Pritts, T.A. Endocytosis of red blood cell microparticles by pulmonary endothelial cells is mediated by rab5. Shock 2018, 49, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Gollub, E.G.; Liu, K.P.; Dayan, J.; Adlersberg, M.; Sprinson, D.B. Yeast mutants deficient in heme biosynthesis and a heme mutant additionally blocked in cyclization of 2,3-oxidosqualene. J. Biol. Chem. 1977, 252, 2846–2854. [Google Scholar]
- Kim, H.J.; Jeong, M.Y.; Parnell, T.J.; Babst, M.; Phillips, J.D.; Winge, D.R. The plasma membrane protein nce102 implicated in eisosome formation rescues a heme defect in mitochondria. J. Biol. Chem. 2016, 291, 17417–17426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossmann, G.; Malinsky, J.; Stahlschmidt, W.; Loibl, M.; Weig-Meckl, I.; Frommer, W.B.; Opekarova, M.; Tanner, W. Plasma membrane microdomains regulate turnover of transport proteins in yeast. J. Cell Biol. 2008, 183, 1075–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mourer, T.; Brault, A.; Labbe, S. Heme acquisition by shu1 requires nbr1 and proteins of the escrt complex in schizosaccharomyces pombe. Mol. Microbiol. 2019, 112, 1499–1518. [Google Scholar] [CrossRef] [PubMed]
- Mourer, T.; Jacques, J.F.; Brault, A.; Bisaillon, M.; Labbe, S. Shu1 is a cell-surface protein involved in iron acquisition from heme in schizosaccharomyces pombe. J. Biol. Chem. 2015, 290, 10176–10190. [Google Scholar] [CrossRef] [Green Version]
- Mourer, T.; Normant, V.; Labbe, S. Heme assimilation in schizosaccharomyces pombe requires cell-surface-anchored protein shu1 and vacuolar transporter abc3. J. Biol. Chem. 2017, 292, 4898–4912. [Google Scholar] [CrossRef] [Green Version]
- Normant, V.; Mourer, T.; Labbe, S. The major facilitator transporter str3 is required for low-affinity heme acquisition in schizosaccharomyces pombe. J. Biol. Chem. 2018, 293, 6349–6362. [Google Scholar] [CrossRef] [Green Version]
- Protchenko, O.; Shakoury-Elizeh, M.; Keane, P.; Storey, J.; Androphy, R.; Philpott, C.C. Role of pug1 in inducible porphyrin and heme transport in saccharomyces cerevisiae. Eukaryot Cell 2008, 7, 859–871. [Google Scholar] [CrossRef] [Green Version]
- Puy, H.; Gouya, L.; Deybach, J.C. Porphyrias. Lancet 2010, 375, 924–937. [Google Scholar] [CrossRef]
- Chen, C.; Samuel, T.K.; Sinclair, J.; Dailey, H.A.; Hamza, I. An intercellular heme-trafficking protein delivers maternal heme to the embryo during development in c. Elegans. Cell 2011, 145, 720–731. [Google Scholar] [CrossRef] [Green Version]
- Bonkovsky, H.L.; Healey, J.F.; Lourie, A.N.; Gerron, G.G. Intravenous heme-albumin in acute intermittent porphyria: Evidence for repletion of hepatic hemoproteins and regulatory heme pools. Am. J. Gastroenterol. 1991, 86, 1050–1056. [Google Scholar] [PubMed]
- Yang, Z.; Philips, J.D.; Doty, R.T.; Giraudi, P.; Ostrow, J.D.; Tiribelli, C.; Smith, A.; Abkowitz, J.L. Kinetics and specificity of feline leukemia virus subgroup c receptor (flvcr) export function and its dependence on hemopexin. J. Biol. Chem. 2010, 285, 28874–28882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korolnek, T.; Zhang, J.; Beardsley, S.; Scheffer, G.L.; Hamza, I. Control of metazoan heme homeostasis by a conserved multidrug resistance protein. Cell Metab. 2014, 19, 1008–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desuzinges-Mandon, E.; Arnaud, O.; Martinez, L.; Huche, F.; Di Pietro, A.; Falson, P. Abcg2 transports and transfers heme to albumin through its large extracellular loop. J. Biol. Chem. 2010, 285, 33123–33133. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, P.; Ross, D.D.; Nakanishi, T.; Bailey-Dell, K.; Zhou, S.; Mercer, K.E.; Sarkadi, B.; Sorrentino, B.P.; Schuetz, J.D. The stem cell marker bcrp/abcg2 enhances hypoxic cell survival through interactions with heme. J. Biol. Chem. 2004, 279, 24218–24225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saison, C.; Helias, V.; Ballif, B.A.; Peyrard, T.; Puy, H.; Miyazaki, T.; Perrot, S.; Vayssier-Taussat, M.; Waldner, M.; Le Pennec, P.Y.; et al. Null alleles of abcg2 encoding the breast cancer resistance protein define the new blood group system junior. Nat. Genet. 2012, 44, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, Y.; Ishii, T.; Kohda, T.; Ishizuka, M.; Yamazaki, K.; Nishimura, Y.; Tanaka, T.; Dan, S.; Nakajima, M. Mechanistic study of ppix accumulation using the jfcr39 cell panel revealed a role for dynamin 2-mediated exocytosis. Sci. Rep. 2019, 9, 8666. [Google Scholar] [CrossRef] [Green Version]
- Dutta, R.; Zhang, T.Y.; Kohnke, T.; Thomas, D.; Linde, M.; Gars, E.; Stafford, M.; Kaur, S.; Nakauchi, Y.; Yin, R.; et al. Enasidenib drives human erythroid differentiation independently of isocitrate dehydrogenase 2. J. Clin. Investig. 2020. [Google Scholar] [CrossRef]
- Robey, R.W.; To, K.K.; Polgar, O.; Dohse, M.; Fetsch, P.; Dean, M.; Bates, S.E. Abcg2: A perspective. Adv. Drug Deliv. Rev. 2009, 61, 3–13. [Google Scholar] [CrossRef]
- Donegan, R.K.; Moore, C.M.; Hanna, D.A.; Reddi, A.R. Handling heme: The mechanisms underlying the movement of heme within and between cells. Free Radic. Biol. Med. 2019, 133, 88–100. [Google Scholar] [CrossRef]
- Chakravarti, R.; Aulak, K.S.; Fox, P.L.; Stuehr, D.J. Gapdh regulates cellular heme insertion into inducible nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2010, 107, 18004–18009. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Novelli, J.; Jiang, D.; Dailey, H.A.; Landmann, F.; Ford, L.; Taylor, M.J.; Carlow, C.K.; Kumar, S.; Foster, J.M.; et al. Interdomain lateral gene transfer of an essential ferrochelatase gene in human parasitic nematodes. Proc. Natl. Acad. Sci. USA 2013, 110, 7748–7753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinclair, J.; Hamza, I. Lessons from bloodless worms: Heme homeostasis in c. Elegans. Biometals 2015, 28, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Braz, G.R.; Coelho, H.S.; Masuda, H.; Oliveira, P.L. A missing metabolic pathway in the cattle tick boophilus microplus. Curr. Biol. 1999, 9, 703–706. [Google Scholar] [CrossRef] [Green Version]
- Heggland, E.I.; Eichner, C.; Stove, S.I.; Martinez, A.; Nilsen, F.; Dondrup, M. A scavenger receptor b (cd36)-like protein is a potential mediator of intestinal heme absorption in the hematophagous ectoparasite lepeophtheirus salmonis. Sci. Rep. 2019, 9, 4218. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.Y.; Hamm, J.; Xu, X.; Genschmer, K.; Zhong, M.; Lebensburger, J.; Marques, M.B.; Kerby, J.D.; Pittet, J.F.; Gaggar, A.; et al. Absorbance and redox based approaches for measuring free heme and free hemoglobin in biological matrices. Redox Biol. 2016, 9, 167–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fronticelli, C.; Bucci, E. Acetone extraction of heme from myoglobin and hemoglobin at acid ph. Biochim. Biophys. Acta 1963, 78, 530–531. [Google Scholar] [CrossRef]
- Thomas, J.; Weinstein, J.D. Measurement of heme efflux and heme content in isolated developing chloroplasts. Plant Physiol. 1990, 94, 1414–1423. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, P.R.; Gorman, N.; Jacobs, J.M. Measurement of heme concentration. Curr. Protoc. Toxicol. 1999, 00. [Google Scholar] [CrossRef]
- Woods, J.S.; Simmonds, P.L. Hplc methods for analysis of porphyrins in biological media. Curr. Protoc. Toxicol. 2001, 7, 8.9.1–8.9.17. [Google Scholar] [CrossRef]
- Paul, K.; Theorell, H.; Akeson, A. The molar light absorption of pyridine ferroprotoporphyrin (pyridine haemochromogen). Acta Chem. Scand 1953, 7, 1284–1287. [Google Scholar] [CrossRef]
- Barr, I.; Guo, F. Pyridine hemochromagen assay for determining the concentration of heme in purified protein solutions. Bio-protocol 2015, 5, e1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuhrhop, J.-H.; Smith, K.M. Laboratory Methods in Porphyrin and Metalloporphyrin Research; Elsevier Science & Technology: Amsterdam, The Netherlands, 1975. [Google Scholar]
- Marcero, J.R.; Piel Iii, R.B.; Burch, J.S.; Dailey, H.A. Rapid and sensitive quantitation of heme in hemoglobinized cells. Biotechniques 2016, 61, 83–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blake, R.C.I.; Griff, M.N. In situ spectroscopy on intact leptospirillum ferrooxidans reveals that reduced cytochrome 579 is an obligatory intermediate in the aerobic iron respiratory chain. Front. Microbiol. 2012, 3, 136. [Google Scholar] [CrossRef] [Green Version]
- Correia, M.A.; Sinclair, P.R.; De Matteis, F. Cytochrome p450 regulation: The interplay between its heme and apoprotein moieties in synthesis, assembly, repair, and disposal. Drug Metab. Rev. 2011, 43, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Guengerich, F.P.; Martin, M.V.; Sohl, C.D.; Cheng, Q. Measurement of cytochrome p450 and nadph-cytochrome p450 reductase. Nat. Protoc. 2009, 4, 1245–1251. [Google Scholar] [CrossRef] [Green Version]
- Hanna, D.A.; Hu, R.; Kim, H.; Martinez-Guzman, O.; Torres, M.P.; Reddi, A.R. Heme bioavailability and signaling in response to stress in yeast cells. J. Biol. Chem. 2018, 293, 12378–12393. [Google Scholar] [CrossRef] [Green Version]
- Dick, R.; Murray, B.P.; Reid, M.J.; Correia, M.A. Structure--function relationships of rat hepatic tryptophan 2,3-dioxygenase: Identification of the putative heme-ligating histidine residues. Arch. Biochem. Biophys. 2001, 392, 71–78. [Google Scholar] [CrossRef]
- Nelp, M.T.; Kates, P.A.; Hunt, J.T.; Newitt, J.A.; Balog, A.; Maley, D.; Zhu, X.; Abell, L.; Allentoff, A.; Borzilleri, R.; et al. Immune-modulating enzyme indoleamine 2,3-dioxygenase is effectively inhibited by targeting its apo-form. Proc. Natl. Acad. Sci. USA 2018, 115, 3249–3254. [Google Scholar] [CrossRef] [Green Version]
- Atamna, H.; Brahmbhatt, M.; Atamna, W.; Shanower, G.A.; Dhahbi, J.M. Apohrp-based assay to measure intracellular regulatory heme. Metallomics 2015, 7, 309–321. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Yang, M.; Wegner, S.V.; Zhao, J.; Zhu, R.; Wu, Y.; He, C.; Chen, P.R. A genetically encoded fret sensor for intracellular heme. ACS Chem. Biol. 2015, 10, 1610–1615. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Sligar, S.G.; Wand, A.J. Solution structure of apocytochrome b562. Nat. Struct. Biol. 1994, 1, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Abshire, J.R.; Rowlands, C.J.; Ganesan, S.M.; So, P.T.; Niles, J.C. Quantification of labile heme in live malaria parasites using a genetically encoded biosensor. Proc. Natl. Acad. Sci. USA 2017, 114, E2068–E2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strommen, D.P.; Nakamoto, K. Resonance raman spectroscopy. J. Chem. Educ. 1977, 54, 474. [Google Scholar] [CrossRef]
- Keren, S.; Zavaleta, C.; Cheng, Z.; De La Zerda, A.; Gheysens, O.; Gambhir, S. Noninvasive molecular imaging of small living subjects using raman spectroscopy. Proc. Nat. Acad. Sci. USA 2008, 105, 5844–5849. [Google Scholar] [CrossRef] [Green Version]
- Bonifacio, A.; Finaurini, S.; Krafft, C.; Parapini, S.; Taramelli, D.; Sergo, V. Spatial distribution of heme species in erythrocytes infected with plasmodium falciparum by use of resonance raman imaging and multivariate analysis. Anal. Bioanal. Chem. 2008, 392, 1277–1282. [Google Scholar] [CrossRef]
- Boyer, D.; Tamarat, P.; Maali, A.; Lounis, B.; Orrit, M. Photothermal imaging of nanometer-sized metal particles among scatterers. Science 2002, 297, 1160–1163. [Google Scholar] [CrossRef]
- Lu, S.; Min, W.; Chong, S.; Holtom, G.R.; Xie, X.S. Label-free imaging of heme proteins with two-photon excited photothermal lens microscopy. Appl. Phys. Lett. 2010, 96, 113701. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.J.; Yuan, X.; Li, J.; Dong, P.; Hamza, I.; Cheng, J.-X. Label-free imaging of heme dynamics in living organisms by transient absorption microscopy. Anal. Chem. 2018, 90, 3395–3401. [Google Scholar] [CrossRef]
- Wei, L.; Min, W. Pump-probe optical microscopy for imaging nonfluorescent chromophores. Anal. Bioanal. Chem. 2012, 403, 2197–2202. [Google Scholar] [CrossRef]
- Min, W.; Lu, S.; Chong, S.; Roy, R.; Holtom, G.R.; Xie, X.S. Imaging chromophores with undetectable fluorescence by stimulated emission microscopy. Nature 2009, 461, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Domingue, S.R.; Bartels, R.A.; Chicco, A.J.; Wilson, J.W. Transient absorption imaging of hemes with 2-color, independently tunable visible-wavelength ultrafast source. Biomed. Opt. Express 2017, 8, 2807–2821. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
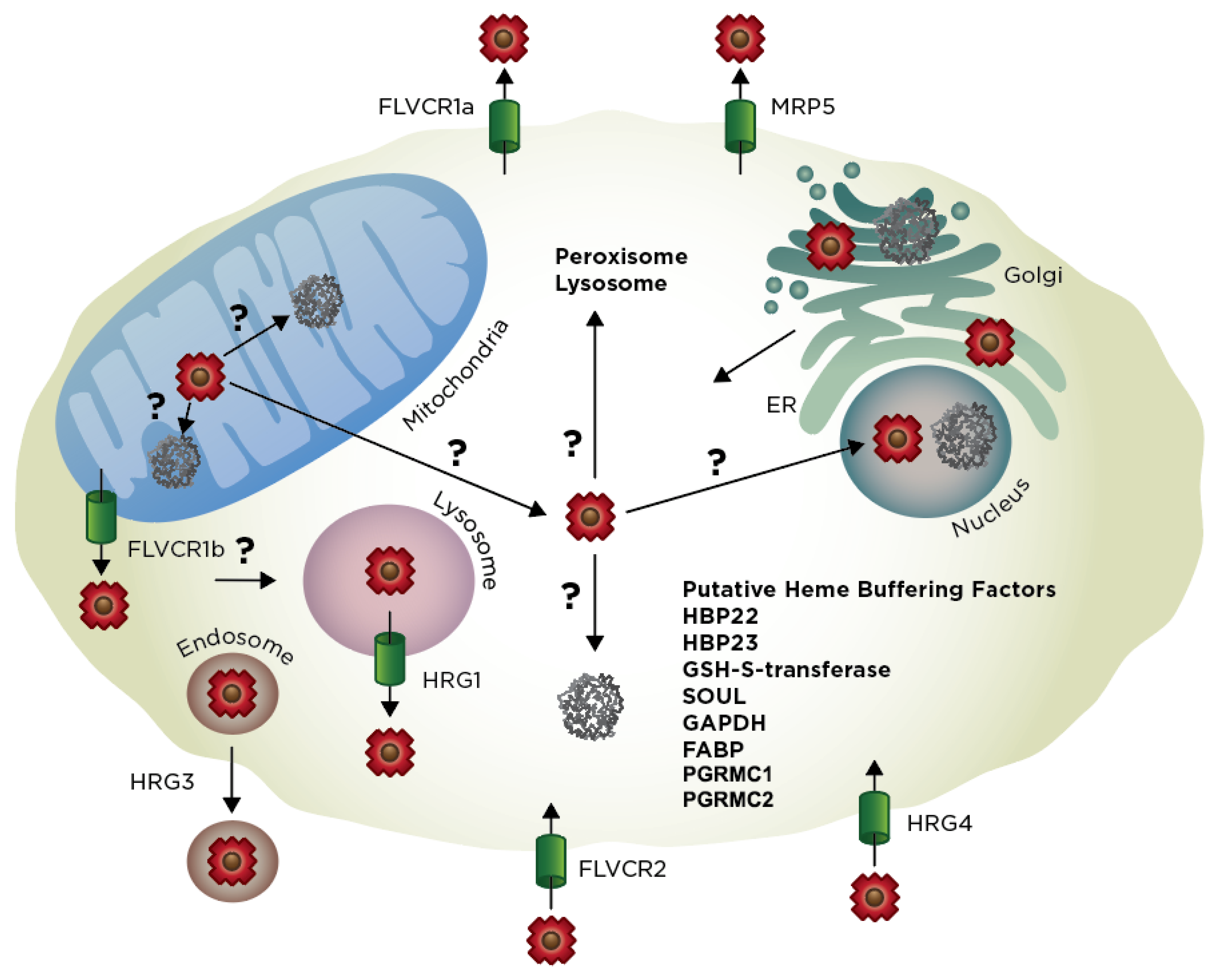{kind=link}
| Heme Homeostatic Process | Enzyme | Saccharomyces cerevisiae | Caenorhabditis elegans | Homo sapiens |
|---|---|---|---|---|
| Heme Synthesis | 5-aminolevulinic acid synthase | Hem1 | ✖ | ALAS1/ALAS2 |
| Porphobilinogen synthase | Hem2 | ✖ | PBGS | |
| Hydroxymethylbilane synthase | Hem3 | ✖ | HMBS | |
| Uroporphyrinogen synthase | Hem4 | ✖ | UROS | |
| Uroporphyrinogen decarboxylase | Hem12 | ✖ | UROD | |
| Coproporphyrinogen oxidase | Hem13 | ✖ | CPOX | |
| Protoporphyrinogen oxidase | Hem14 | ✖ | PPOX | |
| Ferrochelatase | Hem15 | fecl-1 | FECH | |
| Heme Degradation | Heme oxygenase | Hmx1 | ? | Hmox1/Hmox2 |
| Heme Import | FLVCR2 | ✖ | ✔ | ✔ |
| HRG4 | ✖ | ✔ | ✖ | |
| Heme Export | FLVCR1 | ✖ | ✖ | ✔ |
| MRP5 | ✖ | mrp-5 | ABCC5 | |
| Pug1 | ✔ | ✖ | ✖ | |
| HRG3 | ✖ | ✔ | ✖ | |
| Heme Trafficking | PGRMC1/2 | Dap1 | vem-1 | PGRMC1/2 |
| GAPDH | Tdh1/2/3 | gpd1/2/3/4 | GAPDH | |
| HRG1 | ✖ | ✔ | ✔ |
| Approaches | Methods | Advantages | Disadvantages |
|---|---|---|---|
| In Situ Label Free Imaging | Transient Absorption Microscopy Resonance Raman Imaging 2 Photon Photothermal Lens Microscopy | Subcellular resolution (<1 μm) Non-invasive Can probe heme dynamics in living cells | Signals dominated by most abundant and/or highly absorbing species Low-throughput Requires specialized equipment/expertise |
| In Situ Imaging of Labile Heme using Fluorescent Heme Sensors | HS1 CISDY-9 CHY | Subcellular resolution (<1 μm) Direct probe of labile “bioavailable” heme Can probe heme dynamics in living cells High-throughput | May perturb heme homeostasis Possible selection bias depending on the nature of the sensor Extended time resolved studies precluded by photobleaching |
| Assays for Endogenous Markers of Heme Bioavailability | Horseradish Peroxidase Tryptophan 2,3 Dioxygenase (TDO) Indoleamine-2,3-Dioxygenase (IDO) Cytochrome P450 Catalase Transcription Factors | Measurement of heme accessible to endogenous hemoproteins No genetic perturbations | Disruption of cells and tissues Time consuming Difficult to get fast time resolution |
| Assays for Total Heme | HPLC | Resolve different heme types | Time consuming Disruption of cells and tissues Low-throughput |
| Porphyrin Fluorescence | nM sensitivity High-throughput | Disruption of cells and tissues | |
| UV/vis Absorbance Spectroscopy | CLARiTY | Sensitive measurements in turbid samples Possible to measure heme and hemoproteins in intact cells | Signals dominated by most abundant and/or highly absorbing species Low-throughput Requires specialized equipment |
| Pyridine Hemochromagen | Broadly accessible Inexpensive | Disruption of cells and tissues |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swenson, S.A.; Moore, C.M.; Marcero, J.R.; Medlock, A.E.; Reddi, A.R.; Khalimonchuk, O. From Synthesis to Utilization: The Ins and Outs of Mitochondrial Heme. Cells 2020, 9, 579. https://doi.org/10.3390/cells9030579
Swenson SA, Moore CM, Marcero JR, Medlock AE, Reddi AR, Khalimonchuk O. From Synthesis to Utilization: The Ins and Outs of Mitochondrial Heme. Cells. 2020; 9(3):579. https://doi.org/10.3390/cells9030579
Chicago/Turabian StyleSwenson, Samantha A., Courtney M. Moore, Jason R. Marcero, Amy E. Medlock, Amit R. Reddi, and Oleh Khalimonchuk. 2020. "From Synthesis to Utilization: The Ins and Outs of Mitochondrial Heme" Cells 9, no. 3: 579. https://doi.org/10.3390/cells9030579
APA StyleSwenson, S. A., Moore, C. M., Marcero, J. R., Medlock, A. E., Reddi, A. R., & Khalimonchuk, O. (2020). From Synthesis to Utilization: The Ins and Outs of Mitochondrial Heme. Cells, 9(3), 579. https://doi.org/10.3390/cells9030579