Transcription Factors in Cancer: When Alternative Splicing Determines Opposite Cell Fates




Abstract
:1. Introduction
1.1. Alternative Splicing
1.2. Transcription Factors
1.3. Deregulation of Transcription Factors in Cancer
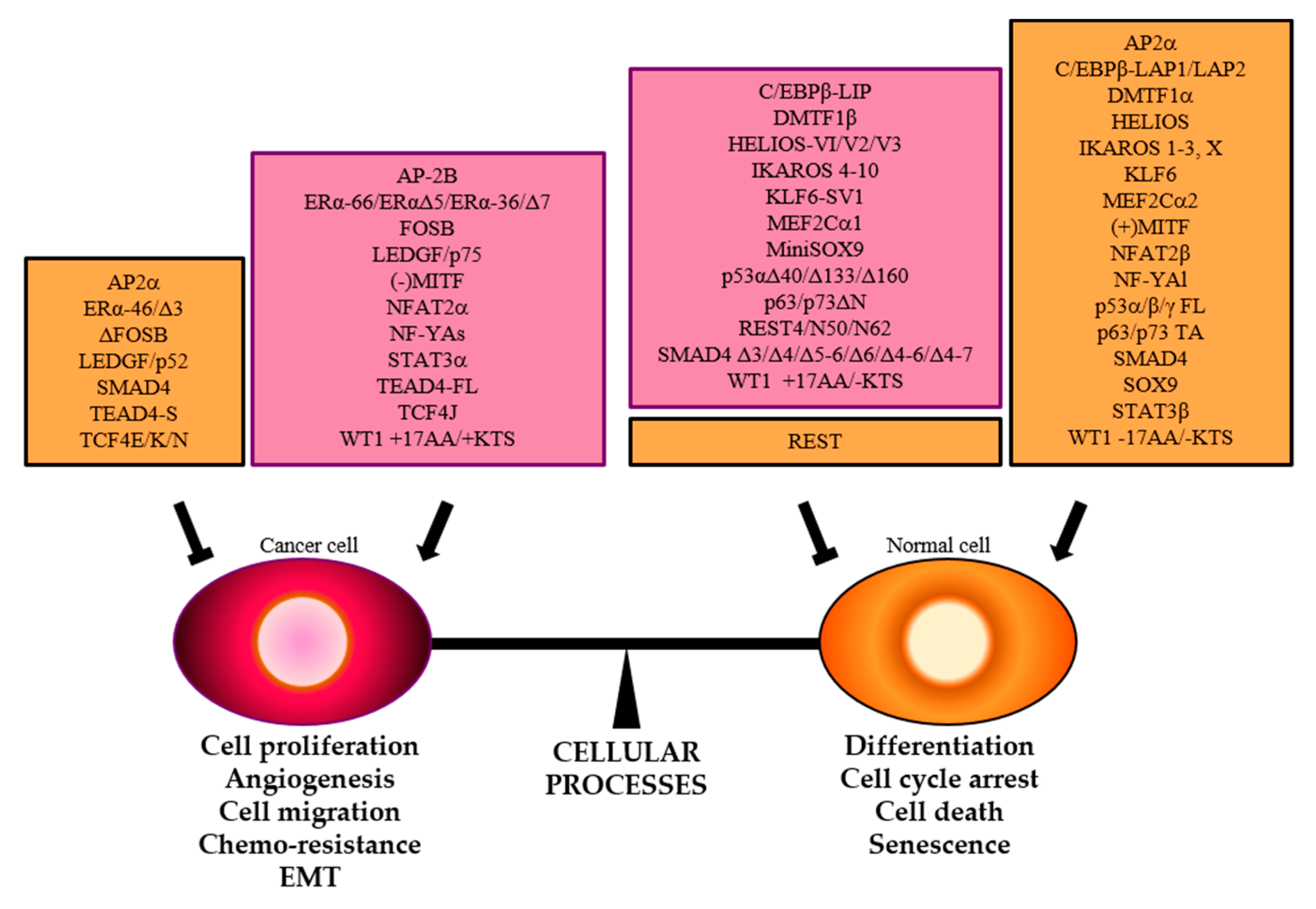
2. TFs Splice Variants that Directly Control Different Transcriptional Programs
2.1. Nuclear Transcription Factor Y (NF-Y)
2.2. Signal Transducer and Activator of Transcription 3 (STAT3)
2.3. T Cell Factor 4 (TCF4)
2.4. Wilm’s Tumor 1 (WT1)
3. TFs Splice Variants that Differently Control the Same set of Genes
3.1. Myocyte Enhancer Factor-2C (MEF2C)
3.2. Melanocyte Transcription Factor (MITF)
3.3. Nuclear Factor of Activated T Cells 1 (NFAT2/ NFATC1)
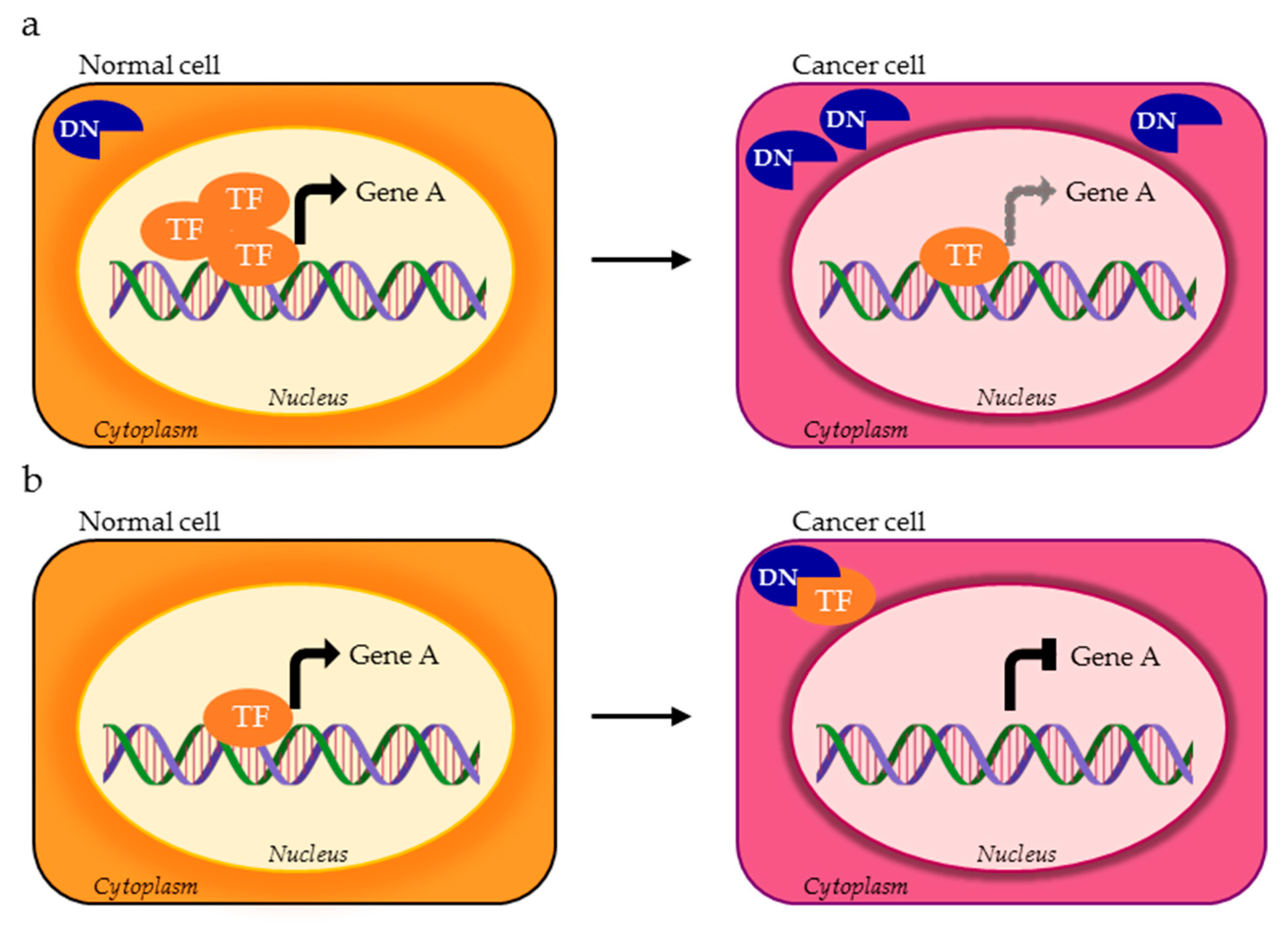
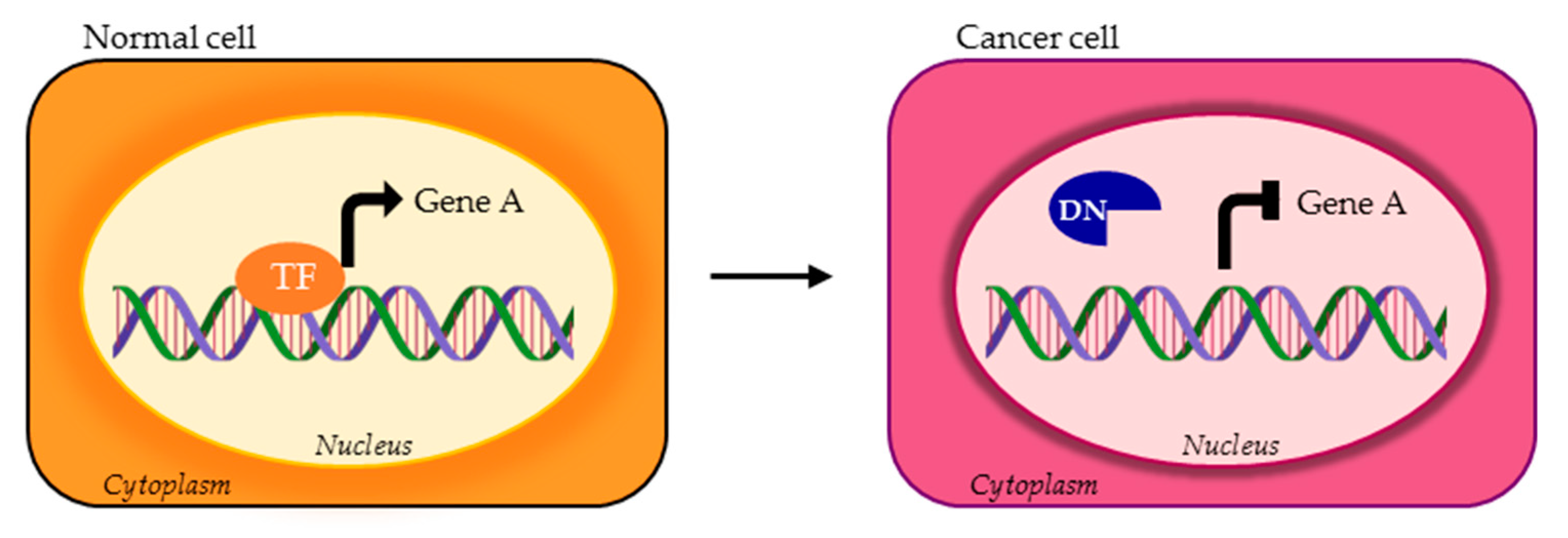
4. TFs Splice Variants with Dominant Negative Activity that Hamper TF Physiological Function
4.1. DNs with Cellular Mislocalization
4.1.1. Ikaros Family Zinc Finger Protein HELIOS
4.1.2. Krüppel-like Factor 6 (KLF6)
4.2. DNs Impaired in DNA Binding Ability
4.2.1. Ikaros Family Zinc Finger Protein 1 (IKAROS)
4.2.2. TEA Domain Family Member 4 (TEAD4)
4.3. DNs with Altered Regultory Ability
4.3.1. CCAAT-Enhancer Binding Protein β (C/EBPβ)
4.3.2. Lens Epithelium-Derived Growth Factor (LEDGF)
4.3.3. RE1 Silencing Transcription Factor (REST)
4.3.4. SMAD Family Member 4 (SMAD4)
4.3.5. Estrogen Receptor Alpha (ERα)
5. Conclusions and Prospects
Author Contributions
Funding
Conflicts of Interest
References
- Park, E.; Pan, Z.; Zhang, Z.; Lin, L.; Xing, Y. The Expanding Landscape of Alternative Splicing Variation in Human Populations. Am. J. Hum. Genet. 2018, 102, 11–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Yan, L.; Zeng, J.; Zhou, H.; Liu, H.; Yu, G.; Yao, W.; Chen, K.; Ye, Z.; Xu, H. Pan-cancer analysis of clinical relevance of alternative splicing events in 31 human cancers. Oncogene 2019, 38, 6678–6695. [Google Scholar] [CrossRef] [PubMed]
- El Marabti, E.; Younis, I. The cancer spliceome: Reprograming of alternative splicing in cancer. Front. Mol. Biosci. 2018, 5, 80. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Hoyos, L.; Knorr, K.; Abdel-Wahab, O. Aberrant RNA Splicing in Cancer. Annu. Rev. Cancer Biol. 2019, 3, 167–185. [Google Scholar] [CrossRef]
- Faustino, N.A.; Cooper, T.A. Pre-mRNA splicing and human disease. Genes Dev. 2003, 17, 419–437. [Google Scholar] [CrossRef] [Green Version]
- Pajares, M.J.; Ezponda, T.; Catena, R.; Calvo, A.; Pio, R.; Montuenga, L.M. Alternative splicing: An emerging topic in molecular and clinical oncology. Lancet Oncol. 2007, 8, 349–357. [Google Scholar] [CrossRef]
- Luscombe, N.M.; Austin, S.E.; Berman, H.M.; Thornton, J.M. An overview of the structures of protein-DNA complexes. Genome Biol. 2000. [Google Scholar] [CrossRef] [Green Version]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef]
- Cusanovich, D.A.; Pavlovic, B.; Pritchard, J.K.; Gilad, Y. The Functional Consequences of Variation in Transcription Factor Binding. PLoS Genet. 2014, 10, e1004226. [Google Scholar] [CrossRef]
- Spitz, F.; Furlong, E.E.M. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef]
- Lee, T.I.; Young, R.A. Transcriptional regulation and its misregulation in disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, M.; Manley, J.L. Transcriptional repression of eukaryotic promoters. Cell 1989, 59, 405–408. [Google Scholar] [CrossRef]
- Payankaulam, S.; Li, L.M.; Arnosti, D.N. Transcriptional repression: Conserved and evolved features. Curr. Biol. 2010, 20, R764–R771. [Google Scholar] [CrossRef] [Green Version]
- Reiter, F.; Wienerroither, S.; Stark, A. Combinatorial function of transcription factors and cofactors. Curr. Opin. Genet. Dev. 2017, 43, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [Green Version]
- Garraway, L.A.; Lander, E.S. Lessons from the cancer genome. Cell 2013, 153, 17–37. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Chen, R. Understanding aberrant RNA splicing to facilitate cancer diagnosis and therapy. Oncogene 2019, 39, 2231–2242. [Google Scholar] [CrossRef]
- Sveen, A.; Kilpinen, S.; Ruusulehto, A.; Lothe, R.A.; Skotheim, R.I. Aberrant RNA splicing in cancer; Expression changes and driver mutations of splicing factor genes. Oncogene 2016, 35, 2413–2427. [Google Scholar] [CrossRef]
- Biamonti, G.; Infantino, L.; Gaglio, D.; Amato, A. An Intricate Connection between Alternative Splicing and Phenotypic Plasticity in Development and Cancer. Cells 2019, 9, 34. [Google Scholar] [CrossRef] [Green Version]
- David, C.J.; Manley, J.L. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev. 2010, 24, 2343–2364. [Google Scholar] [CrossRef] [Green Version]
- Danan-Gotthold, M.; Golan-Gerstl, R.; Eisenberg, E.; Meir, K.; Karni, R.; Levanon, E.Y. Identification of recurrent regulated alternative splicing events across human solid tumors. Nucleic Acids Res. 2015, 43, 5130–5144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurtner, A.; Manni, I.; Piaggio, G. NF-Y in cancer: Impact on cell transformation of a gene essential for proliferation. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, H.; Elemento, O.; Tavazoie, S. Revealing Global Regulatory Perturbations across Human Cancers. Mol. Cell 2009, 36, 900–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolfini, D.; Minuzzo, M.; Pavesi, G.; Mantovani, R. The Short Isoform of NF-YA Belongs to the Embryonic Stem Cell Transcription Factor Circuitry. Stem Cells 2012, 30, 2450–2459. [Google Scholar] [CrossRef]
- Basile, V.; Baruffaldi, F.; Dolfini, D.; Belluti, S.; Benatti, P.; Ricci, L.; Artusi, V.; Tagliafico, E.; Mantovani, R.; Molinari, S.; et al. NF-YA splice variants have different roles on muscle differentiation. Biochim. Biophys. Acta Gene Regul. Mech. 2016, 1859, 627–638. [Google Scholar] [CrossRef] [Green Version]
- Cicchillitti, L.; Corrado, G.; Carosi, M.; Dabrowska, M.E.; Loria, R.; Falcioni, R.; Cutillo, G.; Piaggio, G.; Vizza, E. Prognostic role of NF-YA splicing isoforms and Lamin A status in low grade endometrial cancer. Oncotarget 2017, 8, 7935–7945. [Google Scholar] [CrossRef] [Green Version]
- Dolfini, D.; Andrioletti, V.; Mantovani, R. Overexpression and alternative splicing of NF-YA in breast cancer. Sci. Rep. 2019, 9, 12955. [Google Scholar] [CrossRef]
- Bezzecchi, E.; Ronzio, M.; Dolfini, D.; Mantovani, R. NF-YA overexpression in lung cancer: LUSC. Genes 2019, 10, 937. [Google Scholar] [CrossRef] [Green Version]
- Bezzecchi, E.; Ronzio, M.; Semeghini, V.; Andrioletti, V.; Mantovani, R.; Dolfini, D. NF-YA Overexpression in Lung Cancer: LUAD. Genes 2020, 11, 198. [Google Scholar] [CrossRef] [Green Version]
- Inghirami, G.; Chiarle, R.; Simmons, W.J.; Piva, R.; Schlessinger, K.; Levy, D.E. New and old functions of STAT3: A pivotal target for individualized treatment of cancer. Cell Cycle 2005, 4, 1131–1133. [Google Scholar] [CrossRef]
- Maritano, D.; Sugrue, M.L.; Tininini, S.; Dewilde, S.; Strobl, B.; Fu, X.P.; Murray-Tait, V.; Chiarle, R.; Poli, V. The STAT3 isoforms α and β have unique and specific functions. Nat. Immunol. 2004, 5, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Ng, I.H.W.; Ng, D.C.H.; Jans, D.A.; Bogoyevitch, M.A. Selective STAT3-α or -β expression reveals spliceform-specific phosphorylation kinetics, nuclear retention and distinct gene expression outcomes. Biochem. J. 2012, 447, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Park, O.K.; Schaefer, L.K.; Wang, W.; Schaefer, T.S. Dimer stability as a determinant of differential DNA binding activity of Stat3 isoforms. J. Biol. Chem. 2000, 275, 32244–32249. [Google Scholar] [CrossRef] [Green Version]
- Aziz, M.H.; Hafeez, B.B.; Sand, J.M.; Pierce, D.B.; Aziz, S.W.; Dreckschmidt, N.E.; Verma, A.K. Protein kinase C mediates Stat3Ser727 phosphorylation, Stat3-regulated gene expression, and cell invasion in various human cancer cell lines through integration with MAPK cascade (RAF-1, MEK1/2, and ERK1/2). Oncogene 2010, 29, 3100–3109. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.-Z.; Kong, X.-J.; Banerjee, A.; Muniraj, N.; Pandey, V.; Steiner, M.; Perry, J.K.; Zhu, T.; Liu, D.-X.; Lobie, P.E. STAT3α Is Oncogenic for Endometrial Carcinoma Cells and Mediates the Oncogenic Effects of Autocrine Human Growth Hormone. Endocrinology 2010, 151, 4133–4145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, W.J.; Jiang, H.; Wu, D.L.; Zheng, J.H. Early responses of the STAT3 pathway to platinum drugs are associated with cisplatin resistance in epithelial ovarian cancer. Brazilian J. Med. Biol. Res. 2013, 46, 650–658. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, L.K.; Ren, Z.; Fuller, G.N.; Schaefer, T.S. Constitutive of Stat3α in brain tumors: Localization to tumor endothelial cells and activation by the endothelial tyrosine kinase receptor (VEGFR-2). Oncogene 2002, 21, 2058–2065. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, A.; White, S.M.; Schaefer, T.S.; Ball, E.D.; Dyer, K.F.; Tweardy, D.J. Granulocyte colony-stimulating factor activation of Stat3α and Stat3β in immature normal and leukemic human myeloid cells. Blood 1996, 88, 2442–2449. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, R.L.; Lo, H.W. STAT3 target genes relevant to human cancers. Cancers 2014, 6, 897–925. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, V.N.; Zhou, H.; Partridge, M.A.; Hei, T.K. Inhibition of ataxia telangiectasia mutated kinase activity enhances TRAIL-mediated apoptosis in human melanoma cells. Cancer Res. 2009, 69, 3510–3519. [Google Scholar] [CrossRef] [Green Version]
- Niu, G.; Shain, K.H.; Huang, M.; Ravi, R.; Bedi, A.; Dalton, W.S.; Jove, R.; Yu, H. Overexpression of a Dominant-Negative Signal Transducer and Activator of Transcription 3 Variant in Tumor Cells Leads to Production of SolubleFactors That Induce Apoptosis and Cell Cycle Arrest. Cancer Res. 2001, 61, 3276–3280. [Google Scholar] [PubMed]
- Ivanov, V.N.; Bhoumik, A.; Krasilnikov, M.; Raz, R.; Owen-Schaub, L.B.; Levy, D.; Horvath, C.M.; Ronai, Z. Cooperation between STAT3 and c-Jun suppresses Fas transcription. Mol. Cell 2001, 7, 517–528. [Google Scholar] [CrossRef] [Green Version]
- Aigner, P.; Mizutani, T.; Horvath, J.; Eder, T.; Heber, S.; Lind, K.; Just, V.; Moll, H.P.; Yeroslaviz, A.; Fischer, M.J.; et al. STAT3b is a tumor suppressor in acute myeloid leukemia. Blood Adv. 2019, 3, 1989–2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weise, A.; Bruser, K.; Elfert, S.; Wallmen, B.; Wittel, Y.; Wöhrle, S.; Hecht, A. Alternative splicing of Tcf7l2 transcripts generates protein variants with differential promoter-binding and transcriptional activation properties at Wnt/β-catenin targets. Nucleic Acids Res. 2009, 38, 1964–1981. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.C.; Min, L.; Chen, H.; Liu, Y.L. Alternative splicing isoform of T cell factor 4K suppresses the proliferation and metastasis of non-small cell lung cancer cells. Genet. Mol. Res. 2015, 14, 14009–14018. [Google Scholar] [CrossRef]
- Tsedensodnom, O.; Koga, H.; Rosenberg, S.A.; Nambotin, S.B.; Carroll, J.J.; Wands, J.R.; Kim, M. Identification of T-cell factor-4 isoforms that contribute to the malignant phenotype of hepatocellular carcinoma cells. Exp. Cell Res. 2011, 317, 920–931. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Song, J.; Wu, Y.; Yao, S.; Xu, G.Z.; Diao, B. Expression and functional analysis of TCF4 isoformsin human glioma cells. Mol. Med. Rep. 2018, 17, 6023–6027. [Google Scholar]
- He, G.; Guan, X.; Chen, X.; Wang, Y.; Luo, C.; Zhang, B. Expression and splice variant analysis of human TCF4 transcription factor in esophageal cancer. J. Cancer 2015, 6, 333–341. [Google Scholar] [CrossRef] [Green Version]
- Valenta, T.; Lukas, J.; Korinek, V. HMG box transcription factor TCF-4’s interaction with CtBP1 controls the expression of the Wnt target Axin2/Conductin in human embryonic kidney cells. Nucleic Acids Res. 2003, 31, 2369–2380. [Google Scholar] [CrossRef]
- Koga, H.; Tsedensodnom, O.; Tomimaru, Y.; Walker, E.J.; Lee, H.C.; Kim, K.M.; Yano, H.; Wands, J.R.; Kim, M. Loss of the SxxSS Motif in a Human T-Cell Factor-4 Isoform Confers Hypoxia Resistance to Liver Cancer: An Oncogenic Switch in Wnt Signaling. PLoS ONE 2012, 7, e39981. [Google Scholar] [CrossRef] [Green Version]
- Kennell, J.A.; O’Leary, E.E.; Gummow, B.M.; Hammer, G.D.; MacDougald, O.A. T-Cell Factor 4N (TCF-4N), a Novel Isoform of Mouse TCF-4, Synergizes with -Catenin To Coactivate C/EBP and Steroidogenic Factor 1 Transcription Factors. Mol. Cell. Biol. 2003, 23, 5366–5375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanouchi, K.; Ohta, T.; Liu, Z.; Oji, Y.; Sugiyama, H.; Shridhar, V.; Matsumura, S.; Takahashi, T.; Takahashi, K.; Kurachi, H. The Wilms’ tumor gene WT1 −17AA/−KTS splice variant increases tumorigenic activity through up-regulation of vascular endothelial growth factor in an in vivo ovarian cancer model. Transl. Oncol. 2014, 7, 580–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Han, Y.; Saiz, S.; Minden, M.D. Review a tumor suppressor and oncogene: The WT1 story. Leukemia 2007, 21, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Hammes, A.; Guo, J.K.; Lutsch, G.; Leheste, J.R.; Landrock, D.; Ziegler, U.; Gubler, M.C.; Schedl, A. Two splice variants of the wilms’ tumor 1 gene have distinct functions during sex determination and nephron formation. Cell 2001, 106, 319–329. [Google Scholar] [CrossRef] [Green Version]
- Baudry, D.; Hamelin, M.; Cabanis, M.-O.; Fournet, J.-C.; Tournade, M.-F.; Sarnacki, S.; Junien, C.; Jeanpierre, C. WT1 Splicing Alterations in Wilms’ Tumors. Clin. Cancer Res. 2000, 6, 3957–3965. [Google Scholar] [PubMed]
- Baudry, D.; Faussillon, M.; Cabanis, M.O.; Rigolet, M.; Zucker, J.M.; Patte, C.; Sarnacki, S.; Boccon-Gibod, L.; Junien, C.; Jeanpierre, C. Changes in WT1 splicing are associated with a specific gene expression profile in Wilms’ tumour. Oncogene 2002, 21, 5566–5573. [Google Scholar] [CrossRef] [Green Version]
- Kramarzova, K.; Stuchly, J.; Willasch, A.; Gruhn, B.; Schwarz, J.; Cermak, J.; MacHova-Polakova, K.; Fuchs, O.; Stary, J.; Trka, J.; et al. Real-time PCR quantification of major Wilms tumor gene 1 (WT1) isoforms in acute myeloid leukemia, their characteristic expression patterns and possible functional consequences. Leukemia 2012, 26, 2086–2095. [Google Scholar] [CrossRef] [Green Version]
- Siehl, J.M.; Reinwald, M.; Heufelder, K.; Menssen, H.D.; Keilholz, U.; Thiel, E. Expression of Wilms’ tumor gene 1 at different stages of acute myeloid leukemia and analysis of its major splice variants. Ann. Hematol. 2004, 83, 745–750. [Google Scholar] [CrossRef]
- Burwell, E.A.; McCarty, G.P.; Simpson, L.A.; Thompson, K.A.; Loeb, D.M. Isoforms of Wilms’ tumor suppressor gene (WT1) have distinct effects on mammary epithelial cells. Oncogene 2007, 26, 3423–3430. [Google Scholar] [CrossRef]
- Loeb, D.M.; Evron, E.; Patel, C.B.; Sharma, P.M.; Niranjan, B.; Buluwela, L.; Weitzman, S.A.; Korz, D.; Sukumar, S. Wilms’ tumor suppressor gene (WT1) is expressed in primary breast tumors despite tumor-specific promoter methylation. Cancer Res. 2001, 61, 921–925. [Google Scholar]
- Potthoff, M.J.; Olson, E.N. MEF2: A central regulator of diverse developmental programs. Development 2007, 134, 4131–4140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Giorgio, E.; Hancock, W.W.; Brancolini, C. MEF2 and the tumorigenic process, hic sunt leones. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Infantino, V.; Convertini, P.; Menga, A.; Iacobazzi, V. MEF2C exon α: Role in gene activation and differentiation. Gene 2013, 531, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, S.; Faralli, H.; Yao, Z.; Rakopoulos, P.; Palii, C.; Cao, Y.; Singh, K.; Liu, Q.C.; Chu, A.; Aziz, A.; et al. Tissue-specific splicing of a ubiquitously expressed transcription factor is essential for muscle differentiation. Genes Dev. 2013, 27, 1247–1259. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Zhu, B.; Davie, J. Alternative splicing of MEF2C pre-mRNA controls its activity in normal myogenesis and promotes tumorigenicity in rhabdomyosarcoma cells. J. Biol. Chem. 2015, 290, 310–324. [Google Scholar] [CrossRef] [Green Version]
- Baruffaldi, F.; Montarras, D.; Basile, V.; de Feo, L.; Badodi, S.; Ganassi, M.; Battini, R.; Nicoletti, C.; Imbriano, C.; Musarò, A.; et al. Dynamic phosphorylation of the myocyte enhancer factor 2Cα1 splice variant promotes skeletal muscle regeneration and hypertrophy. Stem Cells 2017, 35, 725–738. [Google Scholar] [CrossRef]
- Goding, C.R. Mitf from neural crest to melanoma: Signal transduction and transcription in the melanocyte lineage. Genes Dev. 2000, 14, 1712–1728. [Google Scholar]
- Bismuth, K.; Maric, D.; Arnheiter, H. MITF and cell proliferation: The role of alternative splice forms. Pigment Cell Res. 2005, 18, 349–359. [Google Scholar] [CrossRef] [Green Version]
- Salti, G.I.; Manougian, T.; Farolan, M.; Shilkaitis, A.; Majumdar, D.; Das Gupta, T.K. Micropthalmia transcription factor: A new prognostic marker in intermediate-thickness cutaneous malignant melanoma. Cancer Res. 2000, 60, 5012–5016. [Google Scholar]
- Pogenberg, V.; Ögmundsdóttir, M.H.; Bergsteinsdóttir, K.; Schepsky, A.; Phung, B.; Deineko, V.; Milewski, M.; Steingrímsson, E.; Wilmanns, M. Restricted leucine zipper dimerization and specificity of DNA recognition of the melanocyte master regulator MITF. Genes Dev. 2012, 26, 2647–2658. [Google Scholar] [CrossRef] [Green Version]
- Carreira, S.; Goodall, J.; Aksan, I.; La Rocca, S.A.; Galibert, M.D.; Denat, L.; Larue, L.; Goding, C.R. Mitf cooperates with Rb1 and activates p21 Cip1 expression to regulate cell cycle progression. Nature 2005, 433, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Loercher, A.E.; Tank, E.M.H.; Delston, R.B.; Harbour, J.W. MITF links differentiation with cell cycle arrest in melanocytes by transcriptional activation of INK4A. J. Cell Biol. 2005, 168, 35–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Primot, A.; Mogha, A.; Corre, S.; Roberts, K.; Debbache, J.; Adamski, H.; Dreno, B.; Khammari, A.; Lesimple, T.; Mereau, A.; et al. ERK-regulated differential expression of the Mitf 6a/b splicing isoforms in melanoma. Pigment Cell Melanoma Res. 2010, 23, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Rengarajan, J.; Mittelstadt, P.R.; Mages, H.W.; Gerth, A.J.; Kroczek, R.A.; Ashwell, J.D.; Glimcher, L.H. Sequential involvement of NFAT and Egr transcription factors in FasL regulation. Immunity 2000, 12, 293–300. [Google Scholar] [CrossRef] [Green Version]
- Caetano, M.S.; Vieira-de-Abreu, A.; Teixeira, L.K.; Werneck, M.B.F.; Barcinski, M.A.; Viola, J.P.B. NFATC2 transcription factor regulates cell cycle progression during lymphocyte activation: Evidence of its involvement in the control of cyclin gene expression. FASEB J. 2002, 16, 1940–1942. [Google Scholar] [CrossRef]
- Ranger, A.M.; Gerstenfeld, L.C.; Wang, J.; Kon, T.; Bae, H.; Gravallese, E.M.; Glimcher, M.J.; Glimcher, L.H. The nuclear factor of activated T cells (NFAT) transcription factor NFATp (NFATc2) is a repressor of chondrogenesis. J. Exp. Med. 2000, 191, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Chuvpilo, S.; Jankevics, E.; Tyrsin, D.; Akimzhanov, A.; Moroz, D.; Jha, M.K.; Schulze-Luehrmann, J.; Santner-Nanan, B.; Feoktistova, E.; König, T.; et al. Autoregulation of NFATc1/A expression facilitates effector T cells to escape from rapid apoptosis. Immunity 2002, 16, 881–895. [Google Scholar] [CrossRef] [Green Version]
- Robbs, B.K.; Cruz, A.L.S.; Werneck, M.B.F.; Mognol, G.P.; Viola, J.P.B. Dual Roles for NFAT Transcription Factor Genes as Oncogenes and Tumor Suppressors. Mol. Cell. Biol. 2008, 28, 7168–7181. [Google Scholar] [CrossRef] [Green Version]
- Levin-Gromiko, U.; Koshelev, V.; Kushnir, P.; Fedida-Metula, S.; Voronov, E.; Fishman, D. Amplified lipid rafts of malignant cells constitute a target for inhibition of aberrantly active NFAT and melanoma tumor growth by the aminobisphosphonate zoledronic acid. Carcinogenesis 2014, 35, 2555–2566. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, M.K.; Deane, N.G.; Zhu, J.; An, H.; Mima, S.; Wang, X.; Padmanabhan, S.; Shi, Z.; Prodduturi, N.; Ciombor, K.K.; et al. Nuclear factor of activated T-cell activity is associated with metastatic capacity in colon cancer. Cancer Res. 2014, 74, 6947–6957. [Google Scholar] [CrossRef] [Green Version]
- Viola, J.P.B.; Carvalho, L.D.S.; Fonseca, B.P.F.; Teixeira, L.K. NFAT transcription factors: From cell cycle to tumor development. Brazilian J. Med. Biol. Res. 2005, 38, 335–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucena, P.I.; Faget, D.V.; Pachulec, E.; Robaina, M.C.; Klumb, C.E.; Robbs, B.K.; Viola, J.P.B. NFAT2 isoforms differentially regulate gene expression, cell death and transformation through alternative N-terminal domains. Mol. Cell. Biol. 2015, 36, 119–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabayashi, T.; Ishimaru, F.; Takata, M.; Kataoka, I.; Nakase, K.; Kozuka, T.; Tanimoto, M. Characterization of the short isoform of Helios overexpressed in patients with T-cell malignancies. Cancer Sci. 2007, 98, 182–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Liu, W.; Li, Y.; Liu, P.; Li, S.; Dou, D.; Wang, Y.; Yang, R.; Xiang, R.; Liu, F. Alternative Splice Variants Modulates Dominant-Negative Function of Helios in T-Cell Leukemia. PLoS ONE 2016, 11, e0163328. [Google Scholar] [CrossRef] [Green Version]
- DiFeo, A.; Martignetti, J.A.; Narla, G. The role of KLF6 and its splice variants in cancer therapy. Drug Resist. Updat. 2009, 12, 1–7. [Google Scholar] [CrossRef]
- Li, D.; Yea, S.; Li, S.; Chen, Z.; Narla, G.; Banck, M.; Laborda, J.; Tan, S.; Friedman, J.M.; Friedman, S.L.; et al. Krüppel-like factor-6 promotes preadipocyte differentiation through histone deacetylase 3-dependent repression of DLK1. J. Biol. Chem. 2005, 280, 26941–26952. [Google Scholar] [CrossRef] [Green Version]
- Ito, G.; Uchiyama, M.; Kondo, M.; Mori, S.; Usami, N.; Maeda, O.; Kawabe, T.; Hasegawa, Y.; Shimokata, K.; Sekido, Y. Krüppel-like factor 6 is frequently down-regulated and induces apoptosis in non-small cell lung cancer cells. Cancer Res. 2004, 64, 3838–3843. [Google Scholar] [CrossRef] [Green Version]
- Narla, G.; Heath, K.E.; Reeves, H.L.; Li, D.; Giono, L.E.; Kimmelman, A.C.; Glucksman, M.J.; Narla, J.; Eng, F.J.; Chan, A.M.; et al. KLF6, a candidate tumor suppressor gene mutated in prostate cancer. Science 2001, 294, 2563–2566. [Google Scholar] [CrossRef]
- Narla, G.; Kremer-Tal, S.; Matsumoto, N.; Zhao, X.; Yao, S.; Kelley, K.; Tarocchi, M.; Friedman, S.L. In vivo regulation of p21 by the Kruppel-like factor 6 tumor-suppressor gene in mouse liver and human hepatocellular carcinoma. Oncogene 2007, 26, 4428–4434. [Google Scholar] [CrossRef] [Green Version]
- Slavin, D.A.; Koritschoner, N.P.; Prieto, C.C.; López-Díaz, F.J.; Chatton, B.; Bocco, J.L. A new role for the Kruppel-like transcription factor KLF6 as an inhibitor of c-Jun proto-oncoprotein function. Oncogene 2004, 23, 8196–8205. [Google Scholar] [CrossRef]
- Benzeno, S.; Narla, G.; Allina, J.; Cheng, G.Z.; Reeves, H.L.; Banck, M.S.; Odin, J.A.; Diehl, J.A.; Germain, D.; Friedman, S.L. Cyclin-dependent kinase inhibition by the KLF6 tumor suppressor protein through interaction with cyclin D1. Cancer Res. 2004, 64, 3885–3891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narla, G.; DiFeo, A.; Reeves, H.L.; Schaid, D.J.; Hirshfeld, J.; Hod, E.; Katz, A.; Isaacs, W.B.; Hebbring, S.; Komiya, A.; et al. A germline DNA polymorphism enhances alternative splicing of the KLF6 tumor suppressor gene and is associated with increased prostate cancer risk. Cancer Res. 2005, 65, 1213–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiFeo, A.; Feld, L.; Rodriguez, E.; Wang, C.; Beer, D.G.; Martignetti, J.A.; Narla, G. A functional role for KLF6-SV1 in lung adenocarcinoma prognosis and chemotherapy response. Cancer Res. 2008, 68, 965–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narla, G.; Difeo, A.; Yao, S.; Banno, A.; Hod, E.; Reeves, H.L.; Qiao, R.F.; Camacho-Vanegas, O.; Levine, A.; Kirschenbaum, A.; et al. Targeted inhibition of the KLF6 splice variant, KLF6 SV1, suppresses prostate cancer cell growth and spread. Cancer Res. 2005, 65, 5761–5768. [Google Scholar] [CrossRef] [Green Version]
- Difeo, A.; Huang, F.; Sangodkar, J.; Terzo, E.A.; Leake, D.; Narla, G.; Martignetti, J.A. KLF6-SV1 is a novel antiapoptotic protein that targets the BH3-only protein NOXA for degradation and whose inhibition extends survival in an ovarian cancer model. Cancer Res. 2009, 69, 4733–4741. [Google Scholar] [CrossRef] [Green Version]
- McConnell, B.B.; Yang, V.W. Mammalian Krüppel-Like factors in health and diseases. Physiol. Rev. 2010, 90, 1337–1381. [Google Scholar] [CrossRef]
- Hatami, R.; Sieuwerts, A.M.; Izadmehr, S.; Yao, Z.; Qiao, R.F.; Papa, L.; Look, M.P.; Smid, M.; Ohlssen, J.; Levine, A.C.; et al. KLF6-SV1 Drives Breast Cancer Metastasis and Is Associated with Poor Survival. Sci. Transl. Med. 2013, 5, 169ra12. [Google Scholar] [CrossRef] [Green Version]
- Narla, G.; DiFeo, A.; Fernandez, Y.; Dhanasekaran, S.; Huang, F.; Sangodkar, J.; Hod, E.; Leake, D.; Friedman, S.L.; Hall, S.J.; et al. KLF6-SV1 overexpression accelerates human and mouse prostate cancer progression and metastasis. J. Clin. Investig. 2008, 118, 2711–2721. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, M.; Arechederra, M.; Ávila, M.A.; Berasain, C. Splicing alterations contributing to cancer hallmarks in the liver: Central role of dedifferentiation and genome instability. Transl. Gastroenterol. Hepatol. 2018, 3, 84. [Google Scholar] [CrossRef]
- DiFeo, A.; Narla, G.; Martignetti, J.A. Emerging roles of Kruppel-like factor 6 and Kruppel-like factor 6 splice variant 1 in ovarian cancer progression and treatment. Mt. Sinai J. Med. 2009, 76, 557–566. [Google Scholar] [CrossRef] [Green Version]
- Hilger-Eversheim, K.; Moser, M.; Schorle, H.; Buettner, R. Regulatory roles of AP-2 transcription factors in vertebrate development, apoptosis and cell-cycle control. Gene 2000, 260, 1–12. [Google Scholar] [CrossRef]
- Jean, D.; Gershenwald, J.E.; Huang, S.; Luca, M.; Hudson, M.J.; Tainsky, M.A.; Bar-Eli, M. Loss of AP-2 results in up-regulation of MCAM/MUC18 and an increase in tumor growth and metastasis of human melanoma cells. J. Biol. Chem. 1998, 273, 16501–16508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gershenwald, J.E.; Sumner, W.; Calderone, T.; Wang, Z.; Huang, S.; Bar-Eli, M. Dominant-negative transcription factor AP-2 augments SB-2 melanoma tumor growth in vivo. Oncogene 2001, 20, 3363–3375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buettner, R.; Kannan, P.; Imhof, A.; Bauer, R.; Yim, S.O.; Glockshuber, R.; Van Dyke, M.W.; Tainsky, M.A. An alternatively spliced mRNA from the AP-2 gene encodes a negative regulator of transcriptional activation by AP-2. Mol. Cell. Biol. 1993, 13, 4174–4185. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Lu, D. The Ikaros family of zinc-finger proteins. Acta Pharm. Sin. B 2016, 6, 513–521. [Google Scholar] [CrossRef] [Green Version]
- Vshyukova, V.; Valochnik, A.; Meleshko, A. Expression of aberrantly spliced oncogenic Ikaros isoforms coupled with clonal IKZF1 deletions and chimeric oncogenes in acute lymphoblastic leukemia. Blood Cells Mol. Dis. 2018, 71, 29–38. [Google Scholar] [CrossRef]
- Nakase, K.; Ishimaru, F.; Avitahl, N.; Dansako, H.; Matsuo, K.; Fujii, K.; Sezaki, N.; Nakayama, H.; Yano, T.; Fukuda, S.; et al. Dominant Negative Isoform of the Ikaros Gene in Patients with Adult B-Cell Acute Lymphoblastic Leukemia. Cancer Res. 2000, 60, 4062–4065. [Google Scholar]
- Yagi, T.; Kano, G.; Imamura, T. High frequency of Ikaros isoform 6 expression in acute myelomonocytic and monocytic leukemias: Implications for up-regulation of the antiapoptotic protein Bcl-XL in leukemogenesis. Blood 2011, 99, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.; He, F.; Chen, M.; Hua, L.; Wang, W.; Jiao, S.; Zhou, Z. DNA-binding mechanism of the Hippo pathway transcription factor TEAD4. Oncogene 2017, 36, 4362–4369. [Google Scholar] [CrossRef]
- Tang, J.-Y.; Yu, C.-Y.; Bao, Y.-J.; Chen, L.; Chen, J.; Yang, S.-L.; Chen, H.-Y.; Hong, J.; Fang, J.-Y. Cell Cycle TEAD4 promotes colorectal tumorigenesis via transcriptionally targeting YAP1 TEAD4 promotes colorectal tumorigenesis via transcriptionally targeting YAP1. Cell Cycle 2018, 17, 102–109. [Google Scholar] [CrossRef] [Green Version]
- Huh, H.D.; Kim, D.H.; Jeong, H.-S.; Park, H.W. Regulation of TEAD Transcription Factors in Cancer Biology. Cells 2019, 8, 600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Ren, F.; Zhang, Q.; Chen, Y.; Wang, B.; Jiang, J. The TEAD/TEF family of transcription factor Scalloped mediates Hippo signaling in organ size control. Dev. Cell 2008, 14, 377–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Yuan, L.; Sun, Y.; Wang, P.; Zhang, H.; Feng, X.; Wang, Z.; Zhang, W.; Yang, C.; Zeng, Y.A.; et al. Glucocorticoid receptor signaling activates TEAD4 to promote breast cancer progression. Cancer Res. 2019, 79, 4399–4411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.; Zeng, Z.; Wei, H.; Wang, Z. Alternative splicing in cancers: From aberrant regulation to new therapeutics. Semin. Cell Dev. Biol. 2018, 75, 13–22. [Google Scholar] [CrossRef]
- Qi, Y.; Yu, J.; Han, W.; Fan, X.; Qian, H.; Wei, H.; Tsai, Y.H.S.; Zhao, J.; Zhang, W.; Liu, Q.; et al. A splicing isoform of TEAD4 attenuates the Hippo-YAP signalling to inhibit tumour proliferation. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Vieler, M.; Sanyal, S. P53 isoforms and their implications in cancer. Cancers 2018, 10, 288. [Google Scholar] [CrossRef] [Green Version]
- Surget, S.; Khoury, M.P.; Bourdon, J.C. Uncovering the role of p53 splice variants in human malignancy: A clinical perspective. Onco Targets. Ther. 2013, 7, 57–67. [Google Scholar]
- Harms, K.; Nozell, S.; Chen, X. The common and distinct target genes of the p53 family transcription factors. C Cell. Mol. Life Sci 2004, 61, 822–842. [Google Scholar] [CrossRef]
- Maglic, D.; Stovall, D.B.; Cline, J.M.; Fry, E.A.; Mallakin, A.; Taneja, P.; Caudell, D.L.; Willingham, M.C.; Sui, G.; Inoue, K. DMP1β, a splice isoform of the tumour suppressor DMP1 locus, induces proliferation and progression of breast cancer. J. Pathol. 2015, 236, 90–102. [Google Scholar] [CrossRef] [Green Version]
- Tian, N.; Li, J.; Shi, J.; Sui, G. From General Aberrant Alternative Splicing in Cancers and Its Therapeutic Application to the Discovery of an Oncogenic DMTF1 Isoform. Int. J. Mol. Sci. 2017, 18, 191. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Mallakin, A.; Frazier, D.P. Dmp1 and tumor suppression. Oncogene 2007, 26, 4329–4335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Samad, R.; Zalzali, H.; Rammah, C.; Giraud, J.; Naudin, C.; Dupasquier, S.; Poulat, F.; Boizet-Bonhoure, B.; Lumbroso, S.; Mouzat, K.; et al. MiniSOX9, a dominant-negative variant in colon cancer cells. Oncogene 2011, 30, 2493–2503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nestler, E.J. δfosB: A transcriptional regulator of stress and antidepressant responses. Eur. J. Pharmacol. 2015, 753, 66–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luft, F.C. C/EBPβ LIP induces a tumor menagerie making it an oncogene. J. Mol. Med. 2014, 93, 39–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nerlov, C. Transcriptional and translational control of C/EBPs: The case for “deep” genetics to understand physiological function. BioEssays 2010, 32, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.W.; Johnson, P.F.; Hennighausen, L.; Sterneck, E. The C/EBPβ transcription factor regulates epithelial cell proliferation and differentiation in the mammary gland. Genes Dev. 1998, 12, 1907–1916. [Google Scholar] [CrossRef] [Green Version]
- Seagroves, T.N.; Krnacik, S.; Raught, B.; Gay, J.; Burgess-Beusse, B.; Darlington, G.J.; Rosen, J.M. C/EBPβ, but not C/EBPα, is essential for ductal morphogenesis, lobuloalveolar proliferation, and functional differentiation in the mouse mammary gland. Genes Dev. 1998, 12, 1917–1928. [Google Scholar] [CrossRef] [Green Version]
- Screpanti, I.; Romani, L.; Musiani, P.; Modesti, A.; Fattori, E.; Lazzaro, D.; Sellitto, C.; Scarpa, S.; Bellavia, D.; Lattanzio, G. Lymphoproliferative disorder and imbalanced T-helper response in C/EBP beta-deficient mice. EMBO J. 1995, 14, 1932–1941. [Google Scholar] [CrossRef]
- Chen, X.; Lu, J.; Zhao, X.; Chen, C.; Qiao, D.; Wang, H.; Yue, X. Role of C/EBP-β in Methamphetamine-Mediated Microglial Apoptosis. Front. Cell. Neurosci. 2019, 13, 366. [Google Scholar] [CrossRef] [Green Version]
- Kajimura, S.; Seale, P.; Kubota, K.; Lunsford, E.; Frangioni, J.V.; Gygi, S.P.; Spiegelman, B.M. Initiation of myoblast to brown fat switch by a PRDM16-C/EBP-β transcriptional complex. Nature 2009, 460, 1154–1158. [Google Scholar] [CrossRef] [Green Version]
- Zahnow, C.A. CCAAT/enhancer-binding protein β: Its role in breast cancer and associations with receptor tyrosine kinases. Expert Rev. Mol. Med. 2009, 11, e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.-H.; Wang, Y.; Qiao, S.; Wang, M.-J.; Chen, F.; Zi, X.-Y.; Li, J.-X.; Zhang, H.-B.; Yu, B.; Hu, Y.-P. Liver-enriched activator protein 1 as an isoform of CCAAT/enhancer-binding protein beta suppresses stem cell features of hepatocellular carcinoma. Cancer Manag. Res. 2018, 10, 873–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, S.; Ebner, J.; Warren, C.B.; Raam, M.S.; Piliang, M.; Billings, S.D.; Maytin, E. V C/EBP transcription factors in human squamous cell carcinoma: Selective changes in expression of isoforms correlate with the neoplastic state. PLoS ONE 2014, 9, e112073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milde-Langosch, K.; Löning, T.; Bamberger, A.M. Expression of the CCAAT/enhancer-binding proteins C/EBPα, C/EBPβ and C/EBPδ in breast cancer: Correlations with clinicopathologic parameters and cell-cycle regulatory proteins. Breast Cancer Res. Treat. 2003, 79, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Kurzejamska, E.; Johansson, J.; Jirström, K.; Prakash, V.; Ananthaseshan, S.; Boon, L.; Fuxe, J.; Religa, P. C/EBPβ expression is an independent predictor of overall survival in breast cancer patients by MHCII/CD4-dependent mechanism of metastasis formation. Oncogenesis 2014, 3, e125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, T.; Cui, M.; Sun, J.; Ge, C.; Zhao, F.; Zhang, L.; Tian, H.; Zhang, L.; Chen, T.; Jiang, G.; et al. Orosomucoid 2 inhibits tumor metastasis and is upregulated by CCAAT/enhancer binding protein β in hepatocellular carcinomas. Oncotarget 2015, 6, 16106–16119. [Google Scholar] [CrossRef] [Green Version]
- Brown-Bryan, T.A.; Leoh, L.S.; Ganapathy, V.; Pacheco, F.J.; Mediavilla-Varela, M.; Filippova, M.; Linkhart, T.A.; Gijsbers, R.; Debyser, Z.; Casiano, C.A. Alternative splicing and caspase-mediated cleavage generate antagonistic variants of the stress oncoprotein LEDGF/p75. Mol. Cancer Res. 2008, 6, 1293–1307. [Google Scholar] [CrossRef] [Green Version]
- Turlure, F.; Maertens, G.; Rahman, S.; Cherepanov, P.; Engelman, A. A tripartite DNA-binding element, comprised of the nuclear localization signal and two AT-hook motifs, mediates the association of LEDGF/p75 with chromatin in vivo. Nucleic Acids Res. 2006, 34, 1653–1665. [Google Scholar] [CrossRef]
- Pradeepa, M.M.; Sutherland, H.G.; Ule, J.; Grimes, G.R.; Bickmore, W.A. Psip1/Ledgf p52 Binds Methylated Histone H3K36 and Splicing Factors and Contributes to the Regulation of Alternative Splicing. PLoS Genet. 2012, 8, e1002717. [Google Scholar] [CrossRef] [Green Version]
- Basu, A.; Sanchez, T.W.; Casiano, C.A. DFS70/LEDGFp75: An Enigmatic Autoantigen at the Interface between Autoimmunity, AIDS, and Cancer. Front. Immunol. 2015, 6, 116. [Google Scholar] [CrossRef] [Green Version]
- Blokken, J.; De Rijck, J.; Christ, F.; Debyser, Z. Protein–protein and protein–chromatin interactions of LEDGF/p75 as novel drug targets. Drug Discov. Today Technol. 2017, 24, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.P.; Fatma, N.; Kimura, A.; Chylack, L.T.; Shinohara, T. LEDGF binds to heat shock and stress-related element to activate the expression of stress-related genes. Biochem. Biophys. Res. Commun. 2001, 283, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Rojas, H.; Banerjee, H.; Cabrera, I.B.; Perez, K.Y.; De León, M.; Casiano, C.A. Expression of the Stress Response Oncoprotein LEDGF/p75 in Human Cancer: A Study of 21 Tumor Types. PLoS ONE 2012, 7, e30132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, D.P.; Ohguro, N.; Kikuchi, T.; Sueno, T.; Reddy, V.N.; Yuge, K.; Chylack, L.T.; Shinohara, T. Lens epithelium-derived growth factor: Effects on growth and survival of lens epithelial cells, keratinocytes, and fibroblasts. Biochem. Biophys. Res. Commun. 2000, 267, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.S.; Myklebust, L.M.; Kjarland, E.; Gjertsen, B.T.; Pendino, F.; Bruserud, Ø.; Døskeland, S.O.; Lillehaug, J.R. LEDGF/p75 has increased expression in blasts from chemotherapy-resistant human acute myelogenic leukemia patients and protects leukemia cells from apoptosis in vitro. Mol. Cancer 2007, 6, 31. [Google Scholar] [CrossRef] [Green Version]
- Leoh, L.S.; Van Heertum, B.; De Rijck, J.; Filippova, M.; Rios-Colon, L.; Basu, A.; Martinez, S.R.; Tungteakkhun, S.S.; Filippov, V.; Christ, F.; et al. The stress oncoprotein LEDGF/p75 interacts with the methyl CpG binding protein MeCP2 and influences its transcriptional activity. Mol. Cancer Res. 2012, 10, 378–391. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, A.; Cleary, M.L. Menin Critically Links MLL Proteins with LEDGF on Cancer-Associated Target Genes. Cancer Cell 2008, 14, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.K.; Gholamalamdari, O.; Jadaliha, M.; Li, X.L.; Lin, Y.-C.; Zhang, Y.; Guang, S.; Hashemikhabir, S.; Tiwari, S.; Zhu, Y.J.; et al. PSIP1/p75 promotes tumorigenicity in breast cancer cells by promoting the transcription of cell cycle genes. Carcinogenesis 2017, 38, 966–975. [Google Scholar] [CrossRef] [Green Version]
- Daugaard, M.; Kirkegaard-Sørensen, T.; Ostenfeld, M.S.; Aaboe, M.; Høyer-Hansen, M.; Ørntoft, T.F.; Rohde, M.; Jäättelä, M. Lens Epithelium-Derived Growth Factor Is an Hsp70-2 Regulated Guardian of Lysosomal Stability in Human Cancer. Cancer Res. 2007, 67, 2559–2567. [Google Scholar] [CrossRef] [Green Version]
- Bruce, A.W.; Donaldson, I.J.; Wood, I.C.; Yerbury, S.A.; Sadowski, M.I.; Chapman, M.; Gottgens, B.; Buckley, N.J. Genome-wide analysis of repressor element 1 silencing transcription factor/neuron-restrictive silencing factor (REST/NRSF) target genes. Proc. Natl. Acad. Sci. USA 2004, 101, 10458–10463. [Google Scholar] [CrossRef] [Green Version]
- Deng, P.; Zuo, Y.; Feng, S.; Li, Z.; Chen, W.; Li, H.; Wang, X. Knockdown of NRSF inhibits cell proliferation of ovarian cancer via activating Hippo pathway. Life Sci. 2018, 215, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Liu, P.; Kong, L.; Fei, X.; Gao, Y.; Wu, T.; Sun, D.; Tan, X. Identification of RE1-Silencing Transcription Factor as a Promoter of Metastasis in Pancreatic Cancer. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Wagoner, M.P.; Gunsalus, K.T.W.; Schoenike, B.; Richardson, A.L.; Friedl, A.; Roopra, A. The transcription factor REST is lost in aggressive breast cancer. PLoS Genet. 2010, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumder, S. REST in good times and bad: Roles in tumor suppressor and oncogenic activities. Cell Cycle 2006, 5, 1929–1935. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, Z.; Tang, X.; Zeng, L.; Fan, X.; Li, Z. Molecular mechanisms and potential prognostic effects of REST and REST4 in glioma (Review). Mol. Med. Rep. 2017, 16, 3707–3712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coulson, J.M.; Edgson, J.L.; Woll, P.J.; Quinn, J.P. A splice variant of the neuron-restrictive silencer factor repressor is expressed in small cell lung cancer: A potential role in derepression of neuroendocrine genes and a useful clinical marker. Cancer Res. 2000, 60, 1840–1844. [Google Scholar]
- Chen, G.-L.; Miller, G.M. Alternative REST Splicing Underappreciated. eNeuro 2018, 5. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.-L.; Miller, G.M. Extensive Alternative Splicing of the Repressor Element Silencing Transcription Factor Linked to Cancer. PLoS ONE 2013, 8, e62217. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.R.; Che, N.; Lovnicki, J.M.; Dong, X. Development of neuroendocrine prostate cancers by the Ser/Arg repetitive matrix 4-mediated RNA splicing network. Front. Oncol. 2018, 8, 93. [Google Scholar] [CrossRef] [Green Version]
- Palm, K.; Metsis, M.; Timmusk, T. Neuron-specific splicing of zinc finger transcription factor REST/NRSF/XBR is frequent in neuroblastomas and conserved in human, mouse and rat. Mol. Brain Res. 1999, 72, 30–39. [Google Scholar] [CrossRef]
- Morikawa, M.; Koinuma, D.; Miyazono, K.; Heldin, C.H. Genome-wide mechanisms of Smad binding. Oncogene 2013, 32, 1609–1615. [Google Scholar] [CrossRef] [PubMed]
- Ullah, I.; Sun, W.; Tang, L.; Feng, J. Roles of SmAds family and alternative splicing variants of Smad4 in different cancers. J. Cancer 2018, 9, 4018–4028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Z.; Wu, C.-J.; Chu, G.C.; Xiao, Y.; Ho, D.; Zhang, J.; Perry, S.R.; Labrot, E.S.; Wu, X.; Lis, R.; et al. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature 2011, 470, 269–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haeger, S.M.; Thompson, J.J.; Kalra, S.; Cleaver, T.G.; Merrick, D.; Wang, X.-J.; Malkoski, S.P. Smad4 loss promotes lung cancer formation but increases sensitivity to DNA topoisomerase inhibitors. Oncogene 2016, 35, 577–586. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Mishra, L.; Deng, C.-X. The role of TGF-β/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Wang, Y.; Zeng, Z.; Qiao, L.; Zhuang, L.; Gao, Q.; Ma, D.; Huang, X. Smad4 deletion in blood vessel endothelial cells promotes ovarian cancer metastasis. Int. J. Oncol. 2017, 50, 1693–1700. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Mao, C.; Teng, Y.; Li, W.; Zhang, J.; Cheng, X.; Li, X.; Han, X.; Xia, Z.; Deng, H.; et al. Targeted disruption of Smad4 in mouse epidermis results in failure of hair follicle cycling and formation of skin tumors. Cancer Res. 2005, 65, 8671–8678. [Google Scholar] [CrossRef] [Green Version]
- Pierreux, C.E.; Nicolas, F.J.; Hill, C.S. Transforming Growth Factor beta -Independent Shuttling of Smad4 between the Cytoplasm and Nucleus. Mol. Cell. Biol. 2000, 20, 9041–9054. [Google Scholar] [CrossRef] [Green Version]
- Lazzereschi, D.; Nardi, F.; Turco, A.; Ottini, L.; D’Amico, C.; Mariani-Costantini, R.; Gulino, A.; Coppa, A. A complex pattern of mutations and abnormal splicing of Smad4 is present in thyroid tumours. Oncogene 2005, 24, 5344–5354. [Google Scholar] [CrossRef] [Green Version]
- Kageyama, H.; Seki, N.; Yamada, S.; Sakiyama, S.; Nakagawara, A. DPC4 splice variants in neuroblastoma. Cancer Lett. 1998, 122, 187–193. [Google Scholar] [CrossRef]
- De Winter, J.P.; Roelen, B.A.J.; Ten Dijke, P.; Van Der Burg, B.; Van Den Eijnden-Van Raaij, A.J.M. DPC4 (SMAD4) mediates transforming growth factar-β1 (TGF-β1) induced growth inhibition and transcriptional response in breast tumour cells. Oncogene 1997, 14, 1891–1899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chantalat, E.; Boudou, F.; Laurell, H.; Palierne, G.; Houtman, R.; Melchers, D.; Rochaix, P.; Filleron, T.; Stella, A.; Burlet-Schiltz, O.; et al. The AF-1-deficient estrogen receptor ERα46 isoform is frequently expressed in human breast tumors. Breast Cancer Res. 2016, 18, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herynk, M.H.; Fuqua, S.A.W. Estrogen receptor mutations in human disease. Endocr. Rev. 2004, 25, 869–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Ren, J.; Wei, J.; Chong, C.C.N.; Yang, D.; He, Y.; Chen, G.G.; Lai, P.B.S. Alternative splicing of estrogen receptor alpha in hepatocellular carcinoma. BMC Cancer 2016, 16, 926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa, E.; Colantoni, A.; Cammà, C.; Grottola, A.; Buttafoco, P.; Gelmini, R.; Ferretti, I.; Manenti, F. Estrogen receptor classification for hepatocellular carcinoma: Comparison with clinical staging systems. J. Clin. Oncol. 2003, 21, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Bushweller, J.H. Targeting transcription factors in cancer—From undruggable to reality. Nat. Rev. Cancer 2019, 19, 611–624. [Google Scholar] [CrossRef]
- Lambert, M.; Jambon, S.; Depauw, S.; David-Cordonnier, M.H. Targeting transcription factors for cancer treatment. Molecules 2018, 23, 1479. [Google Scholar] [CrossRef] [Green Version]
- Brinkman, B.M.N. Splice variants as cancer biomarkers. Clin. Biochem. 2004, 37, 584–594. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Wang, H.; Chen, Y.; Zhu, Y.; Silberstein, J.L.; Taylor, M.N.; Maughan, B.L.; Denmeade, S.R.; et al. Clinical significance of androgen receptor splice variant-7 mRNA detection in circulating tumor cells of men with metastatic castration-resistant prostate cancer treated with first & second-line Abiraterone & Enzalutamide. J. Clin. Oncol. 2017, 35, 2149–2156. [Google Scholar]
- Wang, Y.; Bernhardy, A.J.; Cruz, C.; Krais, J.J.; Nacson, J.; Nicolas, E.; Peri, S.; Van Der Gulden, H.; Van Der Heijden, I.; O’Brien, S.W.; et al. The BRCA1-Δ11q alternative splice isoform bypasses germline mutations and promotes therapeutic resistance to PARP inhibition and cisplatin. Cancer Res. 2016, 76, 2778–2790. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.S.; Chan, K.Y.K.; Leung, R.C.Y.; Law, H.K.W.; Leung, T.W.; Ngan, H.Y.S. Enhancement of the radiosensitivity of cervical cancer cells by overexpressing p73α. Mol. Cancer Ther. 2006, 5, 1209–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeifer, D.; Wallin, Å.; Holmlund, B.; Sun, X.F. Protein expression following γ-irradiation relevant to growth arrest and apoptosis in colon cancer cells. J. Cancer Res. Clin. Oncol. 2009, 135, 1583–1592. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Schleithoff, E.S.; Stremmel, W.; Melino, G.; Krammer, P.H.; Schilling, T. One, two, three-p53, p63, p73 and chemosensitivity. Drug Resist. Updat. 2006, 9, 288–306. [Google Scholar] [CrossRef] [PubMed]
- Havens, M.A.; Hastings, M.L. SURVEY AND SUMMARY Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TF | DNA Binding Motif | Physiological Regulated Processes | Splice Variants | TP Splice Variants | AS Domains in TP Splice Variants | Ref. |
|---|---|---|---|---|---|---|
| NF-YA | CCAAT | Cell proliferation and differentiation, metabolism, cell death | NF-YAs NF-YAl | NF-YAs | TAD | [26,27,28,29] |
| STAT3 | TTCC(G=C)GGAA | Cell proliferation, cell death | STAT3α STAT3β | STAT3α | TAD | [31,32,33,34,35,36,37,38,39,40,41,42,43] |
| TCF4 | (A/T)(A/T)CAAAG | Cell proliferation, apoptosis | TCF4 TCF4N/E/M/B/S/K/J | TCF4J | SxxSS motif | [44,45,46,47,48,49,50,51] |
| WT1 | GCGTGGGAGT | Cell proliferation and differentiation, metabolism, apoptosis | -17AA/-KTS -17AA/+KTS +17AA/-KTS +17AA/+KTS | -17AA* +17AA* +17AA/+KTS | TAD DBD | [54,55,56,57,58,59,60] |
| TF | DNA Binding Motif | Physiological Regulated Processes | Splice Variants | TP Splice Variants | AS Domains in TP Splice Variants | Ref. |
|---|---|---|---|---|---|---|
| MEF2C | YTA(A/T)4TAR | Muscle cell proliferation and differentiation | Mef2Cα1 Mef2Cα2 | Mef2Cα1 | Adjacent to DBD | [65] |
| MITF | TCATGTGCT | Melanocyte proliferation and differentiation | (+) MITF (−) MITF | (−) MITF | Adjacent to DBD | [68,69,70,71,72,73] |
| NFAT2 | (A/T)GGAAA | Cell proliferation and differentiation, apoptosis, inflammatory response | NFAT2α NFAT2β | NFAT2α | TAD | [78,82] |
| TF | DNA Binding Motif | Physiological Regulated Processes | Splice Variants | TP Splice Variants | AS Domains in TP Splice Variants | Ref. |
|---|---|---|---|---|---|---|
| AP-2α | GCCNNNGGC | Development, cell growth and differentiation, apoptosis | AP-2α AP-2B | AP-2B | Dimerization domain | [103,104] |
| CEBPβ | T(TG)NNGNAA (TG) | Cell cycle, differentiation, apoptosis and senescence | LAP1 LAP2 LIP | LIP | TAD | [131,132,133,134,135,136] |
| DMTF1 | CCCG(G/T)ATGT | Cell proliferation, apoptosis | DMTF1α/β/γ | DMTF1β | TAD DBD | [119,120,121] |
| ERα | AGGTCANNNTGACCT | Cell proliferation, apoptosis, inflammation | ERα (ERα-66) ERαΔ1 (ERα-46) ERαΔ2/Δ3/Δ4/ Δ5/Δ7 ERα−36 ERα−30 | ERα-66 ERα−36 ERαΔ3/Δ5/Δ7 | TAD DBD LBD | [172,173,174,175] |
| FOSB | TGAC/GTCA | Cell proliferation and differentiation, apoptosis, stress response | FOSB ΔFOSB | FOSB | TAD Degron domain | [123] |
| HELIOS | GGGAA | T-lineage differentiation | HELIOS HELIOS-V1/V2/V3 | HELIOS-V1/V2/V3 | NLS DBD | [84] |
| IKAROS | GGAAA | Hematopoiesis, myelopoiesis, lymphopoiesis | IK1-10 IKX | IK4-10 | DBD | [105,106,107,108] |
| KLF6 | GC box CACC box | Cell proliferation and differentiation, adhesion, tissue repair | KLF6 KLF6-SV1/SV2/SV3 | KLF6-SV1 | DBD | [88,92,93,94,95,96,97,98,99,100] |
| LEDGF | NGAAN T/AGGGG | Neuroepithelial stem cell differentiation and neurogenesis, stress-induced apoptosis, lens epithelial cell growth and differentiation, host-virus interaction | LEDGF/p52 LEDGF/p75 | LEDGF/p75 | IBD CTD | [140,143,144,145,146,147,148,149] |
| p53/p63/p73 | RRRC(A/T)(A/T)GYYY | Cell cycle arrest, cell death, genome stability, cell differentiation, development | p53α/β/γ Δ40/Δ133/ Δ160 p53α/β/γ TA/ΔN p63 TA/ΔN p73 | Δ40p53α Δ133p53α Δ160p53α ΔN p63/p73 | TAD DBD | [116,117,118] |
| REST/NRSF | NT(T/C)AG(A/C)(A/G)CCNN(A/G)G(A/C)(G/S)AG | Cell differentiation | REST REST1/4/5 REST-N50/N62 REST-5FΔ | REST4 REST-N50 REST-N62 | DBD NLS | [153,155,156,157,158,159,160] |
| SMAD4 | GTCTAGAC | Cell proliferation and differentiation | SMAD4 SMAD4 Δ3/ Δ4/Δ5-6/Δ6/ Δ4-6/Δ4-7 | SMAD4 Δ3/ Δ4/Δ5-6/ Δ6/Δ4-6/Δ4-7 | Linker domain TAD | [168,169,170,171] |
| SOX9 | (A/T)(A/T)CAA(A/T)G | Stem cell maintenance and commitment, differentiation, matrix deposition | SOX9 MiniSOX9 | MiniSOX9 | TAD | [122] |
| TEAD4 | CATTCCA | Cell proliferation and differentiation, apoptosis | TEAD-FL TEAD-S | TEAD-FL | DBD | [111,113,114,115] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belluti, S.; Rigillo, G.; Imbriano, C. Transcription Factors in Cancer: When Alternative Splicing Determines Opposite Cell Fates. Cells 2020, 9, 760. https://doi.org/10.3390/cells9030760
Belluti S, Rigillo G, Imbriano C. Transcription Factors in Cancer: When Alternative Splicing Determines Opposite Cell Fates. Cells. 2020; 9(3):760. https://doi.org/10.3390/cells9030760
Chicago/Turabian StyleBelluti, Silvia, Giovanna Rigillo, and Carol Imbriano. 2020. "Transcription Factors in Cancer: When Alternative Splicing Determines Opposite Cell Fates" Cells 9, no. 3: 760. https://doi.org/10.3390/cells9030760
APA StyleBelluti, S., Rigillo, G., & Imbriano, C. (2020). Transcription Factors in Cancer: When Alternative Splicing Determines Opposite Cell Fates. Cells, 9(3), 760. https://doi.org/10.3390/cells9030760